Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly
aggressive malignant disease, and is ranked as the fourth leading
cause of cancer-related mortality with a median survival of 6
months (1). Despite rapid advances
in diagnostic and surgical procedures, PDAC remains one of the most
difficult human malignancies to treat. Thus, identification of key
molecules or pathways specifically expressed in PDAC that are
essential for the growth and survival of cancer cells may provide
novel therapeutic targets and ultimately lead to improved
survival.
MicroRNAs (miRNAs) are a class of small, non-coding
RNAs that play important roles in various biological processes
(2). miRNAs have been discovered as
naturally occurring non-coding RNAs, that control gene expression
via specific sites at the 3′-UTR of target mRNAs, causing
translational repression or degradation (3,4).
Recent evidence has shown that miRNA mutations or mis-expression is
correlated with various human cancers and indicates that miRNAs can
function as tumor suppressors or oncogenes. miR-183 is one member
of the miR-182-183 miRNA cluster located in the 7q31-34 locus
(5). Overexpression of miR-183 has
been noted in human colorectal cancer, and the miR-183-96-182
cluster is frequently amplified in melanoma (6,7).
However, miR-183 was identified as a potential metastasis-inhibitor
in lung cancer cells (8). These
data suggest that the effect of miR-183 as an oncogene is cell-type
dependent.
Although miR-183 and its target have been widely
explored as cancer-related targets for tumors, there is no
information available concerning their relevance in PDAC. The aim
of the present study was to examine the role of miR-183 in PDAC and
to investigate the potential mechanisms involved in miR-183
function in PDAC.
Materials and methods
Clinical samples and cell lines
PDAC tissues were obtained from the Department of
Hepatobiliary Surgery, Xijing Hospital, The Fourth Military Medical
University (Xi’an, China), between January 2003 and September 2008.
Specimens were obtained from patients who had not received
preoperative treatments such as chemotherapy. This study was
approved by the Ethics Committee of The Fourth Military Medical
University and conformed to the ethical guidelines of the 2004
Declaration of Helsinki. Written informed consent was obtained from
each patient or from his/her legal guardians. Before the study was
initiated, histopathological examinations were performed to confirm
that there were enough cancer cells in the tumor samples and that
no cancer cells had contaminated the non-cancerous tissues. Tissues
were snap frozen in liquid nitrogen after surgical resection until
use. The human pancreatic non-tumor cell line (HPDE6c7) and human
PDAC cell lines (ASPC-1, SW1990, BXPC-3, CFPAC-1 and PANC-1) were
cultivated in DMEM supplemented with 10% fetal calf serum (Sigma
Chemical Co., St. Louis, MO, USA). Primary antibodies against
Bim-1, cyclin D1, Cdk2, Cdk4 and GAPDH were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). All secondary antibodies
were obtained from Pierce (Rockford, IL, USA). Bim-1 small
interfering RNA (siRNA) and siRNA controls were obtained from Santa
Cruz Biotechnology. Lipofectamine 2000 was purchased from
Invitrogen (Carlsbad, CA, USA). All other chemicals and solutions
were purchased from Sigma-Aldrich unless otherwise indicated.
Cell transfection
miR-183 mimics, inhibitors (miR-183-AS) and their
respective negative controls (NC) were obtained from GenePharma
Company (Shanghai, China). The day before transfection, cells were
seeded in antibiotic-free medium. Transfections were carried out
using Lipofectamine 2000 (Invitrogen, USA) in accordance with the
manufacturer’s instructions. To monitor transfection efficiency,
fluorescein (FAM) siRNA (GenePharma) was used as control.
Successfully transfected cells were observed with a fluorescence
microscope.
siRNA cell transfection
According to the protocol supplied with the
Lipofectamine 2000, the cells were transfected with either siRNA or
control siRNA. siRNA-transfected cells were seeded into 6-well cell
culture plates at a density of 1×105 cells/well. The
cells were allowed to grow for an additional 24 h and were then
harvested for further analysis.
