Introduction
Hematopoietic stem cell transplantation (HSCT) is a
curative therapy for a variety of diseases, especially for
hematological malignancies and immunodeficiency diseases. However,
graft-versus-host disease (GVHD) is still a major cause of
post-transplant mortality among patients receiving HSCT (1,2). Based
on the time frame and type of organ involvement, GVHD can be
characterized as acute (aGVHD) and chronic (cGVHD). Despite recent
advances in therapies to improve the outcome of aGVHD in patients,
this complication still occurs in 30–50% of all HSCT recipients
(2,3). The pathophysiology of aGVHD is
characterized by the enhanced recognition of host alloantigens by
alloreactive donor T cells, proliferation, differentiation and
secretion of cytokines, host cell apoptosis and tissue damage
(4–6). Studies show an association of both
donor CD4+ and CD8+ T cells with aGVHD
(4). After activation by host
alloantigens, CD4+ T cells differentiate into various T
helper (Th) cell subsets, such as Th1 and Th17 cells that are often
increased in aGVHD and play a role in mediating aGVHD-induced
tissue damage (7), and Th2 cells
that apparently have the protective role in the development of
aGVHD (8). In contrast to
CD4+ T cells, activated CD8+ T cells mainly
differentiate into cytotoxic T lymphocytes (CTLs), and the damage
induced by activated CD8+ T cells in aGVHD primarily
depends on the cytolytic machinery (9–11).
Although the role of different T cell subsets in aGVHD has been
investigated, the key molecular mechanisms that trigger and
maintain abnormality of these T cell subsets remain unclear.
Interleukin-18 (IL-18) is a member of the IL-1
cytokine superfamily and is produced by a variety of cells. The
receptor for IL-18 (IL-18R) consists of a ligand-binding (IL-18Rα)
and a signal-transducing chain (IL-18Rβ), both essential for signal
transduction (12). Increased IL-18
serum levels were reported in patients with aGVHD, and further
studies have shown that IL-18 stimulates the Th1 cell-mediated
immune response, enhances expansion and cytotoxic activity of donor
CD8+ T cells, and increases pro-inflammatory cytokine
secretion in the course of aGVHD (10,13).
In the present study, we evaluated the protective effect of IL-18R
neutralizing antibody (Ab) in an experimental aGVHD model. We
analyzed the changes in the clinical manifestations of aGVHD, Th
cell subsets, systemic inflammation and cell apoptosis that
resulted from anti-IL-18Rα monoclonal antibody (mAb)
administration. We suggest that blocking the interaction of IL-18
with IL-18R may be beneficial for the treatment of aGVHD.
Materials and methods
Animals
The mice were purchased from the Experimental Animal
Center of Yangzhou University (Yangzhou, China). Six- to 8-week-old
female donor C57BL/6 (B6, H-2b) mice and 8- to
10-week-old recipient BALB/c (H-2d) mice were caged in a
pathogen-free controlled environment with a 12-h light/dark cycle.
Animals were fed and provided water ad libitum for ~2 weeks.
All animal experiments were approved by the Animal Ethics Committee
of Yangzhou University.
Induction of aGVHD and immunologic
interventions
aGVHD was induced in mice as previously described
(14). Briefly, the transplantation
day was set as day 0. On day 0, donor C57BL/6 mice were sacrificed
by cervical dislocation, and the femur, tibia and spleen were
harvested and kept in ice-cold phosphate-buffered solution (PBS).
Bone marrow cells (BMCs) were flushed from the femur and tibia, and
a single-cell BMC suspension in PBS was prepared. Donor spleen
cells (SPs) were minced and a single-cell suspension was obtained
by passage of minced spleen through a 75-µm wire mesh
strainer and resuspended in RPMI-1640 medium (Life Technologies,
Carlsbad, CA, USA). Recipient BALB/c mice were divided into 2
groups. Mice in the normal control group (n=6) received no
treatment, while the rest of the animals (n=12) received 7.5 Gy
total body irradiation from a Softex M-150 WE 60Co
source (Softex, Tokyo, Japan). After a 4-h irradiation, the mice
were randomly divided into 2 experimental groups, BS+Ab and BS (6
animals/group), and were injected intravenously with 0.25 ml
RPMI-1640 medium containing 5×106 donor BMCs combined
with 5×105 donor SPs. Recipient mice in the BS+Ab group
also received 10 µg/mouse intraperitoneal injection of
neutralizing mAb against murine IL-18Rα (catalog no. MAB12161;
R&D Systems, Minneapolis, MN, USA) every 2 days. Recipient mice
in the BS group received an intraperitoneal injection of PBS.
Assessment of aGVHD
Recipient mice were monitored daily for the
following clinical manifestations of aGVHD: weight loss, hunched
posture, poor activity, ruffled fur and loss of skin integrity
(14,15). The severity of the above symptoms
was scored from 0 to 2. The sum of the scores for all the symptoms
for each mouse (maximum 10) was used as an index of severity and
progression of aGVHD. Each experiment was repeated 4–5 times
independently.
Flow cytometric analysis
Blood samples were collected from the animals in all
groups weekly starting from day 7 and up to day 35
post-transplantation (P.T.). Th1, Th17 and Th2 in peripheral blood
were detected by intracellular cytokine staining as previously
described (16). Briefly, blood
samples were stimulated with 50 ng/ml phorbol myristate acetate
(PMA) and 750 ng/ml ionomycin (both from Sigma, St. Louis, MO, USA)
in the presence of 10 µg/ml brefeldin A (Life Technologies)
at 37°C for 4 h. The cells were then harvested and surface staining
was performed for 15–20 min with a mixture of the FITC-CD4 and
PE-CD45 antibodies. After being washed with PBS, the cells were
fixed and penetrated with Fix and Perm buffer (An Der Grub,
Austria), followed by staining with Per-CP-conjugated IFN-γ,
IL-17A, IL-4, or IL-6 monoclonal antibodies. All antibodies were
purchased from BD Biosciences (San Jose, CA, USA). All data were
acquired using FACSCalibur flow cytometer and analyzed with
CellQuest software (both from BD Biosciences).
Cytometric bead array and enzyme-linked
immunosorbent assay (ELISA)
The levels of serum cytokines were determined weekly
during a time course of 5 weeks (week 1 to 5 P.T.) using the
Cytometric Bead Array™ Mouse Th1/Th2/Th17 Cytokine kit (BD
Biosciences). Mouse cytokine-specific bead sets and standards were
implemented according to the manufacturer's instructions. The
fluorescence produced by the beads was measured using FACSCalibur
flow cytometer and analyzed with the accompanying software.
Serum levels of IL-18 were assessed by ELISA. For
each sample, 0.1 ml of supernatants and standards was assayed in
duplicates using a mouse IL-18 immunoassay kit from BlueGene
Systems (Shanghai, China) according to the manufacturer's
instructions.
Real-time PCR analysis
Total RNA was extracted using TRIzol (Life
Technologies) and cDNA was synthesized from 1 µg of RNA
using miRcute miRNA cDNA Synthesis kit (Tiangen, Beijing, China).
