Introduction
Hepatitis B virus (HBV) infection results in a
significantly high risk of severe liver diseases such as hepatic
cirrhosis and hepatocellular carcinoma (HCC). The hepatitis B virus
X protein (HBx), which is a 17-kDa non-structural protein
consisting of 154 amino acids, is considered as a key regulatory
viral protein in HBV replication and virus-associated liver
diseases (1).
HBx is a multifunctional protein that regulates
numerous cellular signal transduction pathways and participates in
cell proliferation, differentiation, cell cycle progression,
autophagy and apoptosis (2,3). The specific function of HBx is linked
to its subcellular localization (4). It is generally believed that HBx is
mostly cytoplasmic, with a small fraction in the nucleus and many
groups have reported that the mitochondrion is a major target for
HBx in the cytoplasm (5,6), causing mitochondrial damage by
regulating mitochondrial membrane potential (7), increasing the generation of cellular
reactive oxygen species (ROS) (8,9) and
modulating the opening of the mitochondrial permeability transition
pore (MPTP). Studies in HepG2 cells and primary rat hepatocytes
suggest that MPTP activity is required for HBV replication and the
expression of cell cycle proteins modulated by HBx (10,11).
Previous studies have observed the release of cytochrome c
from purified mitochondria after the induction of MPT and therefore
induced apoptosis (12),
identifying MPTP as an intracellular sensor of oxidants and other
toxins. However, whether HBx-induced apoptosis is related to its
function of modulating MPTP and the specific mechanism have not
been fully understood.
Since apoptosis has been implicated as a vital
mechanism for inflammation and hepatocarcinogenesis (13), a large body of research has tried to
explore the role of HBx in cell apoptosis and its contribution to
HBV-associated HCC. The results of these studies are controversial;
HBx has been shown to induce (14,15),
inhibit (16,17) or have no effect on apoptosis
(18). The discrepancy of the role
of HBx on apoptosis may be due to the different cell types, culture
condition or experimental systems used in different studies. In
addition, some of these studies have demonstrated that HBx did not
induce cell apoptosis itself, but instead sensitized hepatocytes to
a variety of apoptotic signals such as TNF-α, TRAIL, ethanol, Fas
and oxidative stress (19,20). Notably, oxidative stress has been
implicated in DNA damage and apoptosis, contributing to the
pathogenesis of inflammatory diseases and cancer (13,21).
The Bcl-2 protein family plays a pivotal role in
HBx-induced cell apoptosis. They are divided into anti-apoptotic
members (Bcl-2, Bcl-xL and Mcl-1) and pro-apoptotic members (Bax,
Bak and Bid) (22). Bax is one of
the pro-apoptotic members of the Bcl-2 protein family and is
considered as a key factor of the intrinsic apoptosis pathway upon
various stimuli. In non-apoptotic cells, it is in dynamic
equilibrium between the mitochondrion and cytosol. In response to
apoptotic stimulus, Bax changes its conformation, disrupting the
equilibrium and causing Bax to accumulate at the mitochondrion
(23). A previous study in
serum-starved HepG2 cells demonstrated that HBx induced the
translocation of Bax to the mitochondrion (24), suggesting that Bax functions as an
important element in the HBx-induced intrinsic apoptosis pathway.
Other studies also demonstrated that Bax plays an important role in
H2O2-induced apoptosis via its mitochondrial
translocation (25). Recent studies
have confirmed that the translocation of Bax in apoptosis is
dependent on voltage-dependent anion channel (VDAC) 2 (26), which is thought to be a component of
MPTP (27).
In the present study, we reported a possible
function for HBx to modulate MPTP in the hepatic HL-7702 cell line,
and confirmed that this function was associated with oxidative
stress-induced apoptosis through translocation of Bax. These new
findings may have implications for understanding the role of HBx in
HBV-associated inflammation and hepatocarcinogenesis.
Materials and methods
Cell culture and transfection
The human HL-7702 hepatocyte cell line was purchased
from the Shanghai Cell Bank (Shanghai, China). The cells were grown
in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal
bovine serum (FBS) and maintained at 37°C in a humidified
atmosphere composed of 95% air and 5% CO2. Cultures of
HL-7702 cells were transfected with recombinant plasmids using the
transfection reagent Lipofectamine 3000 (Invitrogen, Carlsbad, CA,
USA) according to the manufacturer's protocol.
Plasmids
The recombinant plasmid pGEM-HBV, which expresses a
greater-than-genome-length cDNA of wild-type HBV (ayw) and
pGEM-HBV-HBx, which is identical to HBx-deficient mutant HBV, were
a gift from Professor M.J. Bouchard (Drexel University,
Philadelphia, PA USA) (28,29). The HBx expression plasmid pcDNA3.1-X
was kept in our laboratory. We designed new pcDNA3.1 plasmids
expressing HBx fused to the eight amino acid FLAG epitope using
oligonucleotides containing terminal XbaI or EcoRI
restriction enzyme sites by PCR. The forward oligonucleotide
sequence for HBx-flag was: 5′-GCT^CTAGAGCCACCATGGCTGCTAGGCTGTGCT-3′
and the reverse oligonucleotide sequence was,
5′-GCCTTAA^GTTACTTATCATCGTCGTCCTTGTAGTCGGCAGAGGTGAAAAAG-3′.