Real-time PCR
Total RNA, including miRNAs, was isolated from
prepared liver samples or cells with TRIzol reagent (Invitrogen)
according to the manufacturer’s instructions. Expression of
hsa-miR-183 was analyzed with the miScript System (Qiagen, USA),
which consists of the miScript Reverse Transcription kit, miScript
Primer assays and miScript SYBR-Green PCR kit, according to the
protocol provided by the company. Small nuclear RNA U6 was used for
normalization. For the analysis of B-cell-specific Moloney murine
leukemia virus insertion site 1 (Bmi-1) expression, cDNA was
synthesized using Moloney murine leukaemia virus reverse
transcriptase (Epicentre, Paris, France) as described by the
manufacturer. The housekeeping gene, glyceraldehyde 3-phosphate
dehydrogenase (GAPDH), was used for normalization. The forward
primer for Bmi-1 was 5′-GCTTCAAGATGGCCGCTTG-3′ and the reverse
primer was 5′-TTCTCGTTGTTCGATGCATTTC-3′. The forward primer for
GAPDH was 5′-GCACCGTCAAGGCTGAGAAC-3′ and the reverse primer was
5′-TGGTGAAGACGCCAGTGGA-3′. Real-time PCR was run on the ABI Prism
7700 Sequence Detector (Applied Biosystems, USA). All of the
reactions were run in triplicate. The ΔΔCt method was
used for relative quantification of gene expression to determine
miR-183 and Bmi-1 mRNA expression levels.
Protein extraction and western
blotting
The cells were lysed in lysis buffer [50 mmol/l Tris
(pH 7.5), 100 mmol/l NaCl, 1 mmol/l EDTA, 0.5% NP40, 0.5% Triton
X-100, 2.5 mmol/l sodium orthovanadate, 10 μl/ml protease inhibitor
cocktail and 1 mmol/l PMSF] by incubating for 20 min at 4°C. The
protein concentration was determined using the Bio-Rad assay system
(Bio-Rad, Hercules, CA, USA). Total proteins were fractionated
using SDS-PAGE and transferred onto nitrocellulose membranes. The
membranes were blocked with 5% non-fat dried milk or bovine serum
albumin in 1X TBS buffer containing 0.1% Tween-20 and then
incubated with the appropriate primary antibodies. Horseradish
peroxidase-conjugated anti-rabbit or anti-mouse IgG was used as the
secondary antibody, and the protein bands were detected using the
enhanced chemiluminescence detection system (Amersham Pharmacia
Biotech). Quantification of the western blot analyses was performed
using laser densitometry, and relative protein expression was then
normalized to GAPDH levels.
Colony formation assay
Approximately 3×102 cells from each
treated cell line were plated in 6-well dishes. After 2 weeks,
cells were fixed with 20% methanol and stained with 1% crystal
violet. Colonies consisting of >50 cells were counted per well,
and each experiment was performed in triplicate.
Analysis of cell cycle
Both cell cycle distribution and spontaneous
apoptosis events were detected using a FACSCalibur II sorter and
Cell Quest FACS system (BD Biosciences, San Jose, CA, USA). To
analyze cell cycle distribution, cells were synchronized using
serum starvation for 24 h and stimulated with complete medium for
24 h before being harvested. Cells were fixed with 70% ethanol at
4°C overnight, washed twice with phosphate-buffered saline (PBS),
and resuspended in staining solution (50 lg/ml propidium iodide, 1
mg/ml RNase A, 0.1% Triton X-100 in PBS) for 30 min at 37°C in the
dark.
Cell growth assays
Cell growth was assessed using
3-(4,5-dimethylthiazol-2-y1)-2,5-diphenyltetrazolium bromide (MTT;
Sigma) assays. Briefly, 2,000 cells/well were seeded into 96-well
plates, and cell viability was assayed on days 1–4 following
seeding. Absorption values were determined using an enzyme linked
immunosorbent assay reader (Dasit, Milan, Italy) at 490 nm.
Statistical analysis
Correlations between categorical variables were
analyzed using Pearson’s Chi-square, and two-tailed t-tests were
used for continuous variables. Survival curves were plotted using
the Kaplan-Meier method and were compared using the log-rank test.
All statistical analyses were performed using the SPSS software
package (SPSS, Chicago, IL, USA). A P-value <0.05 was considered
to indicate a statistically significant result.