The PCR primers for the target genes were as follows: IL-18,
5′-tgacaacacgtttactttatacct-3′ (sense) and
5′-cacagccagtcctctacttca-3′ (antisense); Fas,
5′-agtttcatgaacccgcctc-3′ (sense) and 5′-gcagacatgctgtggatctg-3′
(anti-sense); FasL, 5′-ttaaatgggccacactcctc-3′ (sense) and
5′-actccgtgagttcaccaacc-3′ (antisense); and β-actin,
5′-atggaggggaatacagccc-3′ (sense) and 5′-ttctttgcagctccttcgtt-3′
(antisense). mRNA expression levels of the target genes were
analyzed by real-time qPCR using Fast SYBR-Green Master Mix and an
Applied Biosystems® instrument (both from Life
Technologies). qPCR reaction was carried out as follows:
denaturation for 2 min at 94°C, followed by 40 cycles of 30 sec at
94°C, 30 sec at 60°C and 1 min at 2°C. The baseline adjustment
method of the HRM software was used to determine the Ct in each
reaction. All samples were amplified in duplicates, and the mean
was used for further analysis. Gene expression was normalized to
β-actin.
Western blotting
Mice were sacrificed after bone marrow
transplantation (BMT) at indicated times, and the liver and small
intestine were resected aseptically. Partial tissues were
flash-frozen in liquid nitrogen immediately upon dissection, while
the rest of the tissues were fixed with 4% paraformaldehyde
solution for further analysis as described below. Individual frozen
samples of liver and small intestine were homogenized with an
electric hand-held precise tissue homogenizer (PRO200; Pro
Scientific, USA) in a 5X volume of ice-cold homogenization buffer
(Kangchen, Shanghai, China). Protein concentrations were quantified
using a BCA protein assay (Thermo Scientific, Waltham, MA, USA).
Equal amounts of proteins were separated on 12% SDS-PAGE and
transferred to PVDF membranes (Millipore, Bedford, MA, USA).
Membranes were blocked with 5% non-fat milk in Tris-buffered saline
(TBS) for 3 h at room temperature and then incubated with p38MAPK
and phospho-38MAPK antibodies (Cell Signaling Technology, Danvers,
MA, USA) overnight at 4°C followed by incubation with
HRP-conjugated anti-rabbit or anti-mouse IgG (Sigma). Detection was
performed with the enhanced chemiluminescence reagent (Millipore).
GAPDH was used as a loading control. Densitometry was performed to
compare protein expression among the 3 groups using Bio-Rad
Quantity One software (Bio-Rad, Hercules, CA, USA).
Histopathology and immunohistochemical
analysis
Liver and small intestine samples were fixed with 4%
paraformaldehyde solution as described above, ethanol-dehydrated
and embedded in paraffin. Tissues were then sectioned and stained
with hematoxylin and eosin (H&E). Scoring was performed as
previously described (0 as normal, 0.5 as focal and rare, 1.0 as
focal and mild, 2.0 as diffuse and mild, 3.0 as diffuse and
moderate, and 4.0 as diffuse and severe). Histological examination
and lesion assessment were independently carried out by two
pathologists in a blinded fashion.
Immunohistochemistry (IHC) was performed using
rabbit polyclonal IL-18, Fas and FasL antibodies (Abcam, Cambridge,
UK), respectively. Briefly, 4- to 5-µm sections were
deparaffinized, and antigen retrieval was performed in citrate
buffer using a microwave oven. Slides were rinsed, treated with 3%
H2O2 in methanol and blocked by goat serum
(Sigma) in Tris-buffer. Slides were incubated overnight at 4°C with
a primary antibody diluted in blocking solution. After rinsing in
Tris-buffer, the slides were incubated with biotin-labeled
secondary anti-rabbit IgG (Abcam). The sections were then incubated
with the ABC-HRP complex (ZSGB-Bio, Beijing, China). Binding sites
were visualized with diaminobenzidine/hydrogen peroxide, followed
by counterstaining with hematoxylin.
TUNEL assay
The apoptotic cells in target organs were detected
using the terminal deoxynucleotidyl transferase-mediated
deoxyuridine triphosphate nick-end labeling (TUNEL) assay (Roche,
Basel, Switzerland). All target tissues were fixed in freshly
prepared 4% paraformaldehyde solution. Tissue sections
(4-µm) were prepared on glass slides. TUNEL assay was
performed according to the manufacturer's instructions. Briefly,
sections were deparaffinized in xylene, dehydrated in ethanol, and
incubated with 20 µg/ml proteinase K for 30 min at room
temperature. Sections were then incubated with TUNEL reaction
mixture for 60 min at 37°C in a humidified atmosphere in the dark
and analyzed under a Olympus BX50 fluorescence microscope (Olympus,
Japan).
Three tissue slices were randomly selected from each
group, and five high-power fields were randomly selected from each
slice. The number of apoptotic and normal cells were counted. The
apoptosis index (AI) for each target organ was calculated using the
following equation: AI (%) = (the number of apoptotic cells/number
of normal cells) × 100%.
Statistical analysis
Experimental data are expressed as means ± SD.
Differences between two groups were analyzed by the two-tailed
t-test. One-way ANOVA was used for comparison of multiple groups. A
p-value ≤0.05 was considered statistically significant. Survival
curves were plotted using the Kaplan-Meier methods. All statistical
analyses were performed by SPSS ver16.0 software.
Results
Clinical manifestations of aGVHD are
alleviated in mice treated with anti-IL-18Rα Ab
To investigate the protective role of IL-18R
blockage in aGVHD, BMCs from C57BL/6 mice were transplanted into
irradiated BALB/c mice as described in Materials and methods.
