Introduction
Autophagy, or autophagocytosis, is from the Greek
auto-, ‘self’, and phagein, ‘to eat’. Autophagy is a
conserved protein degradation pathway that plays important roles in
mammalian cell survival, proliferation and differentiation,
particularly within the hematopoietic system. Diminished autophagic
flux results in the development of a myeloproliferative state or
acute myeloid leukemia (AML) (1).
In AML, there is copy number loss in autophagic genes such as
BECN1 (2,3), ATG7 (1,4) and
ATG5 (1,5). Decreased autophagy and the development
of AML are related. BECN1 is a critical mediator that
influences the onset and progress of autophagy, and there is a
remarkable association between reduced BECN1 expression and
FLT3-internal tandem duplication (ITD) mutation (6). ATG7 and ATG5 play
important roles in autophagy and their loss of function in
hematopoietic stem and progenitor cells (HSPCs) always leads to a
lethal pre-leukemic phenotype in mice (1).
Recently, autophagy pathway inducers have shown
promising effects for treating AML. Mammalian target of rapamycin
(mTOR) signaling is a critical pathway in the biology of several
cancers, including AML. Constitutive activation of the
phosphatidylinositol 3-kinase (PI3K)/mTOR signaling pathway has
been observed in different cancers and leukemias, including chronic
myelogenous leukemia (CML), AML and acute lymphoblastic leukemia
(ALL). The PI3K/mTOR pathway has always been considered a
prosurvival factor in leukemia stem cells and leukemic precursors,
and its inhibition has been regarded as an effective therapeutic
approach (7). MLN0128 is a novel,
recently developed mTOR kinase inhibitor that can disrupt survival
signaling and triggers apoptosis in AML stem and AML progenitor
cells (8). Abnormal mTOR activity
contributes to chemotherapy resistance, and aberrant activation of
the PI3K/mTOR pathway promotes sorafenib resistance in AML cells
(9).
The serine/threonine protein kinase polo-like kinase
1 (PLK1), or serine/threonine-protein kinase 13 (STPK13), regulates
multiple intracellular processes, including DNA replication,
mitosis and stress response. PLK1 is expressed during mitosis and
is overexpressed in multiple cancers, including breast cancer
(10), prostate cancer (11), renal cancer (12) and neuroblastoma (13). PLK1 is also highly expressed in
leukemia cell lines; PLK1 expression in patients with AML is
significantly higher than in normal progenitors (14). Furthermore, PLK1 expression in
normal or untransformed cells is much lower than in cancer cells,
which renders PLK1 a suitable anticancer target (15,16).
Downregulating or inhibiting the kinase activity of PLK1 induces
cell cycle arrest and apoptosis in most cancer cell types in
vitro and in vivo (17–20).
The potential of PLK1 inhibitors as cancer therapeutics has been
investigated widely. The PLK1 inhibitor volasertib has shown
considerable promise in clinical studies of AML, having reached
phase III trials (21,22). Other PLK1 inhibitors, including
GSK461364A, TKM-080301, GW843682, purpurogallin and poloxin are in
early clinical development (23).
To date, the molecular function of PLK1 in AML cell
autophagy is unclear. In our study, the autophagy-related effect of
PLK1 was evaluated in AML cells to characterize its preclinical
efficacy.
Materials and methods
Animal and human rights statement
The studies have been performed in accordance with
the ethical standards as laid down in the 1964 Declaration of
Helsinki and its later amendments. Ethical approval was provided by
the Children's Hospital of Soochow University Ethics Committee
(nos. SUEC2008-011 and SUEC2000-021).
Cell and culture conditions
Leukemia cell lines HL-60 and K562 were obtained
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). NB4 cell line (gifts from Hematology Institute of Soochow
University). All cell lines were maintained at 37°C in the
RPMI-1640 (Gibco Life Technologies, Carlsbad, CA, USA) supplemented
with 10% fetal bovine serum (FBS; Invitrogen Life Technologies,
Carlsbad, CA, USA). Sixty-nine inhibitors ABT-263, ABT-737, YM155,
SK1-I, SKI-5C, 17-AAG, XAV-939, AC220, tosedostat (CHR2797),
VER-50589, FH535, G-749, BV-6 (apoptosis and anti-apoptosis);
rapamycin, valproic acid, 3-methyladenine (3-MA), BEZ235, HS-173,
pilaralisib (autophagy); SP600125, elesclomol, BAY 11–7082,
ipatasertib, SB202190, PD98059, LY294002, INCB018424, SH-4-54,
AT13148, JNK inhibitor IX, PX-478 2HCl (oxidative stress and MAPK
pathway); BI 2536, PF-3758309, nutlin-3, MI-773, YH239-EE, XL-413,
MLN0905, SBE13 HCL, RO3280, volasertib, nutlin-3b (cell cycle);
JIB-04, GSK J1, GSK J4, GSK 126, LBH589, SGC-CBP30, 4SC-202
(histone modification); KPT-276, KPT-330, KPT-185, KPT-335 (CRM1);
CW069, TAPI-1, INH6, ODM-201, ESI-09, EW-7197, GDC-0623, AZD6738,
LY3009120, SB-3CT, INH1, XMD8-92, LY2584702, ML323 (other targets).
Tosedostat (CHR2797) was purchased from Molbase Chemicals
(Shanghai, China). Other inhibitors were purchased all from Selleck
Chemicals (West Paterson, NJ, USA) and was dissolved in dimethyl
sulfoxide (DMSO) (cat. no. D4540; Sigma-Aldrich, St. Louis, MO,
USA).
Expression of hLC3 in leukemia cells
with UBC-LC3-GFP lentivirus
UBC-LC3-GFP lentivirus was purchased from Shanghai
Genechem Co., Ltd. (Shanghai, China) (24). Lentivirus infection was according to
the manufacturer (Shanghai GeneChem Co, Ltd.) at a final
concentration of 100–200 multiplicity of infection (MOI).