Results
Low expression of miR-183 is associated
with clinical progression of PDAC
To investigate the clinical role of miR-183 during
pancreatic carcinogenesis, we analyzed its expression level in PDAC
and compared it with clinicopathological features of the patients
with PDAC. As shown in Table I, low
expression of miR-183 in PDAC was significantly associated with
tumor grade, metastasis and TNM stage (P<0.05). These results
revealed a correlation between miR-183 expression and PDAC invasion
and proliferation. However, there was no correlation between the
expression of miR-183 and the other clinical features such as
gender and age (P>0.05 for all comparisons). Furthermore,
Kaplan-Meier survival analysis demonstrated that patients harboring
low expression of miR-183 had a significantly reduced overall
survival when compared with patients with a high level of miR-183
expression (P<0.001, log-rank test; Fig. 1). These observations indicate that
low expression of miR-183 is associated with PDAC clinical
progression and may play a negative role in PDAC.
 | Table IAssociation of microRNA-183 expression
and clinicopathologic factors of the PDAC patients. |
Table I
Association of microRNA-183 expression
and clinicopathologic factors of the PDAC patients.
| Tumor
characteristics | n | miR-183 | P-value | χ2 |
|---|
|
|---|
| Low | High |
|---|
| All cases | 91 | 47 | 44 | | |
| Gender |
| Male | 56 | 29 | 27 | 0.974 | 0.001 |
| Female | 35 | 18 | 17 | | |
| Age (years) |
| ≤50 | 43 | 19 | 24 | 0.452 | 0.566 |
| >50 | 48 | 23 | 25 | | |
| Tumor grade
(differentiation) |
| Well | 27 | 22 | 5 | <0.001 | 13.682 |
| Moderately or
poorly | 64 | 25 | 39 | | |
| Metastasis |
| No | 57 | 22 | 35 | 0.001 | 10.407 |
| Yes | 34 | 25 | 9 | | |
| TNM stage |
| I and II | 20 | 1 | 19 | <0.001 | 22.337 |
| III and IV | 71 | 46 | 25 | | |
An inverse correlation exists between
miR-183 and Bmi-1