Recipient mice were then divided into two experimental groups. The
BS+Ab group received intraperitoneal injection of 10 µg
neutralizing anti-IL-18Rα mAb, while animals in the control BS
group were treated with an intraperitoneal injection of PBS. The
characteristics of aGVHD, such as mean weight loss, clinical score,
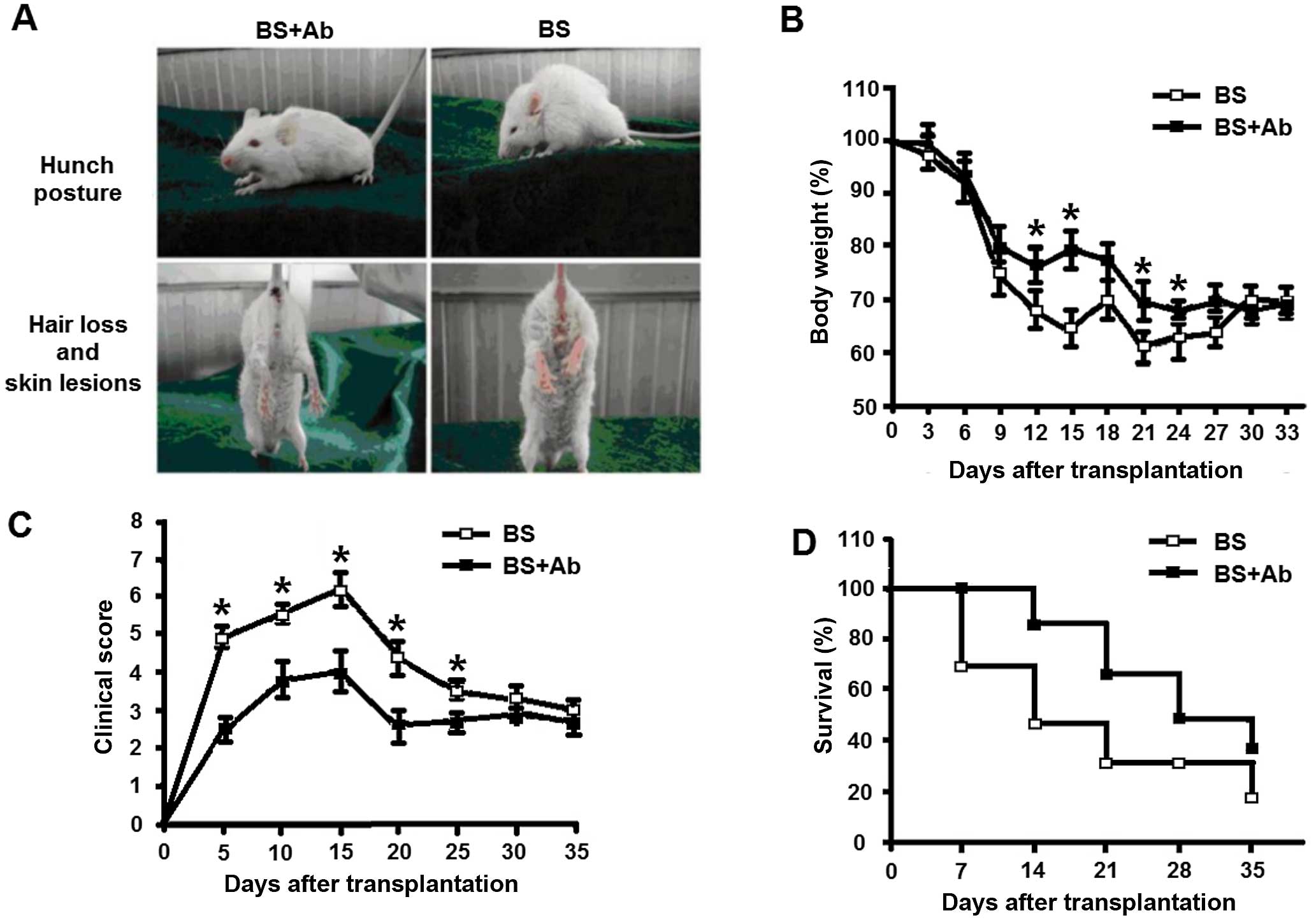
and survival rate were assessed in each group. Seven days after
transplantation, the mice in both groups exhibited characteristic
clinical symptoms of aGVHD, including hunch posture, hair loss and
skin lesions (Fig. 1A). All
irradiated mice lost weight that peaked on day 14 P.T., with
significantly less weight loss observed in the BS+Ab group
(compared to the BS group (19±1.5 and 27±2.4%, respectively,
P<0.05, Fig. 1B). During the
entire observation period of 35 days, mice in the BS+Ab group had a
significantly lower clinical score than the BS group, with the
difference reaching its maximum at 14 P.T. (P<0.05, Fig. 1C). We detected a markedly higher
survival rate of mice in the BS+Ab group compared to the BS group
(Fig. 1D). Additionally, the
manifestations of aGVHD in the BS+Ab group were milder than those
in the BS group until the end of the study (35 days P.T.). Taken
together, these results indicated that blocking the interaction of
IL-18 with IL-18R by anti-IL-18Rα mAb attenuated the clinical
manifestations of aGVHD in the mouse animal model.
Anti-IL-18Rα Ab administration decreases
systemic inflammation in the aGVHD mouse model
Systemic inflammation is associated with the
expression of Th cell subsets (17). We analyzed Th subsets in the
peripheral blood of the BS+Ab and BS aGVHD mice at various
time-points using FACS analysis. In both groups, the percentage of
Th1 cells increased reaching its peak at day 21 P.T. (Fig. 2A). BS+Ab mice exhibited
significantly reduced levels of Th1 cells 7 and 14 days P.T. as
compared to the BS group (P<0.05). This difference was no longer
observed at 21–35 days P.T., as the percentage of Th1 cells in both
groups gradually decreased to pre-transplantation levels (Fig. 2A). Different dynamic changes were
observed in the levels of Th2 and Th17 cells. The BS+Ab group
exhibited a markedly lower percentage of Th2 cells in the
peripheral blood when compared to the BS group on day 21 and 35
(Fig. 2B, P<0.05), while no
significant difference was detected at earlier time-points. Levels
of Th17 cells rose slowly in both groups during the same period,
with the percentage of Th17 cells being lower in the BS+Ab group
than that in the BS group on day 7 P.T., but higher on days 21 and
35 P.T. (Fig. 2C). Taken together,
these results suggest that blocking IL-18R had differential effects
on the differentiation of the various Th subsets.
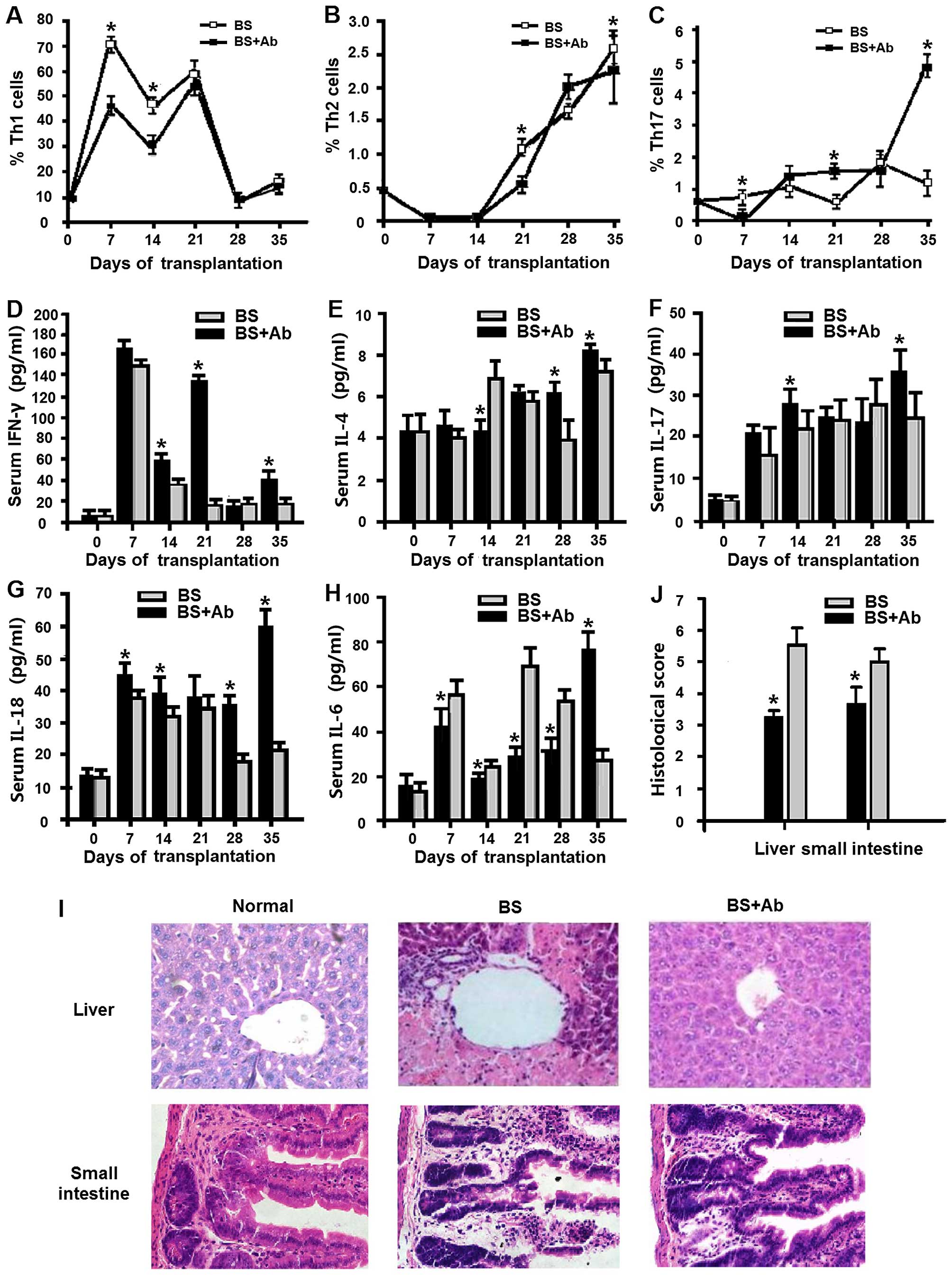 | Figure 2Effect of anti-IL-18Rα mAb
administration on Th cell subsets, pro-inflammatory cytokines and
histological scores in the aGVHD mice. (A–C) Peripheral blood
levels of Th1 (A), Th2 (B) and Th17 (C) cell subsets in the BS+Ab
and BS experimental groups were measured by flow cytometry at
different time-points. Serum levels of IFN-γ (D), IL-4 (E), IL-17A
(F) and IL-6 (H) at different time-points were detected by
cytometric bead array, and IL-18 levels (G) were measured by ELISA.