Aggregation of LC3 was observed when the autophagy occurred in
cells. LC3-GFP fusion protein is diffused in the cytoplasm when the
cells occur autophagy. LC3-GFP fusion protein translocates to
autophagic membranes, forming a bright green fluorescence spots
under the fluorescence confocal microscopy (Olympus; Olympus
Corporation, Tokyo, Japan) when the cells are autophagic, a spot is
equivalent to an autophagosome thus can be counted to evaluate the
level of autophagy activity.
Transmission electron microscopy
(TEM)
The presence of autophagosomes in TEM is the
standard for detecting autophagy. TEM analysis protocol was
according to a previous report (25). Briefly, leukemia cells grown on
6-well plates were treated with RO3280, BI2536 or the same volume
of DMSO and 4 h later were harvested and washed with
phosphate-buffered saline (PBS) prior to fixing in fixative buffer.
Subsequently, cells were collected and suspended with 2.5%
glutaraldehyde. Then treated with 2% osmium tetroxide in 0.1 M
sodium cacodylate buffer, and embedded in resin. Finally, the
samples were sliced and processed for TEM analysis (Hitachi
electron microscope H-600; Hitachi, Ltd., Tokyo, Japan).
Interfering expression of PLK1 in
leukemia cells with RNAi lentivirus
RNAi lentivirus for PLK1 was purchased from Shanghai
GeneChem Co, Ltd. RNAi products consist of target-specific
lentivirus designed to knock down PLK1 expres- sion sequences are:
5′-CCGAGTTATTCATCGAGAC-3′. The control sequences are:
5′-TTCTCCGAACGTGTCACGT-3′. Lentivirus infection was according to
the manufacturer (Shanghai Genechem Co., Ltd.) at a final
concentration of 100–200 MOI. PLK1 interference efficiency was
measured by western blotting at 3 days after transfection. The rest
of the cells were harvested for further analysis.
Cell proliferation analysis
Leukemia cells (2×104) were seeded in
96-well plates overnight and incubated with DMSO, or increasing
concentrations of RO3280, BI2536 24 to 96 h. Each drug
concentration was performed replicated 2 times. Then, 10 µl Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) solution was added to each well, incubated at 37°C
for 2 to 4 h and the optical density (OD) values were measured at
450 nm using a scanning multi-well spectrophotometer (Bio-Rad Model
550; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Apoptosis assay
Apoptosis assay was according to the manual
operation of BD Annexin V staining kit (cat. no. 556420; BD
Biosciences, Franklin Lakes, NJ USA). Briefly, cells were washed 2
times with cold PBS and then were resuspend in 1X binding buffer at
a concentration of ~1×106 cells/ml, and 100 µl of the
solution (~1×105 cells) was transfered to a 5-ml culture
tube. Annexin V and PI (5 µl/test) was added. Cells were gently
mixed and incubated for 15 min at room temperature in the dark.
Four hundred microliters of 1X binding buffer was added to each
tube. Cells were analyzed by flow cytometry within 1 h (26).
Western blotting
For western blotting, protocol was introduced as
described (26). Cellular proteins
were blocked and then probed with antibodies against PARP (cat. no.
9542S, 1:1,000), PLK1 (cat. no. 4535S, 1:1,000), beclin-1 (cat. no.
3495S, 1:1,000), SQSTM1/p62 (cat. no. 8025S, 1:1,000), LC3A/B (cat.
no. 4108S, 1:1,000), mTOR (cat. no. 2983S, 1:1,000), phospho-mTOR
(Ser2448) (cat. no. 5536S, 1:1,000), adenosine
monophosphate-activated protein kinase (AMPK)α (cat. no. 5831S,
1:1,000), phospho-AMPKα1 (Ser485) (cat. no. 2537S, 1:1,000),
phospho-AMPKα1 (Thr172) (cat. no. 2537S, 1:1,000), Unc-51-like
kinase 1 (ULK1) (cat. no. 6439S, 1:1,000), phospho-ULK1 (Ser317)
(cat. no. 12753S, 1:1,000), phospho-ULK1 (Ser757) (cat. no. 14202S,
1:1,000), phospho-ULK1 (Ser555) (cat. no. 5869S, 1:1,000) all these
antibodies were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA) and GAPDH (1:5,000; Sigma-Aldrich). Relative
expression of p-mTOR/mTOR was analyzed with ImageJ software
according to the user guide.
Patients and samples
Bone marrow specimens were obtained at the time of
diagnosis during routine clinical assessment of 140 pediatric
patients with AML, who presented at the Department of Hematology
and Oncology, Children's Hospital of Soochow University between
2008 and 2013. Ethical approval was provided by the Children's
Hospital of Soochow University Ethics Committee (nos. SUEC2008-011
and SUEC2000-021), and informed consent was obtained from the
parents or guardians. AML diagnosis was made in accordance with the
revised French-American-British (FAB) classification. The main
clinical and laboratory features of the patient cohort are
summarized in Table I.
 | Table I.Association of PLK1 expression with
clinico-pathological characteristics in 140 pediatric AML
samples. |
Table I.
Association of PLK1 expression with
clinico-pathological characteristics in 140 pediatric AML
samples.
|
|
| PLK1 expression
(n) |
|
|---|
|
|
|
|
|
|---|
| Clinical
variables | No. of
patients | Low | High | P-value |
|---|
| Gender |
|
|
| 0.888 |
|
Male | 56 | 34 | 22 |
|
|
Female | 84 | 50 | 34 |
|
| Age (years) |
|
|
| 0.334 |
|
<6 | 73 | 41 | 32 |
|
| ≥6 | 67 | 43 | 24 |
|
| Leukocyte
(/µl) |
|
|
| 0.673 |
|
>10,000 | 83 | 51 | 32 |
|
|
≤10,000 | 57 | 33 | 24 |
|
| FAB |
|
|
| 0.027 |
|
M1-M6 | 123 | 78 | 45 |
|
| M7 | 17 | 6 | 11 |
|
| Cytogenetics |
|
|
| 0.382 |
|
Favorable | 70 | 43 | 27 |
|
|
Intermediate | 38 | 25 | 13 |
|
|
Unfavorable | 32 | 16 | 16 |
|
| MRD |
|
|
| 0.038 |
|
<0.25% | 65 | 45 | 20 |
|
|
≥0.25% | 75 | 39 | 36 |
|
Quantitative reverse
transcription-polymerase chain reaction (PCR) for PLK1
Quantitative real-time PCR was performed to
determine the expression levels of PLK1 genes. Total RNA was
reverse transcribed using the Reverse Transcription kit, according
to the manufacturer's instructions (Applied Biosystems, Foster
City, CA, USA). The real-time PCR primers used to quantify GAPDH
expression were forward, 5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse,
5′-AGGGGCCATCCACAGTCTTC-3′; and for PLK1 were forward,
5′-CTCAACACGCCTCATCCTC-3′ and reverse, 5′-GTGCTCGCTCATGTAATTGC-3′.