To address the potential mechanism involved in the
function of miR-183 in PDAC, we first examined the expression of
miR-183 in the pancreatic non-tumor cell line, HPDE6c7, and in the
PDAC cell lines, ASPC-1, SW1990, BXPC-3, CFPAC-1 and PANC-1
(Fig. 2). The results showed that,
in the PDAC cells, the expression of miR-183 was lower than the
level in the non-tumor cells. We also examined the expression of
Bmi-1 in the pancreatic non-tumor cell line and the PDAC cell
lines. The results revealed that, in the PDAC cell lines, the
expression of Bmi-1 was higher than that in the non-tumor cells.
These results indicate that an inverse correlation exists between
miR-183 and Bmi-1.
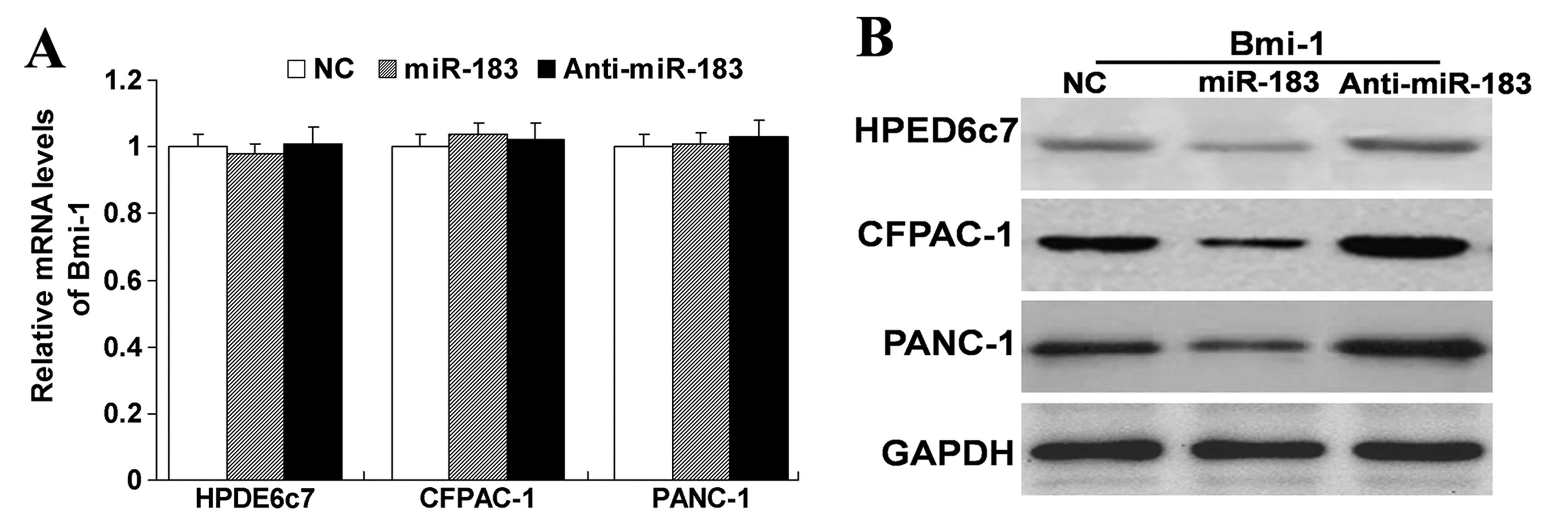
miR-183 regulates the protein expression
of Bmi-1
To further examine the relationship between miR-183
and Bmi-1, we treated the HPED6c7, CFPAC-1 and PANC-1 cells with
miR-183 mimics or inhibitors. As shown in Fig. 3, we found that upregulation of
miR-183 did not affect the mRNA expression of Bmi-1, but did
decrease the protein expression of Bmi-1. In contrast,
downregulation of the expression of miR-183 did not affect the mRNA
expression of Bmi-1, but did increase the protein expression of
Bmi-1. These results indicate that miR-183 has a negative
regulatory effect on Bmi-1. Yet, it was unknown whether this
negative regulation affects the function of cells. Thus, we carry
out the subsequent experiment.
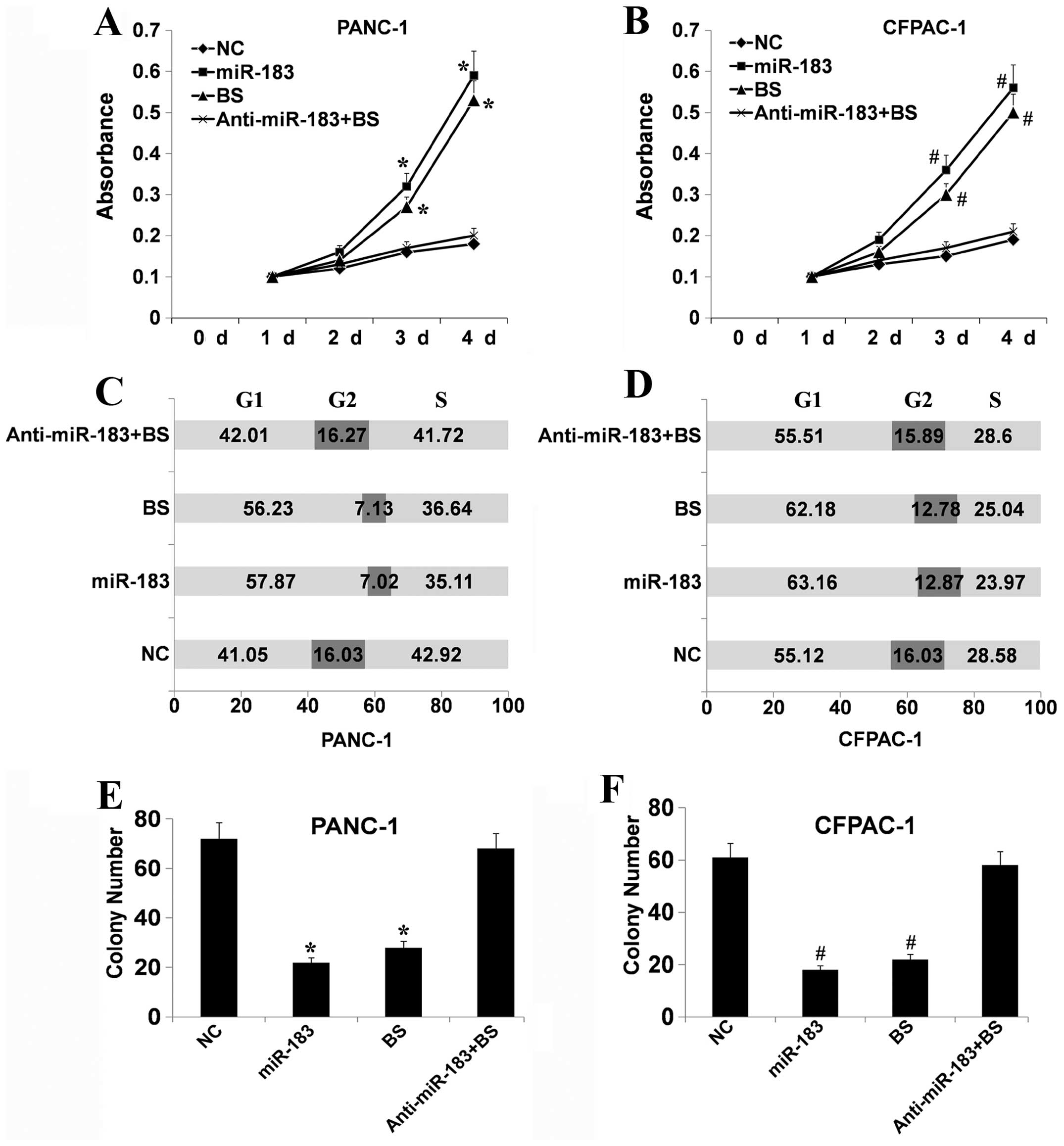
miR-183 regulates the function of cells
via regulating Bmi-1
As known, Bmi-1 regulates the growth and
proliferation of cells. To investigate whether miR-183 regulates
the function of cells via regulation of Bmi-1, PANC-1 and CFPAC-1
cells, two cell lines with a low level of miR-183 and a high level
of Bmi-1 expression, respectively, were treated with miR-183 mimics
or inhibitors and/or Bmi-1 siRNA. Bmi-1 siRNA downregulated the