(I) Representative H&E staining of the liver and small
intestine tissues of the mice in the BS+Ab and BS groups and the
normal control group (untreated animals) on day 14 P.T.
Magnification, ×400. (J) Histological score was measured on day 14
P.T. n=6 in each group, *P<0.05. GVHD,
graft-versus-host disease; aGVHD, acute GVHD; mAb, monoclonal
antibody; Th, T helper; IL-18, interleukin-18; ELISA, enzyme-linked
immunosorbent assay; H&E, hematoxylin and eosin; P.T.,
post-transplantation. |
Serum levels of IFN-γ, IL-17A, IL-4, IL-6 and IL-18
are associated with the inflammatory response (18–20).
We next evaluated serum levels of these cytokines in the BS+Ab and
BS groups of the aGVHD mice at different time-points using
cytometric bead array and ELISA as described in Materials and
methods. Mice in the BS+Ab group exhibited significantly elevated
serum levels of IFN-γ as compared to the BS group (Fig. 2D). Similarly, serum levels of IL-4
and IL-17A in the BS+Ab group were markedly higher on day 35 P.T.
(Fig. 2E and F), while serum IL-18
was elevated in the BS+Ab group during the entire period of
observation (Fig. 2G). On the other
hand, animals in BS+Ab group had significantly lower levels of IL-6
in the first 4 weeks P.T., followed by a dramatic increase in serum
IL-6 concentration on day 35 P.T. (Fig.
2H). Taken together, these results suggest that IL-18R blockade
mediates the inflammatory response through regulation of cytokine
secretion.
The main target organs of aGVHD are the liver and
small intestine (21). We next
addressed the effect of IL-18R blockage by anti-IL-18Rα on
inflammation and necrosis in the tissues of the aGVHD mouse model.
Histopathological analysis of the liver and small intestine showed
that on day 21 P.T. the degree of inflammation and necrosis in the
mice of the BS group was more severe than that in the BS+Ab and
normal control group (untreated animals), as indicated by the
intensity of H&E staining (Fig.
2I). These results correlated with a significantly increased
histopathologic score detected in both the liver and small
intestine of the BS group as compared to the score in the BS+Ab
group (Fig. 2J, P<0.05). Taken
together, these data imply that the administration of anti-IL-18Rα
mAb affects the inflammatory response and the pathological
progression of aGVHD in main target organs.
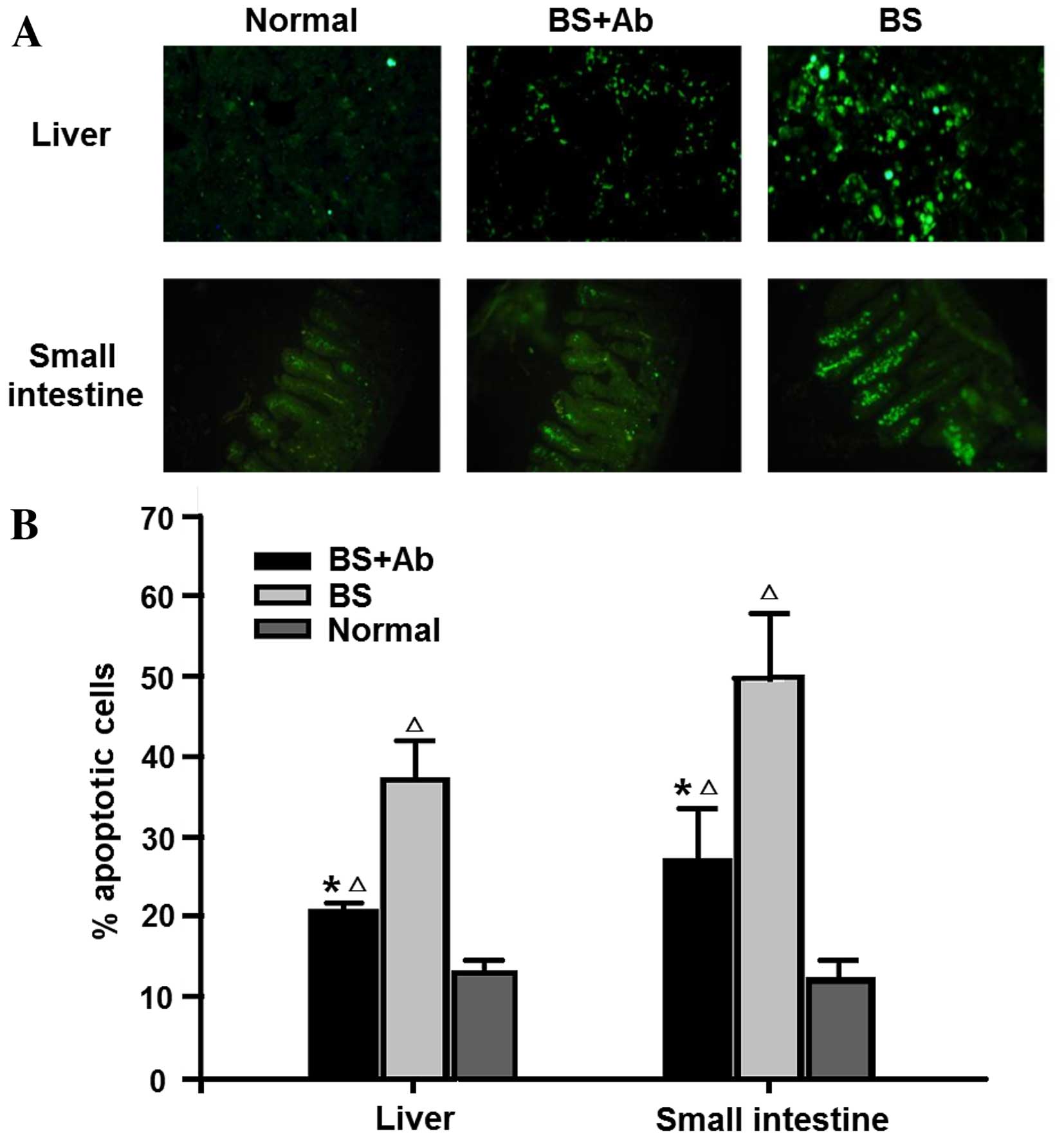
Apoptosis and expression of
apoptosis-related protein are reduced by anti-IL-18Rα Ab
treatment
Numerous studies have shown that the cell apoptosis
rate is increased in experimental GVHD mouse models (22,23).