XbaI or EcoRI enzymes (New England Biolabs, Ipswich,
MA, USA) were used for the digestion of insert and vector, and
digestion products were followed by ligation with T4 DNA ligase
(Promega, Madison, WI, USA).
Western blot analysis
Cells were lysed in RIPA buffer (Beyotime, Shanghai,
China) supplemented with protease inhibitors in ice for 30 min
followed by centrifugation at 12,000 × g for 15 min at 4°C.
Immunoblotting was performed using the anti-cleaved PARP mAb
(1:1,000), anti-Bax mAb (1:1,000; Cell Signaling Technology,
Danvers, MA, USA), anti-cytochrome c mAb (1:500),
anti-caspase-3 mAb (1:1,000; Abcam, Cambridge, MA, USA), anti-HBc
mAb (1:500; Milipore, Billerica, MA, USA), anti-β-actin and
anti-secondary antibodies were purchased from ZSGB-BIO (Beijing,
China).
Isolation of mitochondria and
measurement of COX activity
Cells transfected with recombinant plasmids were
lysed and the mitochondria were isolated using the mitochondrion
isolation kit for mammalian cells (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Protein concentrations were measured using the
bicinchoninic acid (BCA) assay (Beyotime) and COX activity was
determined using the Cytochrome c Oxidase Assay kit (GenMed,
Shanghai, China) as previously described (8,9).
ATP measurement
Cell viability of the transfected cells was assessed
using a luciferase-coupled ATP quantitation assay (Promega).
Transfected cells were dispensed in culture medium at 20,000
cells/100 µl/well into 96-well white/solid-bottom assay plates. The
assay plates were equilibrated at room temperature for 30 min,
followed by the addition of 100 µl/well of CellTiter-Glo reagent
and the contents were mixed for 2 min on an orbital shaker to
induce cell lysis. The assay plates were allowed to incubate at
room temperature for 10 min to stabilize the luminescent signal.
Then, the luminescence intensity of the plates was assessed using a
luminometer (Orion II Microplate Luminometer; Berthold Detection
Systems GmbH, Pforzheim, Germany).
Mitochondrial membrane potential
analysis
The cultured HL-7702 cells were seeded and
transfected in 6-well plate for 36 h, and the mitochondrial
membrane potential was evaluated using the cationic fluorescent dye
Rhodamine-123 (Sigma, St. Louis, MO, USA). Transfected cells were
digested and washed with phosphate-buffered saline (PBS). Diluted
Rhodamine-123 (10 mg Rhodamine-123/ml with serum-free DMEM) was
added to the cultures and incubated at 37°C for 30 min. The cells
were washed and resuspended in PBS for analysis by flow cytometry
(BD Biosciences, San Jose, CA, USA).
Cytosolic calcium imaging assay
Cultured HL-7702 cells were seeded and transfected
in a 24-well plate for 36 h with the addition of 1 µM cyclosporin A
(CsA) (Milipore) or dimethyl sulfoxide (DMSO) for 12 h. Levels of
cytosolic calcium were measured by Flou-4 calcium imaging kit
(Molecular Probes, Eugene, OR, USA) following the manufacturer's
instructions. Fluorescence images were obtained (excitation 494
nm/emission 506 nm) by confocal laser scanning microscopy (Leica
SP5; Leica, Solms, Germany). Relative fluorescence intensity of
each group was measured by LAS AF Lite software (Leica).
Fluorescence assay of ROS
Assay of intracellular ROS was preformed using
2′,7′-dichlorofluorescein diacetate (DCFH-DA; Sigma). Cultured
HL-7702 cells were plated and transfected into 24-well plate for 36
h with or without the following treatment of hydrogen peroxide
(H2O2) at the final concentration of 250 µM
(Sigma). Culture medium was first removed, and the cells were
washed three times with serum-free DMEM, and diluted DCFH-DA (10 µM
DCFH-DA with serum-free DMEM) was added to the cultures and
incubated at 37°C for 30 min at the same end time point. The
fluorescence was measured at 485 nm for excitation and 530 nm for
emission within 60 min by confocal laser scanning microscopy (Leica
SP5). Relative fluorescence intensity of each group was measured by
LAS AF Lite (Leica).
Indirect immunofluorescence assay
Transfected cells were exposed to the indicated
amount of H2O2 (250 µM) for 12 h with or
without pretreatment with 1 µM CsA or DMSO for 30 min. Cells were
transplanted in 24-well plate cell slides and stained for 30 min
with 150 nM MitoTracker Red (Molecular Probes). After treatment,
the cells were fixed with 4% ice paraformaldehyde for 30 min,
permeabilized with 3‰ Triton X-100 (PBST) for 10 min and incubated
in blocking solution (5% donkey serum albumin in PBST) for 45 min.