expression of Bmi-1 (data not shown). We first examined the effect
of the different treatments on cell viability using MTT assays. As
shown in Fig. 4A and B, Bmi-1
depletion and upregulation of miR-183 significantly suppressed the
growth of PANC-1 and CFPAC-1 cells compared with the control cells
(P<0.05). In contrast, downregulation of miR-183 following
downregulation of Bmi-1 had a slight influence on cell viability.
This indicated that miR-183 may participate in cell growth via
regulation of Bmi-1. Flow cytometric analysis showed that the cell
cycle distribution of PANC-1 and CFPAC-1 cells was significantly
affected following Bmi-1 depletion or upregulation of miR-183. As
shown in Fig. 4C and D, there was a
higher proportion of G0–G1 phase cells following Bmi-1 depletion or
upregulation of miR-183 than in the control. A compensatory
decrease in the S and G2/M phase proportions was also detected as
compared with the control in the S and G2/M phases. Yet,
downregulation of miR-183 following silencing of Bmi-1 had a slight
influence. These results suggest that miR-183 can cause Bmi-1
silencing which inhibits the entry of cells into the S phase
therefore suppressing cell growth. Colony formation analysis showed
that following Bmi-1 depletion or upregulation of miR-183, the
PANC-1 and CFPAC-1 cells had a greatly reduced capacity to form
colonies compared with the control cells (Fig. 4E and F). Downregulation of miR-183
following silencing of Bmi-1 had a slight influence. Collectively,
these data showed that a low level of miR-183 expression
effectively suppresses the growth of PDAC cells via the regulation
of Bmi-1. Furthermore, western blotting was performed to explore
the cell cycle regulatory role of miR-183. Following Bmi-1
silencing or upregulation of miR-183, the expression levels of
cyclin D1, cyclin dependent kinase (CDK)2 and CDK4 were decreased
(Fig. 5).