In order to assess the influence of anti-IL-18Rα mAb on cell
apoptosis in aGVHD target organs, we evaluated the percentage of
apoptotic cells in the liver and small intestine of aGVHD mice on
day 14 P.T. by TUNEL assay. The liver and small intestine of the BS
group animals showed increased expression of apoptotic cells as
compared to the normal group (untreated animals) (Fig. 3). On the other hand, administration
of anti-IL-18Rα mAb in the BS+Ab group (experimental group)
markedly reduced TUNEL staining of the liver and small intestine,
suggesting that blocking IL-18R inhibited aGVHD-induced cell
apoptosis.
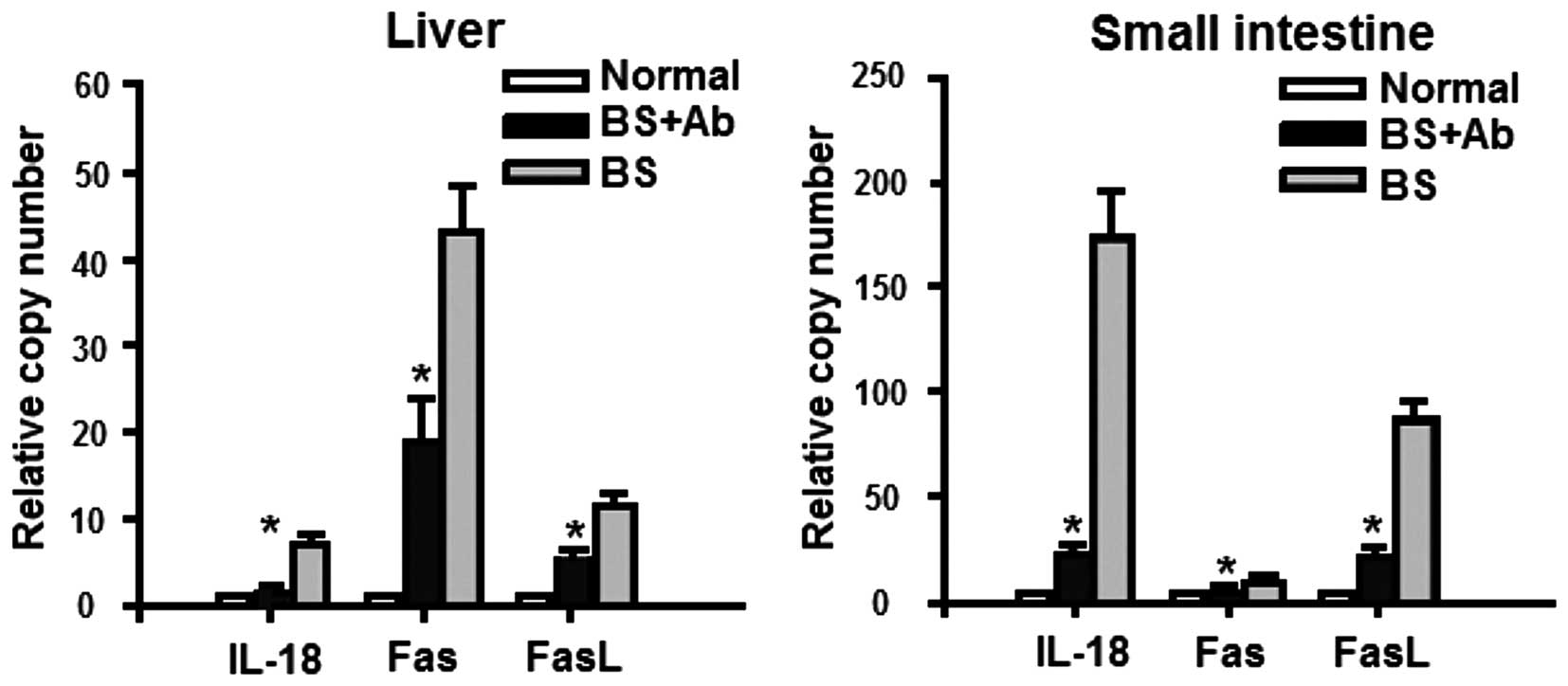
Next, we addressed the mechanism of the effect of
anti-IL-18Rα mAb on cell apoptosis in aGVHD. Since the activity of
IL-18 in different organ tissues is determined by the infiltration
of IL-18 into these tissues, we analyzed mRNA and protein levels of
IL-18 in the liver and small intestine on day 14 P.T. using qPCR
and histopathological analysis, respectively. The expression of
IL-18 mRNA was markedly increased in the BS group when compared to
the level in the normal and BS+Ab group (Fig. 4). These results correlated with the
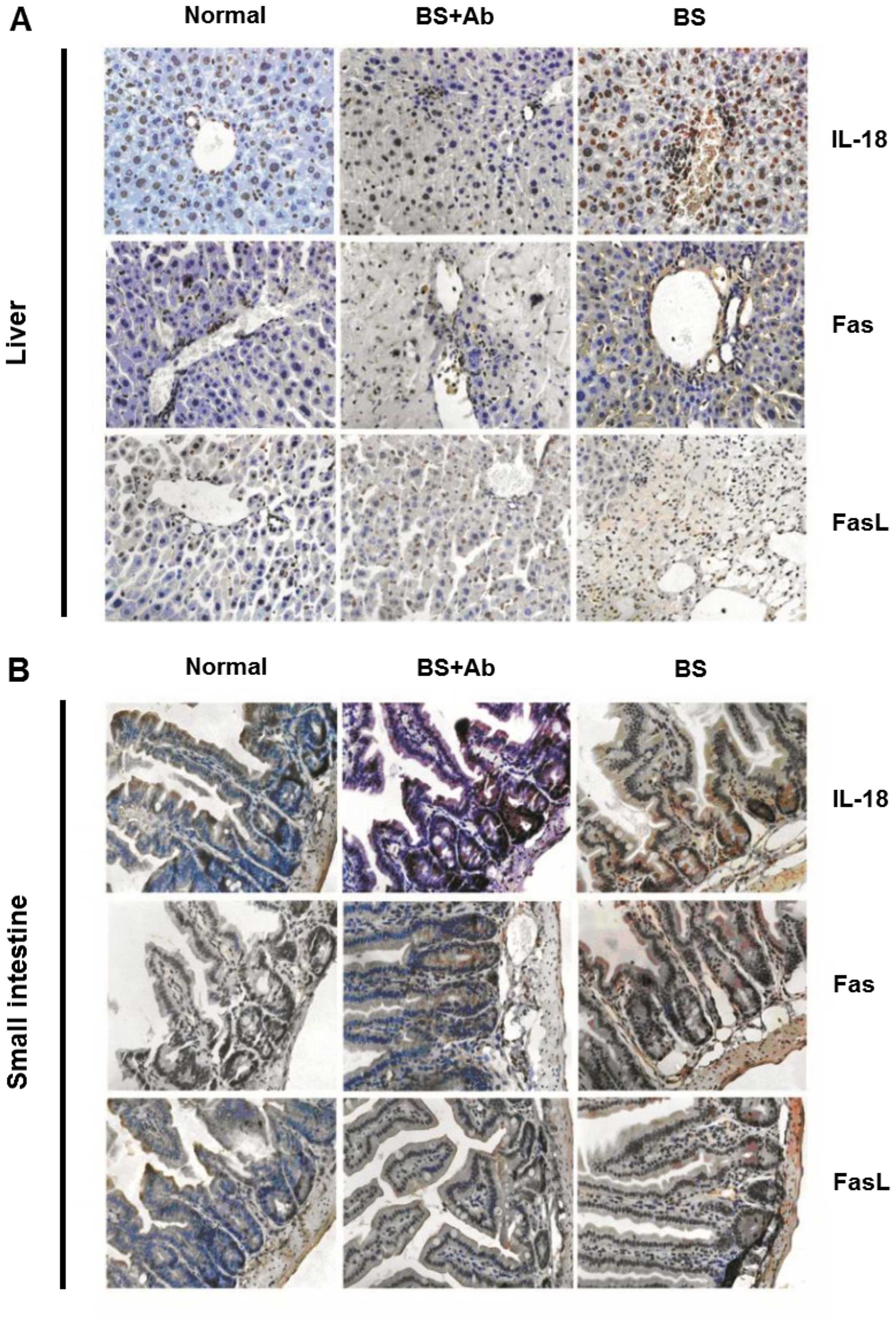
increased IL-18 protein expression in the affected tissues of the
BS group as compared to the BS+Ab and normal control animals
(Fig. 5). Increased expression of
Fas and FasL serves as a reliable marker of apoptosis and has been
implicated in GVHD pathogenesis (23,24).
To determine whether these apoptosis-related proteins were affected
by anti-IL-18Rα treatment, we analyzed the expression of Fas and