Expression of PLK1 was normalized to endogenous GAPDH
expression.
Statistical analysis
Each experimental condition was performed three
times, and these replicates are presented in the results. All
values are presented as means ± SEM. Student's paired t-test was
applied to reveal statistical significance. P-values <0.05 were
considered significant. Statistical analyses were performed using
SPSS software for Windows (version 11.5; SPSS, Inc., Chicago, IL,
USA).
Results
Screening of novel autophagy inducers
of AML cells
As autophagy pathway inducers have shown promising
effects for treating AML, NB4 AML cells overexpressing LC3-GFP
fusion protein were screened with 69 inhibitors (1 µM each; Selleck
Chemicals, Houston, TX, USA) for 4 to 12 h before analysis. LC3
aggregation in the cells was observed during autophagy. LC3-GFP
translocation to the autophagic membranes formed bright green
fluorescent spots under fluorescence confocal microscopy when the
cells underwent autophagy; one spot was equivalent to an
autophagosome, which were counted to evaluate the level of
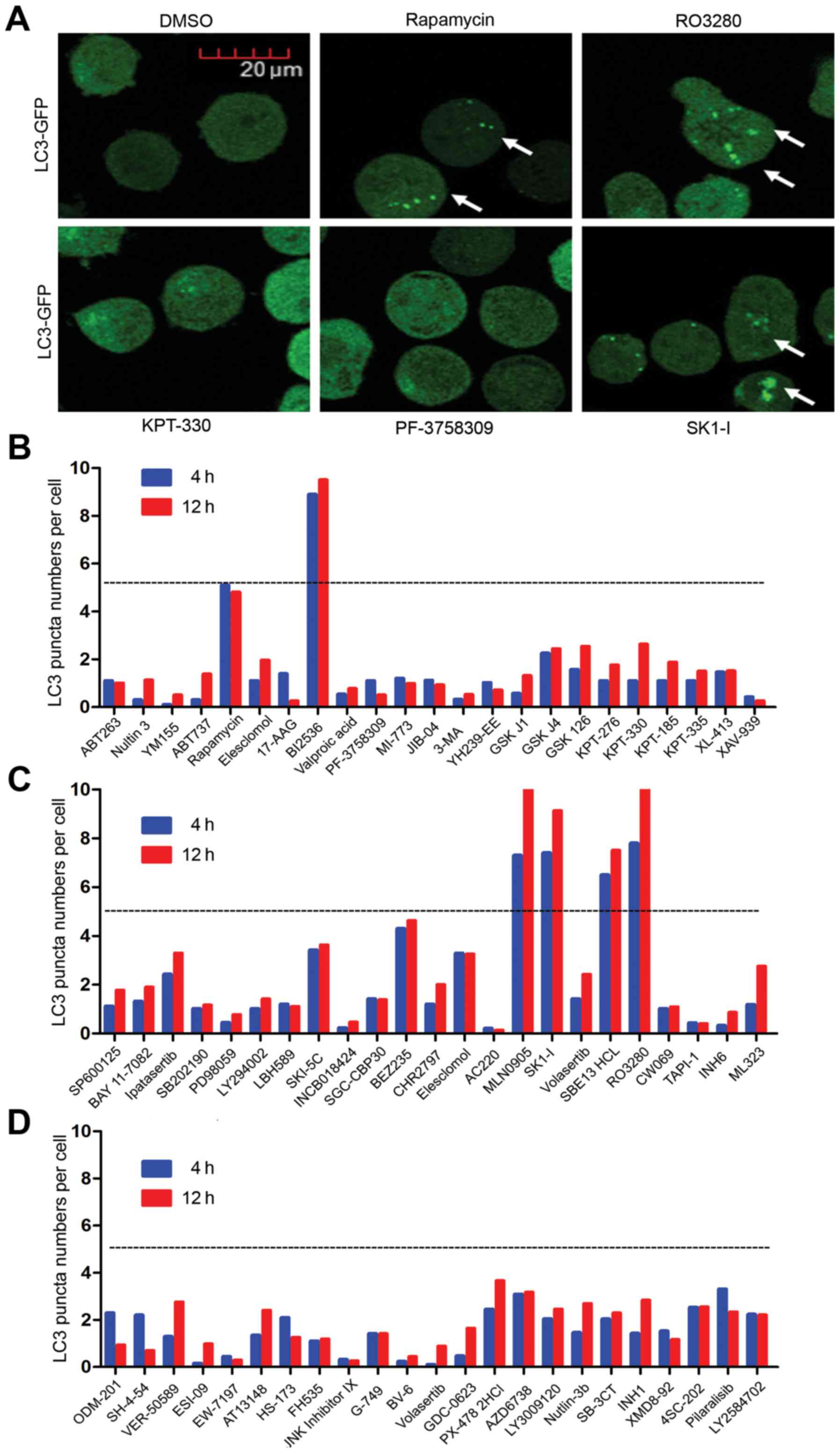
autophagy activity (Fig. 1A).