Discussion
The results of the present study revealed that
miR-183 contributes to PDAC cell growth and proliferation via
regulation of Bmi-1. These results elucidate the potential
mechanisms and further confirm the importance and complexity of
miR-183 in PDAC. To the best of our knowledge, this report is the
first to describe the correlation of the miR-183-Bmi-1 pathway with
PDAC cell growth and proliferation.
MicroRNAs (miRNAs) are small RNAs processed from
endogenous transcripts that function to mediate
post-transcriptional silencing of complementary target genes.
miRNAs are a class of small non-coding regulatory RNA molecules,
with a profound impact on various biological processes (2,9,10). It
has been reported that miRNAs are aberrantly expressed in most
types of cancer where they are considered to play significant roles
by regulating the expression of various tumor suppressors and
oncogenes (11,12). miR-183 is a member of an miRNA
family (miR-183, miR-182 and miR-96) that is clustered within 2–4
kb at chromosome 7q32. miRNAs from this locus are dysregulated in a
variety of tumors such as hepatic and colorectal, as well as in
leukemia, lung and breast cancer (6,13–15).
miR-183 family members have been shown to be upregulated in
colorectal and hepatic tumors, as well as in leukemia and breast
cancer (6,13–15).
In contrast, miR-183 has been shown to be downregulated and
inversely correlated with invasive and metastatic abilities in
pulmonary giant cell (8) and breast
cancer (16). These studies
demonstrated that the expression profiling of miR-183 was
tissue-specific and that it may have divergent functions depending
on the tumor tissue or cell type.
Our results indicated that low levels of miR-183
expression in PDAC tissues are correlated with tumor grade,
metastasis and TNM stage, each of which is an indicator of advanced
tumor status. These results strongly suggest that miR-183 plays a
key role in the progression of human PDAC. Kaplan-Meier analysis of
the survival curves from patients in the present study showed a
significantly worse overall survival rate for patients whose tumors
had low miR-183 expression levels (log-rank test, P<0.001),
indicating that low levels of miR-183 may serve as a marker of poor
prognosis for patients with PDAC. To identify the potential role of
miR-183 in PDAC, we compared the miR-183 expression level in a
pancreatic non-tumor cell line and in PDAC cell lines,
respectively. The results showed that, in the PDAC cell lines, the
expression of miR-183 was lower than that in the non-tumor cell
line. These results were similar to the results noted in the PDAC
tissues. This indicates that miR-183 is a tumor suppressor in
PDAC.
Since the potential mechanism of miR-183 as a tumor
suppressor in PDAC was unknown, we explored the potential
mechanism. We focused on B-cell-specific Moloney murine leukemia
virus insertion site 1 (Bmi-1). Bmi-1 has been predicted to be a
common target of miR-183 (17).
Bmi-1, a member of the Polycomb family of proteins, which suppress
the transcription of their target genes via an epigenetic mechanism
(18–20), was originally identified as an
oncogene cooperating with c-Myc in a murine lymphomagenesis model
(21). Subsequent research
identified the essential role of Bmi-1 in embryonic development and
the maintenance of self-renewal of both normal and malignant human
mammary stem cells (22). Bmi-1
also regulates cellular processes including cell cycle progression,
apoptosis and senescence as well as immortalization (23) and induces telomerase activity
(24). In addition, there is
accumulating evidence that Bmi-1 is overexpressed in a variety of
human malignant neoplasms, such as melanoma (25) and HCC (26–28).
Our previous study showed that Bmi-1 was overexpressed in PDAC cell
lines and was associated with an unfavorable prognosis for patients
with PDAC. When Bmi-1 was downregulated, cell growth was suppressed
as a result of cell cycle arrest, and susceptibility to apoptosis
was enhanced (29). Furthermore,
Bmi-1 was found to be involved in tumor development and progression
and is associated with a poor prognosis (30). For example, Bmi-1 expression is
significantly correlated with nodal involvement, distant metastasis
and clinical stage of colon and gastric cancers (31,32).