FasL in the liver and small intestine of aGVHD mice on day 14 P.T.
We found that the mRNA expression levels of both Fas and FasL were
significantly lower in the BS+Ab group than levels in the BS group
(Fig. 4, P<0.05), and similar to
expression levels detected in the untreated control group.
Similarly, histopathological analysis of the liver and small
intestine tissues of the BS+Ab animals showed decreased protein
levels of Fas and FasL as compared to the BS experimental group
(Fig. 5). Together these data
revealed that administration of IL-18Rα mAb may decrease IL-18
infiltration into the liver and small intestine, as well as
diminish Fas and FasL levels in these target organs in aGVHD
mice.
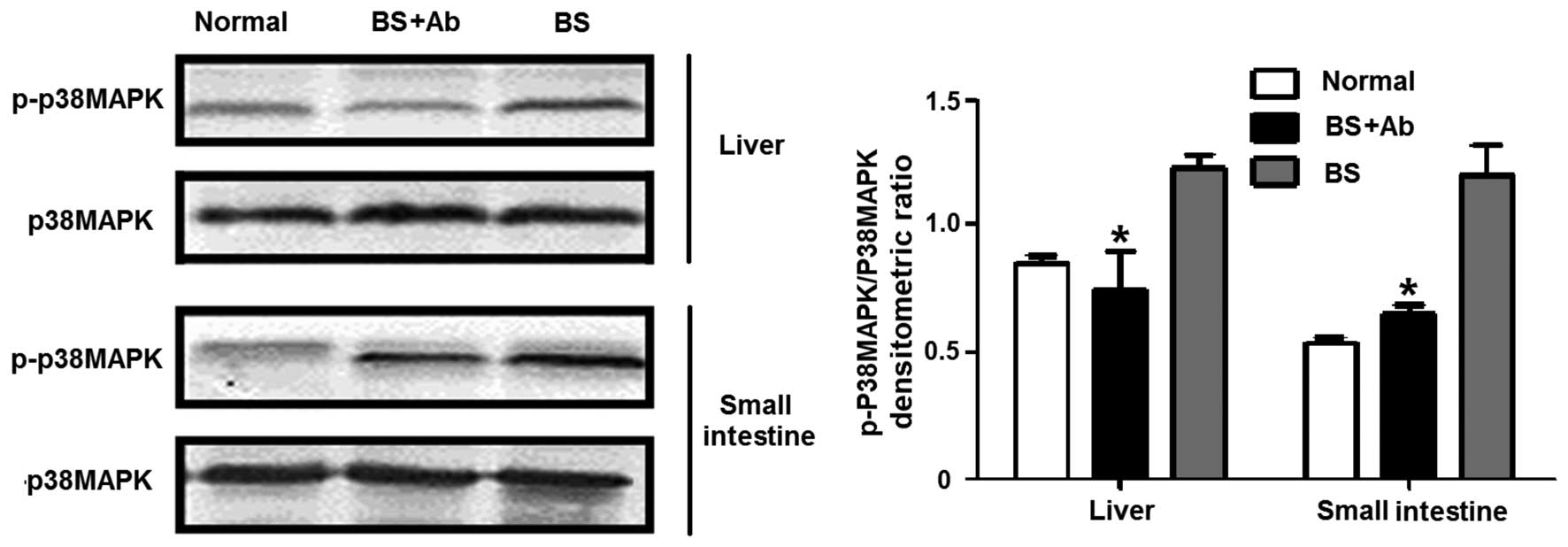
Anti-IL-18Rα Ab treatment reduces
apoptosis-related p38MAPK phosphorylation
Previous studies suggest that the p38MAPK signaling
pathway is involved in cell apoptosis mediated by Fas/FasL
(25). Since anti-IL-18Rα mAb
administration clearly diminished Fas and FasL levels in the liver
and small intestine of the aGVHD animals, we next analyzed the
effect of IL-18 blockage on the levels of p38MAPK activity in the
liver and small intestine of the aGVHD animals by assessing p38MAPK
phosphorylation on day 14 P.T. As shown in Fig. 6, the levels of p38MAPK
phosphorylation were markedly increased in the liver and small
intestine of the BS group comparing to levels in the normal group
on day 14 P.T. (P<0.05), while IL-18R blockage with anti-IL-18Rα
mAb (BS+Ab group) led to a decrease in p38MAPK phosphorylation to
levels comparable to the untreated control animals (P<0.05
compared to the BS group).