Autophagy activity was calculated as autophagy spots per number of
cells. Our screening experiment showed that several inhibitors had
promising autophagy inducer effects. Autophagy activity >5 in
the inhibitors BI2536, MLN0905, SK1-I, SBE13 HCL and RO3280
(Fig. 1B-D). Rapamycin was used as
the positive control, where its autophagy activity was ~5. As
reported previously, SK1-I is a novel inhibitor of sphingosine
kinase 1 (SPHK1) (27), an
evolutionarily conserved lipid kinase responsible for converting
sphingosine to sphingosine-1-phosphate (S1P). SPHK1 is
overexpressed in many cancers and therefore serves as a cancer
prognostic biomarker. BI2536, MLN0905, SBE13 HCL and RO3280 are all
PLK1 inhibitors. PLK1 is always highly expressed in leukemic cell
lines and is overexpressed in a majority of samples from patients
with AML compared with normal progenitors (14). Yet, its molecular function in the
autophagy of AML cells is still unclear.
PLK1 inhibitors induce AML cell
autophagy
To confirm the induction of autophagy by the PLK1
inhibitors, we treated NB4 cells with 200 and 500 nM RO3280 or
BI2536 for 4 h before analysis (Fig.
2A), and found that autophagy activity levels were increased
(500 nM BI2536: 9.73±1.89 vs. DMSO: 0.73±0.45, p=0.011; 500 nM
RO3280: 11.63±2.48 vs. DMSO: 0.27±0.15, p=0.015). LC3 is converted
from the inactive form (LC3-I) to the cleaved form (LC3-II) during
autophagy, rendering LC3 a specific marker for detecting autophagy.
Western blotting showed more LC3-II in NB4 cells treated with
RO3280 and BI2536. More LC3-II was observed in the 100–500 nM
RO3280 group and in the 200–500 nM BI2536 group (Fig. 2B). Beclin-1 upregulation and
SQSTM1/p62 downregulation was also observed in the PLK1 inhibitor
treatment groups. TEM was used to examine the autophagosomes in
RO3280-treated cells (Fig. 2C).
Compared with the DMSO control group, there were more
autophagosomes in RO3280-treated NB4 cells. These results are
consistent with the autophagy activity analysis involving LC3-GFP.
Detecting autophagy is related to correct timing. We treated NB4
cells with 0.5 µM RO3280 or BI2536 continuously 4 to 12 h, and
observed their induction of autophagy at different time-points
(Fig. 3A). There was more LC3-II in
the NB4 cells treated with 0.5 µM RO3280 or BI2536. These results
suggest that PLK1 inhibitors induce significant AML cell
autophagy.
PLK1 inhibition by RNA interference
induces AML cell autophagy
Inhibitors tend to have several candidate targets,
and their molecular function is influenced by other candidate
targets, especially homologous molecules. The molecular function of
PLK1 inhibitors is influenced by the candidate targets PLK2, 3 and
4. To confirm the effect of PLK1, we inhibited its expression with
RNA interference. When PLK1 expression was downregulated, western
blotting detected more LC3-II (Fig.
3B). LC3-GFP analysis showed increased autophagy activity
levels when PLK1 expression was significantly downregulated in the
three leukemia cell lines (Fig.
3C). The autophagy activity was calculated and results were as
follows: NB4 cells, Si-PLK1: 11.23±2.43 vs. Si-Nc: 0.33±0.15
(p=0.015); K562 cells, Si-PLK1: 9.73±1.89 vs. Si-Nc: 0.63±0.49
(p=0.011); HL-60 cells, Si-PLK1: 6.73±0.90 vs. Si-Nc: 0.40±0.10
(p=0.002), showing that PLK1 inhibition by RNA interference
significantly induces AML cell autophagy (Fig. 3C).
PLK1 inhibition induces AML cell
autophagy via mTOR dephosphorylation
We demonstrated that PLK1 inhibition induces
significant leukemia cell autophagy. We analyzed the
phosphorylation of these three kinases in NB4 cells treated with
RO3280 and BI2536, and found that mTOR phosphorylation was reduced
significantly in NB4 cells treated with RO3280 and BI2536 (Fig 4A). The dephosphorylation of mTOR is a
crucial step in the induction of autophagy in eukaryotes. ULK1
phosphorylation at Ser317 was also reduced significantly in NB4
cells treated with RO3280 and BI2536. AMPK levels and AMPK
phosphorylation levels did not exhibit significant changes. To
confirm the effect of PLK1, RNA interference lentivirus
transfection of NB4, K562 and HL-60 cells was used to significantly
downregulate PLK1 expression, which was followed by significantly
reduced mTOR phosphorylation (Fig.
4B). These results imply that PLK1 inhibition induces AML cell
autophagy via mTOR dephosphorylation.
PLK1 is an excellent anticancer target
in pediatric AML
PLK1 inhibition induces significant apoptosis in
several cancer cell types. The relationship between autophagy and
apoptosis following PLK1 inhibition in AML cells remains unclear.
Our analysis showed that for 4 and 8 h, NB4 cell apoptosis was
almost identical between the DMSO group and 200 nM RO3280 group,
suggesting that autophagy occurs earlier than apoptosis when cells
are treated with PLK1 inhibitors. Apoptosis increased significantly
when the NB4 cells were treated with RO3280 for 12 h, and it was
inhibited by 5 mM 3-MA, an autophagy inhibitor (Fig. 5A). Autophagy and apoptosis induced
by PLK1 inhibition are closely related, and the specific molecular
mechanism warrants further study. Proliferation analysis showed
that PLK1 inhibition significantly inhibited NB4 cell proliferation
(Fig. 5B and C).