Then, the cells were incubated with primary antibodies (anti-Bax;
1:100) at 4°C overnight. The fluorescence-labeled secondary
antibodies (donkey anti-rabbit fluorescence-labeled secondary
antibodies; Alexa Fluor® 488-conjugated; 1:500;
Molecular Probes) were added and incubated for 60 min at 37°C in
the dark, and the sections were mounted using Antifade mounting
medium (Beyotime). Fluorescence images were obtained by confocal
laser scanning microscopy (Leica SP5).
Apoptosis assay
Transfected cells were exposed to the indicated
amount of H2O2 at the final concentration of
250 µM for 12 h with or without pretreatment with 1 µM CsA or DMSO
for 30 min. The extent of apoptosis was evaluated using the Annexin
V-FITC apoptosis detection kit (BD Biosciences) by flow cytometric
analysis.
Statistical analysis
Each set of experiments was repeated at least three
times with similar results. The values given are presented as mean
± SD. Statistical analysis was performed using the Student's
t-test. In all cases, P<0.05 was considered to indicate a
statistically significant result.
Results
Intracellular expression of
recombinant plasmids in transfected cells
Cultures of HL-7702 cells transiently transfected
with recombinant pcDNA3.1, HBx-flag, pHBV, pHBV-HBx and HBx-flag
fusion protein and HBcAg were detected by western blotting for
assessment of HBx-flag and pHBV/pHBV-HBx (Fig. 1A).
HBx reduces COX activity to decrease
cell ATP levels and modulate mitochondrial membrane potential
COX is a vital component of the mitochondrial
respiratory chain. After transfections, a significant decrease in
COX activity was noted in the HBx-flag- and pHBV-transfected cells
compared with the activity in the cells transfected with pcDNA3.1
and pHBV-HBx (Fig. 1B). Correlated
with the downregulation of COX activity, we found an obvious
decrease in cell ATP levels in the HBx-flag- and pHBV-transfected
cells vs. that in the cells transfected with pcDNA3.1 and pHBV-HBx
(Fig. 1C). Consistently, flow
cytometric analysis of Rhodamine-123-stained cells was used to
determine the effects of HBx on mitochondrial membrane potential
(Fig. 1D). Results for the four
groups indicated that HBx reduced the mitochondrial membrane
potential in the HL-7702 cells. All the results suggest that since
COX is essential for the generation of cell ATP and maintenance of
mitochondrial membrane potential (30), the reduction in COX activity by HBx
may be the initial factor of HBx-induced mitochondrial dysfunction,
including regulation of MPTP and induction of the mitochondrial
apoptosis pathway.
HBx increases the cytosolic calcium levels via
modulating MPTP in HL-7702 cells. The transfected cells were loaded
with Ca2+ sensitive dye Fluo-4 to monitor the occurrence
of cytosolic calcium levels. As shown in Fig. 2A and B, Fluo-4 fluorescence was
significantly increased in the HBx- and pHBV-expressing cells vs.
that in the control group. The result was consistent with a
previous study in HepG2 cells (11). To determine whether the association
with mitochondria is involved in the HBx-induced elevation of
cytosolic calcium, we next examined the cytosolic calcium levels in
the four groups treated with CsA (1 µM), a specific inhibitor of
MPTP. The data showed that 1 µM CsA partially decreased the
cytosolic calcium concentrations in four groups. Meanwhile,
following treatment with CsA, cytosolic calcium of HBx-flag cells
was reduced to a level similar to pcDNA3.1 cells without CsA.
Similarly, pHBV cells also exhibited a reduction in fluorescence
intensity with CsA to the extent of pHBV-HBx cells, which indicated
that 1 µM CsA significantly blocked the function of HBx in
modulating MPTP.
HBx increases intracellular ROS and
sensitizes HL-7702 cells to oxidative stress
To further evaluate whether susceptibility of
HL-7702 cells under oxidative stress conditions could be disturbed
by HBx, cells with or without H2O2 treatment
were incubated with DCFH-DA and fluorescence was evaluated by
confocal laser scanning microscopy (Fig. 3A). Notably, no apparent change was
noted in the HBx-expressing cells compared with the control group
without treatment of H2O2, while a
significant increase in fluorescence intensity was observed in the
pHBV-expressing cells compared with that in the pHBV-HBx-expressing
cells. However, although increased ROS levels were observed in all
four groups when exposed to H2O2, an
increased number of DCFH-DA-positive cells was found in the
H2O2-exposed HBx-expressing cells and
pHBV-expressing cells than the control groups under the same
condition (Fig. 3B).
HBx induces translocation of Bax to
mitochondria upon exposure to H2O2 via
modulating MPTP
Bax is the key effector of the intrinsic apoptotic
pathway initiated in response to diverse stimuli (23,31).