Together, these results suggest that IL-18 binding
to IL-18Rα induces p38MAPK phosphorylation that is correlated with
increased Fas/FasL expression in the liver and intestine of aGVHD
animals. The suppression of p38MAPK activity and Fas/FasL
expression in aGVHD mice by anti-IL-18Rα mAb administration is
associated with a decrease in cell apoptosis.
Discussion
Previous studies suggest an important role of IL-18
in the pathogenesis of numerous conditions, such as lupus
nephritis, and rheumatoid arthritis (26,27).
IL-18 is overexpressed in patients with aGVHD and in experimental
aGVHD animal models, and plays an important role in the progression
of the disease (28,29). Previous studies have demonstrated
that anti-IL-18Rα antibody administration could efficiently
alleviate lung inflammation and attenuate the development of
necrotizing enterocolitis (30,31).
In the present study we evaluated the possible protective effect of
the anti-IL-18Rα antibody on mice with experimental aGVHD. Our
results confirmed that administration of the anti-IL-18Rα mAb to
irradiated mice after BMC transplantation significantly reduced the
severity of aGVHD symptoms, such as hunch posture, hair loss, skin
lesions and weight loss. Our results therefore suggest that
anti-IL-18Rα therapy could potentially abrogate the biological
effect of IL-18 on the onset of aGVHD and is beneficial for
reducing the clinical symptoms of cGVHD.
aGVHD is often associated with changes in subsets of
Th cells. We found that blocking IL-18R markedly affected the
levels of Th1, Th2 and Th17 subsets in the peripheral blood of
aGVHD animals, but with different dynamics. Anti-IL-18Rα antibody
administration led to a significant but transient decrease in Th1
levels 7 and 14 days P.T., with Th1 levels decreasing gradually to
control levels by day 35 P.T. in all experimental groups. In
contrast, the percentage of Th2 cells in mice treated with
anti-IL-18Rα antibody was similar to the untreated aGVHD animals at
early time-points, but decreased significantly on days 21 and 35
P.T., while Th17 levels were initially decreased in the
anti-IL-18Rα mAb-treated mice as compared to the aGVHD animals.
These animals exhibited a marked increase in the percentage of Th17
cells at later time-points (21 and 35 days P.T.). Different
dynamics in these changes suggest that in aGVHD Th cell subsets may
be mediated by different factors.
In contrast with the decreased levels of Th1 cells
in the anti-IL-18Rα mAb-treated mice with aGVHD, we found elevated
expression of IFN-γ, a cytokine secreted mainly by Th1 cells, in
this experimental group. Although Th1 cells are considered the main
source of IFN-γ, other immune cells also have the potential to
secrete this cytokine (32). It is
possible that blocking IL-18 may influence the function of these
immune cells, leading to increased secretion of IFN-γ. The exact
source of IFN-γ in the anti-IL-18Rα mAb-treated aGVHD mice merits
further investigation.
Administration of anti-IL-18Rα mAb led to
significant attenuation of IL-4 and IL-17A serum concentrations as
compared to the aGVHD animals, but the dynamics of these changes
were different, suggesting that these two subsets of cytokines may
also be mediated by different factors in aGVHD. Notably,
anti-IL-18Rα mAb also induced an increase in the serum level of
IL-18 in the aGVHD mice. We may speculate that the anti-IL-18Rα
antibody may hinder IL-18 from binding to IL-18R, leading to serum
IL-18 accumulation. At the same time, anti-IL-18Rα-treated aGVHD
mice exhibited markedly reduced IL-18 expression in the liver and
small intestine as compared with the untreated aGVHD animals,
suggesting that blocking the interaction of IL-18 with its
receptors prevents the accumulation of IL-18 in organ tissues of
the aGVHD animals.
Previous studies have shown that IL-18 activates
inflammatory cytokines, such as IL-6, to promote the inflammatory
response (33). In this study, we
found that IL-6 was reduced in the aGVHD mice treated with the
anti-IL-18Rα antibody during the first 4 weeks P.T., suggesting
that blockade of IL-18 is able to inhibit the inflammatory
response. Further studies are required to identify the factors
associated with the increased expression of IL-6 on day 35 P.T.
reported in our study.
The effect of the anti-IL-18Rα antibody on
pro-inflammatory cytokine levels in aGVHD mice were correlated with
decreased inflammation and necrosis detected in the liver and small
intestine of animals receiving anti-IL-18Rα therapy, suggesting
that blocking IL-18R may be beneficial in preventing organ tissue
damage.
Studies have shown an association between IL-18 and
cell apoptosis in aGVHD (22). We
were able to significantly reduce the apoptotic rates in the liver
and small intestine of the aGVHD mouse model by interfering with
IL-18/IL-18R binding. This effect of IL-18R blockage was associated
with the inhibition of Fas and FasL expression. Previous studies
suggest that p38MAPK activity mediates Fas/FasL-induced apoptosis
(25). Our studies confirm that
p38MAPK phosphorylation plays a role in mediating aGVHD, and
blocking IL-18R inhibits p38MAPK activation. These results suggest
that inhibition of cell apoptosis by the IL-18Rα antibody is
associated with the downregulation of apoptosis-related proteins
and the p38MAPK signaling pathway.
In conclusion, our study indicated that blocking
IL-18R with the anti-IL-18Rα antibody prevents the pathogenesis in
early stages of experimental aGVHD. As an interference method,
anti-human IL-18Rα may represent a novel strategy for the treatment
of human aGVHD. Additional research concerning the utility of
IL-18R-based therapeutics to prevent and treat aGVHD is
warranted.
Acknowledgments
This study was supported by the Qinglan Project of
Jiangsu Province of China, National Nature Science Foundation of
China (grant no. 81070446) and the Priority Academic Program
Development of Jiangsu Higher Education Institutions (PAPD). We
thank Dr Weidong Du (University Clinic Ulm, Ulm, Germany) for
proofreading the manuscript.
References
|
1
|
Storb R: Allogeneic hematopoietic stem
cell transplantation - yesterday, today, and tomorrow. Exp Hematol.
31:1–10. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferrara JL, Levine JE, Reddy P and Holler
E: Graft-versus-host disease. Lancet. 373:1550–1561. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tobin LM, Healy ME, English K and Mahon
BP: Human mesenchymal stem cells suppress donor CD4(+) T cell
proliferation and reduce pathology in a humanized mouse model of
acute graft-versus-host disease. Clin Exp Immunol. 172:333–348.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reddy P: Pathophysiology of acute
graft-versus-host disease. Hematol Oncol. 21:149–161. 2003.
View Article : Google Scholar
|
|
5
|
Antin JH and Ferrara JL: Cytokine
dysregulation and acute graft-versus-host disease. Blood.
80:2964–2968. 1992.PubMed/NCBI
|
|
6
|
Levine JE, Paczesny S and Sarantopoulos S:
Clinical applications for biomarkers of acute and chronic
graft-versus-host disease. Biol Blood Marrow Transplant. 18(Suppl
1): S116–S124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pan B, Zhang Y, Sun Y, Cheng H, Wu Y, Song
G, Chen W, Zeng L and Xu K: Deviated balance between Th1 and Th17
cells exacerbates acute graft-versus-host disease in mice.