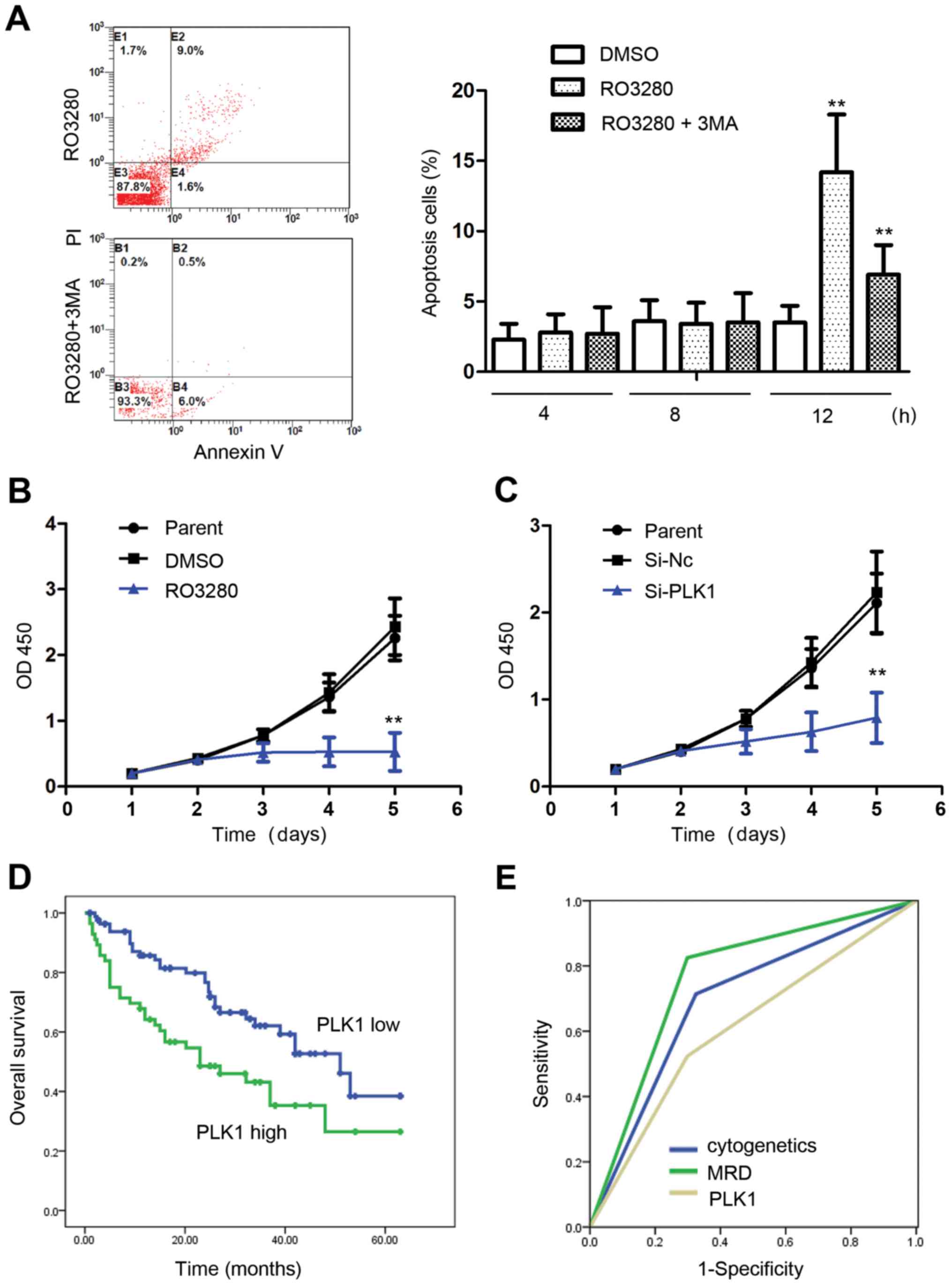 | Figure 5.PLK1 is an excellent anticancer
target for pediatric AML. (A) Analysis of NB4 cell apoptosis
treated with RO3280 or 3-MA. At 4 and 8 h, NB4 cell apoptosis was
almost identical between the DMSO and 200 nM RO3280 group. NB4 cell
apoptosis increased significantly following 12-h treatment with
RO3280, and was inhibited significantly by 5 mM 3-MA. **p<0.01.
(B) Analysis of NB4 cell proliferation following treatment with 200
nM RO3280. (C) Analysis of NB4 cell proliferation following
transfection with PLK1 RNA interference lentivirus at 100 MOI.
*p<0.05, **p<0.01. (D) The prognostic significance of PLK1
expression was assessed in 140 Chinese patients with pediatric AML
with clinical follow-up records. Kaplan-Meier survival analysis
revealed shorter survival in the patients with high PLK1 expression
in tumors (p=0.007). (E) ROC curve analysis showing that the area
under the curve of cytogenetics, MRD, and PLK1 is 0.695, 0.763 and
0.613, respectively. PLK1, polo-like kinase 1; AML, acute myeloid
leukemia; 3-MA, 3-methyladenine; DMSO, dimethyl sulfoxide; MOI,
multiplicity of infection; ROC, receiver operating characteristic;
MRD, minimal residual disease. |
Patients were divided into two groups with average
value of PLK1 expressions (10.32×10−5). Results revealed
that PLK1 expression was related with FAB subtype and minimal
residual disease (MRD; Table I).
Kaplan-Meier survival analysis revealed shorter survival in
patients with high PLK1 expression in tumors (p=0.007; Table II and Fig. 5D). Multivariate analysis revealed
that PLK1 expression is a near independent prognostic factor in
pediatric AML patients (p=0.066; Table III). Receiver operating
characteristic (ROC) curve analysis showed that the area under the
curve for cytogenetic, MRD, and PLK1 expression was 0.695, 0.763
and 0.613, respectively (Fig. 5E).
In conclusion, our results demonstrate that PLK1 is an excellent
anticancer target in pediatric AML.
| Table II.Association of PLK1 expression with
Kaplan-Meier survival in 140 pediatric AML samples. |
Table II.