Evidence indicates that Bax maintains a balance between the cytosol
and mitochondria in the non-apoptotic state, while under the
stimulation of apoptosis, cytosolic Bax changes its formation and
then insert into the mitochondrial outer membrane, forming large
channels at the mitochondrial outer membrane leading to the release
of cytochrome c (32).
Therefore, translocation of Bax from the cytosol to the
mitochondrion may be an essential step in mitochondrial-mediated
apoptosis. HBx has been shown to induce the translocation of Bax
under serum starvation condition in HepG2 cells (24). To explore the state of Bax in
HBx-expressing cells upon H2O2 in normal
hepatocytes, we proceeded with fluorescent staining experiments by
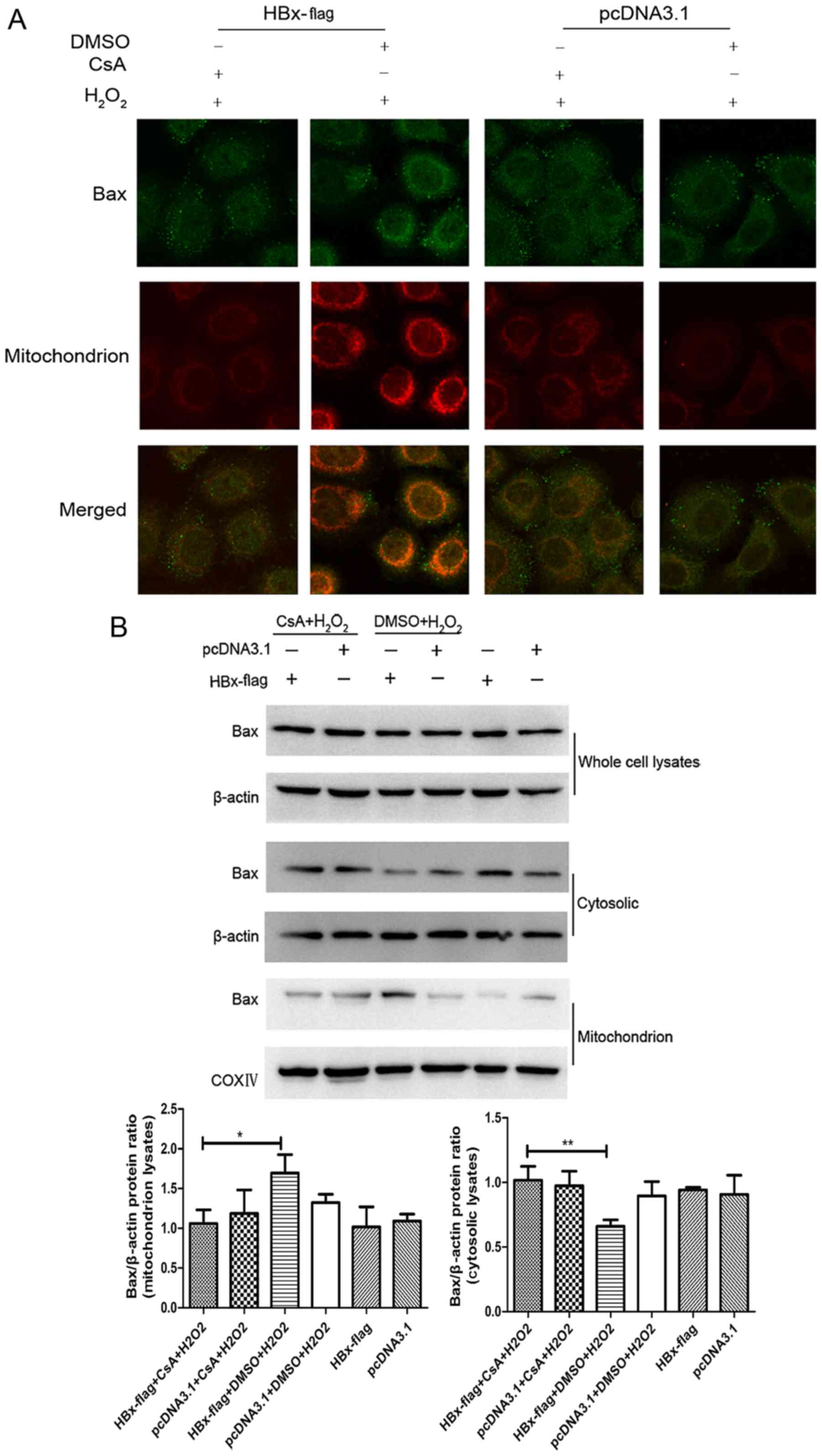
a combination of anti-Bax antibody and MitoTracker Red. As shown in
Fig. 4A, the staining of Bax (green
color) along with mitochondria (red color) was observed (yellow) in
the HBx-flag cells exposed to H2O2 while the
HBx-flag cells without treatment of H2O2 did
not show a visible co-localization of Bax and mitochondria, and
this translocation of Bax was blocked by pretreatment of CsA. In
addition, western blot analysis revealed that the whole protein
levels of Bax did not show an apparent change in each group. While
the expression of Bax protein exhibited a significant decrease in
the cytosol and an increase in the mitochondria in the
HBx-expressing cells treated with H2O2,
pretreatment with CsA blocked the translocation of Bax (Fig. 4B). Both results suggest that HBx
induced translocation of Bax to the mitochondria during oxidative
stress, and MPTP blockage with CsA recovered the balance of Bax
between the mitochondria and the cytosol in the HL-7702 cells.