Overexpression of Bmi-1 has been associated with the invasiveness
of nasopharyngeal carcinomas and was found to be a predictor of
poor survival (33). Inhibition of
Bmi-1 leads to decreased invasion of cervical cancer cells
(34). Taken together, these data
strongly indicate that Bmi-1 contributes to the behavior of cancer
cells. Our study revealed that Bmi-1 expression was inversely
correlated with miR-183. Our findings also demonstrated that Bmi-1
silencing and upregulation of miR-183 had the same effect on
cellular processes including cell cycle progression and growth. In
contrast, downregulation of miR-183 following silencing of Bmi-1
had only a slight influence on cell cycle progression and growth.
These data revealed that low levels of miR-183 expression
effectively suppressed the growth of PDAC cells via regulation of
Bmi-1. Furthermore, western blotting was performed to explore the
cell cycle regulatory role of miR-183. Following Bmi-1 silencing or
upregulation of miR-183, the expression of cyclin D1,
cyclin-dependent kinase (CDK)2 and CDK4 were decreased. It is
reasonable to conclude that alteration of miR-183 expression may
regulate the function of cells by targeting the downregulation of
Bmi-1 expression.
In the present study, we demonstrated that
upregulation of miR-183 has a similar role with inhibition of Bmi-1
in regulating PDAC cellular processes including cell cycle
progression and growth. To the best of our knowledge, this is the
first in vitro study to regulate the progression of PDAC, by
regulation of miR-183 to target the expression of Bmi-1 in PDAC
cells. The findings of this study revealed that by downregulating
the Bmi-1 expression level, miR-183 plays a suppressive role in
cellular processes including cell cycle progression and growth of
PDAC. Our study may provide an important avenue for further
analysis in vivo with the aim to develop a new potential
diagnostic and therapeutic target for the screening and treatment
of PDAC. Further studies are required to fully understand the
regulatory mechanisms of miR-183 and Bmi-1 in PDAC in vitro
and in vivo.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81101820).
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
2
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pillai RS, Bhattacharyya SN, Artus CG, et
al: Inhibition of translational initiation by Let-7 microRNA in
human cells. Science. 309:1573–1576. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zamore PD and Haley B: Ribo-gnome: the big
world of small RNAs. Science. 309:1519–1524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bastian BC, LeBoit PE, Hamm H, Bröcker EB
and Pinkel D: Chromosomal gains and losses in primary cutaneous
melanomas detected by comparative genomic hybridization. Cancer
Res. 58:2170–2175. 1998.PubMed/NCBI
|
|
6
|
Motoyama K, Inoue H, Takatsuno Y, et al:
Over- and under-expressed microRNAs in human colorectal cancer. Int
J Oncol. 34:1069–1075. 2009.PubMed/NCBI
|
|
7
|
Lin WM, Baker AC, Beroukhim R, et al:
Modeling genomic diversity and tumor dependency in malignant
melanoma. Cancer Res. 68:664–673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang G, Mao W and Zheng S: MicroRNA-183
regulates Ezrin expression in lung cancer cells. FEBS Lett.
582:3663–3668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ambros V and Lee RC: Identification of
microRNAs and other tiny noncoding RNAs by cDNA cloning. Methods
Mol Biol. 265:131–158. 2004.PubMed/NCBI
|
|
10
|
Pillai RS, Bhattacharyya SN and Filipowicz
W: Repression of protein synthesis by miRNAs: how many mechanisms?
Trends Cell Biol. 17:118–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caldas C and Brenton JD: Sizing up miRNAs
as cancer genes. Nat Med. 11:712–714. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kefas B, Godlewski J, Comeau L, et al:
microRNA-7 inhibits the epidermal growth factor receptor and the
Akt pathway and is down-regulated in glioblastoma. Cancer Res.
68:3566–3572. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ladeiro Y, Couchy G, Balabaud C, et al:
MicroRNA profiling in hepatocellular tumors is associated with
clinical features and oncogene/tumor suppressor gene mutations.