Association of PLK1 expression with
Kaplan-Meier survival in 140 pediatric AML samples.
| Variables | No. of
patients | Over survival (mean
± SE) | P-value |
|---|
| Cytogenetics |
|
| <0.001 |
|
Favorable | 70 | 48.277±2.857 |
|
|
Intermediate | 38 | 34.785±2.840 |
|
|
Unfavorable | 32 | 13.477±2.245 |
|
| FAB |
|
| 0.001 |
|
M1-M6 | 123 | 40.150±2.344 |
|
| M7 | 17 | 14.120±3.343 |
|
| Leukocyte (µl) |
|
| 0.641 |
|
>10,000 | 83 | 34.611±2.472 |
|
|
≤10,000 | 57 | 36.893±3.431 |
|
| MRD |
|
| <0.001 |
|
<0.25% | 65 | 53.085±2.622 |
|
|
≥0.25% | 75 | 25.061±2.358 |
|
| PLK1
expression |
|
| 0.007 |
| Low
<10.32×10−5 | 84 | 42.253±2.778 |
|
| High
≥10.32×10−5 | 56 | 30.214±3.521 |
|
| Table III.Cox multivariate analysis of PLK1
expression and clinico-pathological features in pediatric AML. |
Table III.
Cox multivariate analysis of PLK1
expression and clinico-pathological features in pediatric AML.
| Variables | Odds ratio | EXP(B) 95% CI | P-value |
|---|
| Cytogenetics |
|
|
|
| Favor.
vs. intermed. and unfavorable | 7.248 | 2.253
(1.247–4.070) | 0.007 |
| MRD |
|
|
|
|
<0.25 vs. ≥0.25% | 11.208 | 3.346
(1.650–6.786) | 0.001 |
| Leukocyte (µl) |
|
|
|
|
>10,000 vs. ≤10,000 | 0.252 | 1.141
(0.681–1.912) | 0.615 |
| FAB
classification |
|
|
|
| M7 vs.
M1-M6 | 6.102 | 2.233
(1.180–4.222) | 0.014 |
| PLK1
expression |
|
|
|
| Low vs.
high | 3.370 | 1.605
(0.968–2.661) | 0.066 |
Discussion
Autophagy functions as a means of managing cellular
stresses and plays a substantial role in tumor cell survival.
Decreased autophagic flux results in the development of a
myeloproliferative state and AML. In AML, there is copy number loss
in the autophagic genes BECN1, ATG7 and ATG5.
AML cells often remain sensitive to autophagy-inducing stimuli,
leading to the idea that harnessing autophagy can be useful in AML
cytotoxic therapy. NB4 AML cells overexpressing LC3-GFP were
treated with 69 inhibitors for 4 to 12 h before analysis, and
several had promising autophagy inducer effects, where the
autophagy activity of BI2536, MLN0905, SK1-I, SBE13 HCL and RO3280
was >5. Very interestingly, these inhibitors all had the same
target, PLK1.
PLK1 is expressed during mitosis and is
overexpressed in multiple cancers, including acute leukemia
(28). PLK1 is also overexpressed
in a majority of samples from patients with AML compared with
normal progenitors. However, the prognostic value of PLK1 in
pediatric AML is still unclear to date. PLK1 supports centrosome
functional maturation in late G2 prophase and the
establishment of the bipolar spindle. These checkpoints, which
occur at G2/M transition, are tightly regulated by
nuclear serine/threonine kinases, including cyclin-dependent
kinases (CDKs), PLKs and Aurora kinases. Evidence suggests that
PLK1 can inhibit the transactivation and proapoptotic functions of
p53 by physical interaction and phosphorylation (29). Overexpression studies of PLK1 in the
NIH3T3 cell line showed that these cells become capable of forming
foci and growing in soft agar, and more importantly, can form
tumors in nude mice due to PLK1 overexpression (30). In our study, Kaplan-Meier survival
analysis revealed shorter survival in patients with high PLK1
expression in tumors. PLK1 inhibition induced significant AML cell
apoptosis. These results suggest that PLK1 is an excellent
anticancer target in pediatric AML. In this study, we demonstrate
that PLK1 inhibition induces AML cell autophagy. Autophagy activity
levels were increased when NB4 cells were treated with the PLK1
inhibitors RO3280 and BI2536. Inhibiting PLK1 expression in NB4,
K562, and HL-60 cells with RNA interference led to more LC3-II, and
autophagy activity was increased when PLK1 expression was
significantly downregulated in these cells. Other members of PLK1
family have been reported with a relationship with autophagy.
Overexpression of PLK3 mediates the degradation of abnormal prion
proteins dependent on chaperone-mediated autophagy (31). PLK2 has a relationship with
α-synuclein which plays an important role in autophagy (32). These results indicate that PLK1
family members may all have an important role in autophay. Also,
this is the first study of autophagy induction in AML cells
following PLK1 inhibition.
To date, the molecular mechanism of PLK1 inhibition
in the induction of AML cell autophagy remains unknown. mTOR, or
FK506-binding protein 12 (FKBP12)-rapamycin- associated protein 1,
is a serine/threonine protein kinase from the family of
PI3K-related kinases. In mammals, amino acid sensing and other
signals such as reactive oxygen species (ROS) and growth factors
regulate the activity of the protein kinases mTOR and AMPK, which
regulate autophagy through inhibitory phosphorylation of ULK1 and
2. Induction of autophagy results in ULK kinase dephosphorylation
and activation. The active ULK relocalizes to the site of
autophagosome initiation, contributing to activation of the
downstream autophagy components. Accordingly, mTOR, AMPK and ULK1
are important regulators of autophagy initiation. As reported
previously, under nutrient sufficiency, high mTOR activity prevents
ULK1 activation by phosphorylating and disrupting the interaction
between ULK1 and AMPK (33–35). This coordinated phosphorylation is
important for ULK1 in autophagy induction. We analyzed the protein
phosphorylation of mTOR, AMPK and ULK1, three important regulators
of autophagy, and found that mTOR phosphorylation was reduced
significantly in NB4 cells treated with RO3280 and BI2536. To
confirm the effect of PLK1 in NB4, K562 and HL-60 cells, mTOR
phosphorylation was also reduced significantly when PLK1 expression
was downregulated using RNA interference. mTOR, a serine/threonine
protein kinase with a molecular size of ~300 kDa, belongs to the
phosphatidylinositol 3-kinase-related kinase (PIKK) family. Its
activity is inhibited under nutrient starvation and other stimuli
of autophagy induction, and its dephosphorylation is a crucial step
in the induction of autophagy in eukaryotes. The PI3K/AKT/mTOR
pathway is very important in cell growth, survival, apoptosis,
angiogenesis and phagocytosis. Disorder of this pathway results in
disease, including cancer, neurological disease and autoimmune
disease. The PI3K/AKT/mTOR pathway is constitutively activated in
~50–80% of AML cases (36), and its
activation is associated with poor prognosis of AML. PI3K/AKT/mTOR
may be an important therapeutic target in AML. Unfortunately,
targeting the mTOR pathway with single-agent rapamycin in clinical
trials was unsuccessful (37). As
an alternative to adding chemotherapy, it is useful to target both
the mTOR pathway and other cancer targets to generate more
effective responses (38,39). As PLK1 inhibitors inhibit the
activity of PLK1 and mTOR, two important pathways in AML, the
present findings imply that PLK1 inhibitors may have good clinical
application in AML. The details of PLK1 regulation of mTOR
phosphorylation are still unknown and warrant further studies.