MPTP blockage with CsA attenuates the
pro-apoptotic effect of HBx in response to oxidative stress
It has been reported that HBx sensitizes cells to
oxidative stress-induced apoptosis (19,20),
while the specific mechanism remains controversial. To investigate
the relationship between HBx and MPTP in oxidative stress-induced
apoptosis, we next examined the effects of an MPTP inhibitor for
its ability to regulate the expression of proteins involved in the
intrinsic apoptosis pathway in H2O2-exposed
HBx-expressing cells by western blot analysis. As shown in Fig. 5A and B, the expression of cleaved
caspase-3 and PARP was markedly decreased as a consequence of the
release of cytosolic cytochrome c in the
H2O2-exposed HBx-expressing cells. We also
found a significant increase in the apoptosis level in the
H2O2-exposed HL-7702-HBx cells by flow
cytometric analysis (Fig. 5C and
D), which was consistent with the immunoblot results.
Additionally, MPTP inhibitor CsA apparently reduced the expression
of cleaved caspase-3 and PARP and blocked the release of cytochrome
c, ultimately preventing the extent of apoptosis.
Discussion
HBx is known to modulate numerous cellular pathways,
including cell cycle regulation, activation of transcription
factors, viral replication, ROS generation and regulation of
apoptosis. Among all these activities, HBx-induced apoptosis is
thought to directly impact the development of HBV-associated liver
damage (3). Given the discrepancy
of the molecular mechanism of HBx in different cells, we usually
conduct our research from the perspectives of a hepatic cell line
and hepatoma cell line, respectively. In the present study, we
explored the molecular mechanism responsible for HBX-induced
apoptosis upon exposure to oxidative stress in HL-7702, a human
normal liver cell line.
MPTP is a large multi-protein complex and composed
of voltage-dependent anion channel (VDAC), adenine nucleotide
translocator (ANT), creatine kinase, hexokinase, the benzodiazapine
receptor and cyclophilin D (27).
It can form a large channel between cytosolic and mitochondrial
matrix. Opening of MPTP in response to numerous mitochondrial
stimuli is correlated with the swelling of mitochondria matrix with
a release in molecules from the mitochondria matrix, including
cytochrome c and calcium, indicating that opening of MPTP
may be an early event in apoptosis (12,33).
Various studies have reported that a fraction of cytosolic HBx
co-localizes with the mitochondria in different cell lines, with
studies in HepG2 cells suggesting that HBx could modulate the onset
of MPTP to increase the level of cytosolic calcium, which
contributes to the elevation of HBV replication, and this activity
of HBx is blocked by treatment with CsA (11), a MPTP inhibitor. This result was
consistent with the present study in HL-7702 cells that HBx has the
ability to modulate MPTP, therefore causing a pronounced increase
in cytosolic calcium levels. Additionally, another study reported
that HBx requires MPTP activity to modulate the levels of cell
cycle regulatory proteins (34).
All these studies suggest that the opening of MPTP may contribute
to the disruption of hepatocyte physiology. Involvement of the MPTP
in HBx may be related to the co-localization of HBx and
mitochondria, while the exact mechanism has not been fully
understood. We previously reported a new HBx-interactive protein
COXIII (8,9,35,36),
which is one of the subunits of cytochrome c oxidase (COX)
residing in the mitochondrial inner membrane in addition to VDAC3.
COX functions as a terminal enzyme of the respiratory chain, and
significantly influences the generation of ATP or depolarization of
the mitochondrial membrane potential (37). The present study demonstrated that
HBx significantly decreased the activity of COX, followed by a
consistent reduction in cell ATP levels and mitochondrial membrane
potential, both of which are responsible for the opening of the
MPTP (38). As we predicted,
cytosolic calcium overload was observed in the HBx-expressing
HL-7702 cells as a consequence of MPTP opening, while the calcium
levels were significantly reduced using CsA. The study in pHBV- and
pHBV-HBx-expressing cells further confirmed these results. Thereby,
it is possible that reduction in COX activity could be an initial
factor in modulating the opening of the MPTP.
Persistent oxidative stress has been suggested to
play a pivotal role in a wide variety of pathophysiological
conditions (13,21). Previous study in our laboratory
showed that HBx expression could enhance ROS production (8,9)
compared with the control group. Other studies suggest that HBx
induces apoptosis by sensitizing cells and tissues to
H2O2 (19).