Hepatology. 47:1955–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Agirre X, Jiménez-Velasco A, San
José-Enériz E, et al: Downregulation of hsa-miR-10a in
chronic myeloid leukemia CD34+ cells increases
USF2-mediated cell growth. Mol Cancer Res. 6:1830–1840. 2008.
|
|
15
|
Mattie MD, Benz CC, Bowers J, et al:
Optimized high-throughput microRNA expression profiling provides
novel biomarker assessment of clinical prostate and breast cancer
biopsies. Mol Cancer. 5:242006. View Article : Google Scholar
|
|
16
|
Lowery AJ, Miller N, Dwyer RM and Kerin
MJ: Dysregulated miR-183 inhibits migration in breast cancer
cells. BMC Cancer. 10:5022010.PubMed/NCBI
|
|
17
|
Wellner U, Schubert J, Burk UC, et al: The
EMT-activator ZEB1 promotes tumorigenicity by repressing
stemness-inhibiting microRNAs. Nat Cell Biol. 11:1487–1495. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jacobs JJ and van Lohuizen M: Polycomb
repression: from cellular memory to cellular proliferation and
cancer. Biochim Biophys Acta. 1602:151–161. 2002.PubMed/NCBI
|
|
19
|
Kondo Y, Shen L, Cheng AS, et al: Gene
silencing in cancer by histone H3 lysine 27 trimethylation
independent of promoter DNA methylation. Nat Genet. 40:741–750.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Raaphorst FM: Deregulated expression of
Polycomb-group oncogenes in human malignant lymphomas and
epithelial tumors. Hum Mol Genet. 14(Spec1): R93–R100. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van Lohuizen M, Verbeek S, Scheijen B,
Wientjens E, van der Gulden H and Berns A: Identification of
cooperating oncogenes in Eμ-myc transgenic mice by provirus
tagging. Cell. 65:737–752. 1991.PubMed/NCBI
|
|
22
|
Liu S, Dontu G, Mantle ID, et al: Hedgehog
signaling and Bmi-1 regulate self-renewal of normal and malignant
human mammary stem cells. Cancer Res. 66:6063–6071. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and Polycomb-group gene
bmi-1 regulates cell proliferation and senescence through
the ink4a locus. Nature. 397:164–168. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dimri GP, Martinez JL, Jacobs JJ, et al:
The Bmi-1 oncogene induces telomerase activity and
immortalizes human mammary epithelial cells. Cancer Res.
62:4736–4745. 2002.
|
|
25
|
Mihic-Probst D, Kuster A, Kilgus S, et al:
Consistent expression of the stem cell renewal factor BMI-1 in
primary and metastatic melanoma. Int J Cancer. 121:1764–1770. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Effendi K, Mori T, Komuta M, Masugi Y, Du
W and Sakamoto M: Bmi-1 gene is upregulated in early-stage
hepatocellular carcinoma and correlates with ATP-binding cassette
transporter B1 expression. Cancer Sci. 101:666–672. 2010.
View Article : Google Scholar
|
|
27
|
Sasaki M, Ikeda H, Itatsu K, et al: The
overexpression of polycomb group proteins Bmi-1 and EZH2 is
associated with the progression and aggressive biological behavior
of hepatocellular carcinoma. Lab Invest. 88:873–882. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang H, Pan K, Zhang HK, et al: Increased
polycomb-group oncogene Bmi-1 expression correlates with poor
prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol.
134:535–541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song W, Tao K, Li H, et al: Bmi-1 is
related to proliferation, survival and poor prognosis in pancreatic
cancer. Cancer Sci. 101:1754–1760. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sparmann A and van Lohuizen M: Polycomb
silencers control cell fate, development and cancer. Nat Rev
Cancer. 6:846–856. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li DW, Tang HM, Fan JW, et al: Expression
level of Bmi-1 oncoprotein is associated with progression and
prognosis in colon cancer. J Cancer Res Clin Oncol. 136:997–1006.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu JH, Song LB, Zhang X, et al: Bmi-1
expression predicts prognosis for patients with gastric carcinoma.
J Surg Oncol. 97:267–272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song LB, Zeng MS, Liao WT, et al: Bmi-1 is
a novel molecular marker of nasopharyngeal carcinoma progression
and immortalizes primary human nasopharyngeal epithelial cells.
Cancer Res. 66:6225–6232. 2006. View Article : Google Scholar
|
|
34
|
Jiang Y, Su B, Meng X, et al: Effect of
siRNA-mediated silencing of Bmi-1 gene expression on HeLa cells.
Cancer Sci. 101:379–386. 2010. View Article : Google Scholar : PubMed/NCBI
|