In conclusion, we demonstrate that PLK1 is an
excellent anticancer target in pediatric AML; its inhibition
induces autophagy of AML cells and results in mTOR
dephosphorylation in AML cells. These results may provide new
insights into the molecular mechanism of PLK1 in autophagy
regulation.
Acknowledgements
This study was supported by grants from National
Natural Science Foundation (81570125, 81370627, 81300423, 81502500,
81501703, 81501840, 81502157, 81501700 and 31500718), Natural
Science Foundation of Jiangsu Province (BK20151207, H201420), Key
Medical Subjects of Jiangsu Province (XK201120), Innovative Team of
Jiangsu Province (LJ201114, LJ201126), Special Clinical Medical
Science and Technology of Jiangsu Province (BL2012050, BL2013014,
BL2012051), Major Scientific and Technological Special Project for
‘Significant New Drugs Creation’ (2012ZX09103301-040). This
manuscript was copy-edited by an English speaking professional with
science background at Elixigen Corporation (Huntington Beach, CA,
USA).
References
|
1
|
Watson AS, Riffelmacher T, Stranks A,
Williams O, De Boer J, Cain K, MacFarlane M, McGouran J, Kessler B,
Khandwala S, et al: Autophagy limits proliferation and glycolytic
metabolism in acute myeloid leukemia. Cell Death Discov.
1:150082015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Radwan SM, Hamdy NM, Hegab HM and
El-Mesallamy HO: Beclin-1 and hypoxia-inducible factor-1α genes
expression: potential biomarkers in acute leukemia patients. Cancer
Biomark. 16:619–626. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu B, Yue QF, Chen Y, Bu FD, Sun CY and
Liu XY: Expression of autophagy related gene BECLIN-1 and number of
autophagic vacuoles in bone marrow mononuclear cells from 40
myelodysplastic syndromes patients and their significance. Zhongguo
Shi Yan Xue Ye Xue Za Zhi. 23:146–149. 2015.(In Chinese).
PubMed/NCBI
|
|
4
|
Piya S, Kornblau SM, Ruvolo VR, Mu H,
Ruvolo PP, McQueen T, Davis RE, Hail N Jr, Kantarjian H, Andreeff
M, et al: Atg7 suppression enhances chemotherapeutic agent
sensitivity and overcomes stroma-mediated chemoresistance in acute
myeloid leukemia. Blood. 128:1260–1269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim Y, Eom JI, Jeung HK, Jang JE, Kim JS,
Cheong JW, Kim YS and Min YH: Induction of cytosine
arabinoside-resistant human myeloid leukemia cell death through
autophagy regulation by hydroxychloroquine. Biomed Pharmacother.
73:87–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zare-Abdollahi D, Safari S, Movafagh A,
Ghadiani M, Tabarraee M, Riazi-Isfahani S, Gorji S, Keyvan L and
Gachkar L: Expression analysis of BECN1 in acute myeloid leukemia:
association with distinct cytogenetic and molecular abnormalities.
Int J Lab Hematol. 38:125–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bertacchini J, Heidari N, Mediani L,
Capitani S, Shahjahani M, Ahmadzadeh A and Saki N: Targeting
PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci.
72:2337–2347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zeng Z, Wang RY, Qiu YH, Mak DH, Coombes
K, Yoo SY, Zhang Q, Jessen K, Liu Y, Rommel C, et al: MLN0128, a
novel mTOR kinase inhibitor, disrupts survival signaling and
triggers apoptosis in AML and AML stem/progenitor cells.
Oncotarget. 7:55083–55097. 2016.PubMed/NCBI
|
|
9
|
Lindblad O, Cordero E, Puissant A,
Macaulay L, Ramos A, Kabir NN, Sun J, Vallon-Christersson J,
Haraldsson K, Hemann MT, et al: Aberrant activation of the
PI3K/mTOR pathway promotes resistance to sorafenib in AML.
Oncogene. 35:5119–5131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maire V, Némati F, Richardson M,
Vincent-Salomon A, Tesson B, Rigaill G, Gravier E, Marty-Prouvost
B, De Koning L, Lang G, et al: Polo-like kinase 1: a potential
therapeutic option in combination with conventional chemotherapy
for the management of patients with triple-negative breast cancer.
Cancer Res. 73:813–823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deeraksa A, Pan J, Sha Y, Liu XD, Eissa
NT, Lin SH and Yu-Lee LY: Plk1 is upregulated in
androgen-insensitive prostate cancer cells and its inhibition leads
to necroptosis. Oncogene. 32:2973–2983. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang G, Zhang Z and Liu Z: Polo-like
kinase 1 is overexpressed in renal cancer and participates in the
proliferation and invasion of renal cancer cells. Tumour Biol.