Notably, ROS accumulation has been shown to upregulate HBx
expression (39). In the present
study, the concentration of H2O2 we used did
not show an apparent change in ROS levels in HBx-expressing cells
or the control group, however, it induced a significant enhancement
of ROS levels in the pHBV-expressing cells compared with the
control group, demonstrating that HBx-induced ROS accumulation in
normal liver cells was dependent on constitutively replicated HBV.
When exposed to H2O2, a pronounced
accumulation of ROS was observed in the HBx- and pHBV-expressing
cells vs. the control groups, suggesting that HBx protein
sensitized HL-7702 cells to oxidative stress. Results from previous
studies showed that exposure to H2O2 caused
the opening of MPTP, resulting in mitochondrial membrane
depolarization and the release of cytochrome c from
mitochondria to the cytosol (12),
while CsA prevents H2O2-induced apoptosis
(40). Accordingly, modulation of
MPTP by HBx may be a potential mechanistic link between HBx and
oxidative stress-induced apoptosis.
Molecules of apoptosis can be released from the
mitochondria through disruption in the outer mitochondrial
membrane, as a consequence of the co-localization of HBx and
mitochondria, pointing out an important role of BcL-2 family
protein which is made up of outer mitochondrial membrane proteins
in the mitochondrial-mediator apoptosis pathway (41). Both of these proteins play a crucial
role in maintaining mitochondrial membrane integrity (42). Various data demonstrated that
pro-apoptotic Bax plays an important role in
H2O2-induced apoptosis via translocation from
cytosol to mitochondria with a release of cytochrome c
(25). In addition, VDAC, which is
a component of MPTP, is required for the efficient translocation of
Bax. During apoptotic stimuli, Bax is dissociated from VDAC2 and
homoligomerizes to form high molecular weight oligomers (26), indicating that Bax translocation is
probably dependent on the status of MPTP. In the present study, we
found a significant co-localization of Bax and mitochondria in the
HBx-expressing HL-7702 cells with a release of cytosolic cytochrome
c and activation of cleaved caspase-3 or PARP when exposed
to H2O2. In HBx-expressing cells pre-treated
with CsA, this translocation of Bax was blocked. Given these
observations, we propose that mitochondrial localization of Bax has
a role in HBx-induced apoptosis during oxidative stress. Moreover,
this mechanism is associated with the function of HBx in modulating
MPTP. Flow cytometric analysis further confirmed that an
intracellular increase in H2O2 had a critical
effect on HBx-induced apoptosis in HL-7702 cells, and this effect
was abrogated by inhibition of MPTP. Our result is consistent with
previous observations that HBx increases the susceptibility of
different hepatocytes to apoptotic stimuli. Nevertheless, evidence
supports an opposite role of HBx, indicating a critical role of HBx
in anti-apoptosis signals. The discrepant consequences of HBx
expression on modulation of apoptosis may result from different
cell lines and different stages of natural HBV infection.
Accordingly, more research using hepatoma cell lines are needed in
the future. HBx-induced apoptosis during H2O2
exposure was significantly reduced with the pre-treatment of CsA,
indicating that modulating of MPTP may contribute to oxidative
stress-induced apoptosis in HBx-expressing cells.
In summary, the present study in HL-7702 cells
demonstrated that HBx enhances the susceptibility of normal
hepatocytes to oxidative stress-induced apoptosis and this effect
is associated with the ability of HBx to modulate MPTP, which is
considered as the major cause of Bax mitochondrial translocation.
This effect of HBx in response to oxidative stress may offer a new
clue that HBx and oxidative signals may work together to upregulate
cellular ROS to a deleterious level, thereby contributing to
HBV-associated inflammation and hepatocarcinogenesis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (81300321), the Key Clinical
Specialty Discipline Construction Program of Fujian, China (Min Wei
Ke Jiao 2012 No. 49), the Young and Middle-Aged Personnel Training
Project of Fujian Province Health Department (2014-ZQN-ZD-9), and
the Natural Science Foundation of Fujian Province, China
(2016J05189). We thank Professor M.J. Bouchard for the pHBV and
pHBV-HBx plasmid.
Glossary
Abbreviations
Abbreviations:
|
Bcl-2
|
B-cell lymphoma-2
|
|
Bax
|
Bcl-2-associated X protein
|
|
COX
|
cytochrome c oxidase
|
|
CsA
|
cyclosporin A
|
|
H2O2
|
hydrogen peroxide
|
|
HBV
|
hepatitis B virus X protein
|
|
HCC
|
hepatocellular carcinoma
|
|
DCFH-DA
|
2,7-dichlorofluorescein diacetate
|
|
HBx
|
hepatitis B virus X protein
|
|
MPTP
|
mitochondrial permeability transition
pore
|
|
PBS
|
phosphate-buffered saline
|
|
ROS
|
reactive oxygen species
|
|
VDAC
|
voltage-dependent anion channel
|
References
|
1
|
Feitelson MA and Lee J: Hepatitis B virus
integration, fragile sites, and hepatocarcinogenesis. Cancer Lett.