Cytokine. 68:69–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tawara I, Maeda Y, Sun Y, Lowler KP, Liu
C, Toubai T, McKenzie AN and Reddy P: Combined Th2 cytokine
deficiency in donor T cells aggravates experimental acute
graft-vs-host disease. Exp Hematol. 36:988–996. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reddy P, Arora M, Guimond M and Mackall
CL: GVHD: A continuing barrier to the safety of allogeneic
transplantation. Biol Blood Marrow Transplant. 15(Suppl 1):
162–168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schmaltz C, Alpdogan O, Horndasch KJ,
Muriglan SJ, Kappel BJ, Teshima T, Ferrara JL, Burakoff SJ and van
den Brink MR: Differential use of Fas ligand and perforin cytotoxic
pathways by donor T cells in graft-versus-host disease and
graft-versus-leukemia effect. Blood. 97:2886–2895. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maeda Y, Levy RB, Reddy P, Liu C,
Clouthier SG, Teshima T and Ferrara JL: Both perforin and Fas
ligand are required for the regulation of alloreactive
CD8+ T cells during acute graft-versus-host disease.
Blood. 105:2023–2027. 2005. View Article : Google Scholar
|
|
12
|
Fremond CM, Togbe D, Doz E, Rose S,
Vasseur V, Maillet I, Jacobs M, Ryffel B and Quesniaux VF: IL-1
receptor-mediated signal is an essential component of
MyD88-dependent innate response to Mycobacterium tuberculosis
infection. J Immunol. 179:1178–1189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chandrasekar B, Vemula K, Surabhi RM,
Li-Weber M, Owen-Schaub LB, Jensen LE and Mummidi S: Activation of
intrinsic and extrinsic proapoptotic signaling pathways in
interleukin-18-mediated human cardiac endothelial cell death. J
Biol Chem. 279:20221–20233. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu Y, Sakamaki S, Kuroda H, Kusakabe T,
Konuma Y, Akiyama T, Fujimi A, Takemoto N, Nishiie K, Matsunaga T,
et al: Prevention of lethal acute graft-versus-host disease in mice
by oral administration of T helper 1 inhibitor, TAK-603. Blood.
97:1123–1130. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cooke KR, Kobzik L, Martin TR, Brewer J,
Delmonte J Jr, Crawford JM and Ferrara JL: An experimental model of
idiopathic pneumonia syndrome after bone marrow transplantation: I.
The roles of minor H antigens and endotoxin. Blood. 88:3230–3239.
1996.PubMed/NCBI
|
|
16
|
Ivanov II, McKenzie BS, Zhou L, Tadokoro
CE, Lepelley A, Lafaille JJ, Cua DJ and Littman DR: The orphan
nuclear receptor RORgammat directs the differentiation program of
proinflammatory IL-17+ T helper cells. Cell.
126:1121–1133. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Y, Tawara I, Toubai T and Reddy P:
Pathophysiology of acute graft-versus-host disease: Recent
advances. Transl Res. 150:197–214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wilson SP and Cassel SL:
Inflammasome-mediated autoinflammatory disorders. Postgrad Med.
122:125–133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miossec P: IL-17 and Th17 cells in human
inflammatory diseases. Microbes Infect. 11:625–630. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nishimoto N and Kishimoto T: Interleukin
6: From bench to bedside. Nat Clin Pract Rheumatol. 2:619–626.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vogelsang GB, Lee L and Bensen-Kennedy DM:
Pathogenesis and treatment of graft-versus-host disease after bone
marrow transplant. Annu Rev Med. 54:29–52. 2003. View Article : Google Scholar
|
|
22
|
Reddy P, Teshima T, Kukuruga M, Ordemann
R, Liu C, Lowler K and Ferrara JL: Interleukin-18 regulates acute
graft-versus-host disease by enhancing Fas-mediated donor T cell
apoptosis. J Exp Med. 194:1433–1440. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wasem C, Frutschi C, Arnold D, Vallan C,
Lin T, Green DR, Mueller C and Brunner T: Accumulation and
activation-induced release of preformed Fas (CD95) ligand during
the pathogenesis of experimental graft-versus-host disease. J
Immunol. 167:2936–2941. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yi T, Zhao D, Lin CL, Zhang C, Chen Y,
Todorov I, LeBon T, Kandeel F, Forman S and Zeng D: Absence of
donor Th17 leads to augmented Th1 differentiation and exacerbated
acute graft-versus-host disease. Blood. 112:2101–2110. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kalina U, Kauschat D, Koyama N,
Nuernberger H, Ballas K, Koschmieder S, Bug G, Hofmann WK, Hoelzer
D and Ottmann OG: IL-18 activates STAT3 in the natural killer cell
line 92, augments cytotoxic activity, and mediates IFN-gamma
production by the stress kinase p38 and by the extracellular
regulated kinases p44erk−1 and p42erk−21. J
Immunol. 165:1307–1313. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Calvani N, Tucci M, Richards HB, Tartaglia
P and Silvestris F: Th1 cytokines in the pathogenesis of lupus
nephritis: The role of IL-18. Autoimmun Rev. 4:542–548. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gracie JA, Forsey RJ, Chan WL, Gilmour A,
Leung BP, Greer MR, Kennedy K, Carter R, Wei XQ, Xu D, et al: A
proinflammatory role for IL-18 in rheumatoid arthritis. J Clin
Invest. 104:1393–1401. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller WP, Srinivasan S,
Panoskaltsis-Mortari A, Singh K, Sen S, Hamby K, Deane T, Stempora
L, Beus J, Turner A, et al: GVHD after haploidentical
transplantation: A novel, MHC-defined rhesus macaque model
identifies CD28− CD8+ T cells as a reservoir
of breakthrough T-cell proliferation during costimulation blockade
and sirolimus-based immunosuppression. Blood. 116:5403–5418. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arnold D, Wasem C, Juillard P, Graber P,
Cima I, Frutschi C, Herren S, Jakob S, Alouani S, Mueller C, et al:
IL-18-independent cytotoxic T lymphocyte activation and IFN-gamma
production during experimental acute graft-versus-host disease. Int
Immunol. 14:503–511. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kang MJ, Homer RJ, Gallo A, Lee CG,
Crothers KA, Cho SJ, Rochester C, Cain H, Chupp G, Yoon HJ, et al:
IL-18 is induced and IL-18 receptor alpha plays a critical role in
the pathogenesis of cigarette smoke-induced pulmonary emphysema and
inflammation. J Immunol. 178:1948–1959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Halpern MD, Khailova L, Molla-Hosseini D,
Arganbright K, Reynolds C, Yajima M, Hoshiba J and Dvorak B:
Decreased development of necrotizing enterocolitis in
IL-18-deficient mice. Am J Physiol Gastrointest Liver Physiol.
294:G20–G26. 2008. View Article : Google Scholar
|
|
32
|
Schroder K, Hertzog PJ, Ravasi T and Hume
DA: Interferon-gamma: An overview of signals, mechanisms and
functions. J Leukoc Biol. 75:163–189. 2004. View Article : Google Scholar
|
|
33
|
Olee T, Hashimoto S, Quach J and Lotz M:
IL-18 is produced by articular chondrocytes and induces
proinflammatory and catabolic responses. J Immunol. 162:1096–1100.
1999.PubMed/NCBI
|