252:157–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murakami S: Hepatitis B virus X protein: A
multifunctional viral regulator. J Gastroenterol. 36:651–660. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rawat S, Clippinger AJ and Bouchard MJ:
Modulation of apoptotic signaling by the hepatitis B virus X
protein. Viruses. 4:2945–2972. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma J, Sun T, Park S, Shen G and Liu J: The
role of hepatitis B virus X protein is related to its differential
intracellular localization. Acta Biochim Biophys Sin. 43:583–588.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huh KW and Siddiqui A: Characterization of
the mitochondrial association of hepatitis B virus X protein, HBx.
Mitochondrion. 1:349–359. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rahmani Z, Huh KW, Lasher R and Siddiqui
A: Hepatitis B virus X protein colocalizes to mitochondria with a
human voltage-dependent anion channel, HVDAC3, and alters its
transmembrane potential. J Virol. 74:2840–2846. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shirakata Y and Koike K: Hepatitis B virus
X protein induces cell death by causing loss of mitochondrial
membrane potential. J Biol Chem. 278:22071–22078. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zou LY, Zheng BY, Fang XF, Li D, Huang YH,
Chen ZX, Zhou LY and Wang XZ: HBx co-localizes with COXIII in
HL-7702 cells to upregulate mitochondrial function and ROS
generation. Oncol Rep. 33:2461–2467. 2015.PubMed/NCBI
|
|
9
|
Zheng BY, Fang XF, Zou LY, Huang YH, Chen
ZX, Li D, Zhou LY, Chen H and Wang XZ: The co-localization of HBx
and COXIII upregulates COX-2 promoting HepG2 cell growth. Int J
Oncol. 45:1143–1150. 2014.PubMed/NCBI
|
|
10
|
Gearhart TL and Bouchard MJ: Replication
of the hepatitis B virus requires a calcium-dependent HBx-induced
G1 phase arrest of hepatocytes. Virology. 407:14–25. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McClain SL, Clippinger AJ, Lizzano R and
Bouchard MJ: Hepatitis B virus replication is associated with an
HBx-dependent mitochondrion-regulated increase in cytosolic calcium
levels. J Virol. 81:12061–12065. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takeyama N, Miki S, Hirakawa A and Tanaka
T: Role of the mitochondrial permeability transition and cytochrome
c release in hydrogen peroxide-induced apoptosis. Exp Cell Res.
274:16–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jaeschke H: Reactive oxygen and mechanisms
of inflammatory liver injury: Present concepts. J Gastroenterol
Hepatol. 26:(Suppl 1). S173–S179. 2011. View Article : Google Scholar
|
|
14
|
Liang X, Liu Y, Zhang Q, Gao L, Han L, Ma
C, Zhang L, Chen YH and Sun W: Hepatitis B virus sensitizes
hepatocytes to TRAIL-induced apoptosis through Bax. J Immunol.
178:503–510. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miao J, Chen GG, Chun SY and Lai PPS:
Hepatitis B virus X protein induces apoptosis in hepatoma cells
through inhibiting Bcl-xL expression. Cancer Lett. 236:115–124.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rawat S and Bouchard MJ: The hepatitis B
virus (HBV) HBx protein activates AKT to simultaneously regulate
HBV replication and hepatocyte survival. J Virol. 89:999–1012.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen L, Zhang X, Hu D, Feng T, Li H, Lu Y
and Huang J: Hepatitis B virus X (HBx) play an anti-apoptosis role
in hepatic progenitor cells by activating Wnt/β-catenin pathway.
Mol Cell Biochem. 383:213–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Madden CRF, Finegold MJ and Slagle BL:
Expression of hepatitis B virus X protein does not alter the
accumulation of spontaneous mutations in transgenic mice. J Virol.
74:5266–5272. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu L, Chen L, Yang G, Li L, Sun H, Chang
Y, Tu Q, Wu M and Wang H: HBx sensitizes cells to oxidative
stress-induced apoptosis by accelerating the loss of Mcl-1 protein
via caspase-3 cascade. Mol Cancer. 10:432011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim WH, Hong F, Jaruga B, Zhang ZS, Fan
SJ, Liang TJ and Gao B: Hepatitis B virus X protein sensitizes
primary mouse hepatocytes to ethanol- and TNF-alpha-induced
apoptosis by a caspase-3-dependent mechanism. Cell Mol Immunol.
2:40–48. 2005.PubMed/NCBI
|
|
21
|
Brady NR, Hamacher-Brady A, Westerhoff HV
and Gottlieb RA: A wave of reactive oxygen species (ROS)-induced
ROS release in a sea of excitable mitochondria. Antioxid Redox
Signal. 8:1651–1665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Breckenridge DG and Xue D: Regulation of
mitochondrial membrane permeabilization by BCL-2 family proteins
and caspases. Curr Opin Cell Biol. 16:647–652. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Westphal D, Dewson G, Czabotar PE and
Kluck RM: Molecular biology of Bax and Bak activation and action.
Biochim Biophys Acta. 1813:521–531. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim HJ, Kim SY, Kim J, Lee H, Choi M, Kim
JK and Ahn JK: Hepatitis B virus X protein induces apoptosis by
enhancing translocation of Bax to mitochondria. IUBMB Life.
60:473–480. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahmad KAIK, Iskandar KB, Hirpara JL,
Clement MV and Pervaiz S: Hydrogen peroxide-mediated cytosolic
acidification is a signal for mitochondrial translocation of Bax
during drug-induced apoptosis of tumor cells. Cancer Res.
64:7867–7878. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma SB, Nguyen TN, Tan I, Ninnis R, Iyer S,
Stroud DA, Menard M, Kluck RM, Ryan MT and Dewson G: Bax targets
mitochondria by distinct mechanisms before or during apoptotic cell
death: A requirement for VDAC2 or Bak for efficient Bax apoptotic
function. Cell Death Differ. 21:1925–1935. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lemasters JJ, Nieminen AL, Qian T, Trost
LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA,
Brenner DA, et al: The mitochondrial permeability transition in
cell death: A common mechanism in necrosis, apoptosis and
autophagy. Biochim Biophys Acta. 1366:177–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Melegari M, Scaglioni P-P and Wands JR:
Cloning and characterization of a novel hepatitis B virus × binding
protein that inhibits viral replication. J Virol. 72:1737–1743.
1998.PubMed/NCBI
|
|
29
|
Scaglioni PP, Melegari M and Wands JR:
Posttranscriptional regulation of hepatitis B virus replication by
the precore protein. J Virol. 71:345–353. 1997.PubMed/NCBI
|
|
30
|
Siletsky SA and Konstantinov AA:
Cytochrome c oxidase: Charge translocation coupled to
single-electron partial steps of the catalytic cycle. Biochim
Biophys Acta. 1817:476–488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li H, Wang B, Zhu C, Feng Y, Wang S,
Shahzad M, Hu C, Mo M, Du F and Yu X: 17β-estradiol impedes
Bax-involved mitochondrial apoptosis of retinal nerve cells induced
by oxidative damage via the phosphatidylinositol 3-kinase/Akt
signal pathway. J Mol Neurosci. 50:482–493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schellenberg B, Wang P, Keeble JA,
Rodriguez-Enriquez R, Walker S, Owens TW, Foster F, Tanianis-Hughes
J, Brennan K, Streuli CH, et al: Bax exists in a dynamic
equilibrium between the cytosol and mitochondria to control
apoptotic priming. Mol Cell. 49:959–971. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang JC and Cortopassi GA: Induction of
the mitochondrial permeability transition causes release of the
apoptogenic factor cytochrome c. Free Radic Biol Med. 24:624–631.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Clippinger AJ, Gearhart TL and Bouchard
MJ: Hepatitis B virus X protein modulates apoptosis in primary rat
hepatocytes by regulating both NF-kappaB and the mitochondrial
permeability transition pore. J Virol. 83:4718–4731. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li D, Wang XZ, Yu JP, Chen ZX, Huang YH
and Tao QM: Cytochrome c oxidase III interacts with hepatitis B
virus X protein in vivo by yeast two-hybrid system. World J
Gastroenterol. 10:2805–2808. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang XZ, Li D, Tao QM, Lin N and Chen ZX:
A novel hepatitis B virus X-interactive protein: Cytochrome c
oxidase III. J Gastroenterol Hepatol. 21:711–715. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mkaouar-Rebai E, Ellouze E, Chamkha I,
Kammoun F, Triki C and Fakhfakh F: Molecular-clinical correlation
in a family with a novel heteroplasmic Leigh syndrome missense
mutation in the mitochondrial cytochrome c oxidase III gene. J
Child Neurol. 26:12–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tsujimoto Y, Nakagawa T and Shimizu S:
Mitochondrial membrane permeability transition and cell death.
Biochim Biophys Acta. 1297–1300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang JH, Yun C, Kim S, Lee JH, Yoon G, Lee
MO and Cho H: Reactive oxygen species modulates the intracellular
level of HBx viral oncoprotein. Biochem Biophys Res Commun.
310:32–39. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sugano N, Ito K and Murai S: Cyclosporin A
inhibits H2O2-induced apoptosis of human
fibroblasts. FEBS Lett. 447:274–276. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Harris MH and Thompson CB: The role of the
Bcl-2 family in the regulation of outer mitochondrial membrane
permeability. Cell Death Differ. 7:1182–1191. 2000. View Article : Google Scholar : PubMed/NCBI
|