34:1887–1894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ackermann S, Goeser F, Schulte JH, Schramm
A, Ehemann V, Hero B, Eggert A, Berthold F and Fischer M: Polo-like
kinase 1 is a therapeutic target in high-risk neuroblastoma. Clin
Cancer Res. 17:731–741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Renner AG, Dos Santos C, Recher C, Bailly
C, Créancier L, Kruczynski A, Payrastre B and Manenti S: Polo-like
kinase 1 is overexpressed in acute myeloid leukemia and its
inhibition preferentially targets the proliferation of leukemic
cells. Blood. 114:659–662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Du XL, Wang CJ, Lin DC, Ruan X,
Feng YB, Huo YQ, Peng H, Cui JL, Zhang TT, et al: Reciprocal
activation between PLK1 and Stat3 contributes to survival and
proliferation of esophageal cancer cells. Gastroenterology.
142:521–530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Behren A, Mühlen S, Sanhueza GA Acuna,
Schwager C, Plinkert PK, Huber PE, Abdollahi A and Simon C:
Phenotype-assisted transcriptome analysis identifies FOXM1
downstream from Ras-MKK3-p38 to regulate in vitro cellular
invasion. Oncogene. 29:1519–1530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Valsasina B, Beria I, Alli C, Alzani R,
Avanzi N, Ballinari D, Cappella P, Caruso M, Casolaro A, Ciavolella
A, et al: NMS-P937, an orally available, specific small-molecule
polo-like kinase 1 inhibitor with antitumor activity in solid and
hematologic malignancies. Mol Cancer Ther. 11:1006–1016. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hikichi Y, Honda K, Hikami K, Miyashita H,
Kaieda I, Murai S, Uchiyama N, Hasegawa M, Kawamoto T, Sato T, et
al: TAK-960, a novel, orally available, selective inhibitor of
polo-like kinase 1, shows broad-spectrum preclinical antitumor
activity in multiple dosing regimens. Mol Cancer Ther. 11:700–709.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen S, Bartkovitz D, Cai J, Chen Y, Chen
Z, Chu XJ, Le K, Le NT, Luk KC, Mischke S, et al: Identification of
novel, potent and selective inhibitors of polo-like kinase 1.
Bioorg Med Chem Lett. 22:1247–1250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gumireddy K, Reddy MV, Cosenza SC,
Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J and Reddy
EP: ON01910, a non-ATP-competitive small molecule inhibitor of
Plk1, is a potent anticancer agent. Cancer Cell. 7:275–286. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Talati C, Griffiths EA, Wetzler M and Wang
ES: Polo-like kinase inhibitors in hematologic malignancies. Crit
Rev Oncol Hematol. 98:200–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hao Z and Kota V: Volasertib for AML:
clinical use and patient consideration. Onco Targets Ther.
8:1761–1771. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gutteridge RE, Ndiaye MA, Liu X and Ahmad
N: Plk1 inhibitors in cancer therapy: from laboratory to clinics.
Mol Cancer Ther. 15:1427–1435. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Feng L, Ma Y, Sun J, Shen Q, Liu L, Lu H,
Wang F, Yue Y, Li J, Zhang S, et al: YY1-MIR372-SQSTM1 regulatory
axis in autophagy. Autophagy. 10:1442–1453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang KF, Yang H, Jiang WQ, Li S and Cai
YC: Puquitinib mesylate (XC-302) induces autophagy via inhibiting
the PI3K/AKT/mTOR signaling pathway in nasopharyngeal cancer cells.
Int J Mol Med. 36:1556–1562. 2015.PubMed/NCBI
|
|
26
|
Wang NN, Li ZH, Zhao H, Tao YF, Xu LX, Lu
J, Cao L, Du XJ, Sun LC, Zhao WL, et al: Molecular targeting of the
oncoprotein PLK1 in pediatric acute myeloid leukemia: RO3280, a
novel PLK1 inhibitor, induces apoptosis in leukemia cells. Int J
Mol Sci. 16:1266–1292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Price MM, Oskeritzian CA, Falanga YT,
Harikumar KB, Allegood JC, Alvarez SE, Conrad D, Ryan JJ, Milstien
S and Spiegel S: A specific sphingosine kinase 1 inhibitor
attenuates airway hyperresponsiveness and inflammation in a mast
cell-dependent murine model of allergic asthma. J Allergy Clin
Immunol. 131:501–511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gjertsen BT and Schöffski P: Discovery and
development of the polo-like kinase inhibitor volasertib in cancer
therapy. Leukemia. 29:11–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X and Erikson RL: Polo-like kinase
(Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad
Sci USA. 100:5789–5794. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Malumbres M and Barbacid M: Cell cycle
kinases in cancer. Curr Opin Genet Dev. 17:60–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang H, Tian C, Sun J, Chen LN, Lv Y, Yang
XD, Xiao K, Wang J, Chen C, Shi Q, et al: Overexpression of PLK3
mediates the degradation of abnormal prion proteins dependent on
chaperone-mediated autophagy. Mol Neurobiol. Jun 25–2016.(Epub
ahead of print). View Article : Google Scholar
|
|
32
|
Kim T, Mehta SL, Kaimal B, Lyons K,
Dempsey RJ and Vemuganti R: Poststroke Induction of α-synuclein
mediates ischemic brain damage. J Neurosci. 36:7055–7065. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Petherick KJ, Conway OJ, Mpamhanga C,
Osborne SA, Kamal A, Saxty B and Ganley IG: Pharmacological
inhibition of ULK1 kinase blocks mammalian target of rapamycin
(mTOR)-dependent autophagy. J Biol Chem. 290:287262015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan XY, Tian C, Wang H, Xu Y, Ren K, Zhang
BY, Gao C, Shi Q, Meng G, Zhang LB, et al: Activation of the
AMPK-ULK1 pathway plays an important role in autophagy during prion
infection. Sci Rep. 5:147282015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Basu S, Rajakaruna S, Reyes B, Van
Bockstaele E and Menko AS: Suppression of MAPK/JNK-MTORC1 signaling
leads to premature loss of organelles and nuclei by autophagy
during terminal differentiation of lens fiber cells. Autophagy.
10:1193–1211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Evangelisti C, Evangelisti C, Bressanin D,
Buontempo F, Chiarini F, Lonetti A, Soncin M, Spartà A, McCubrey JA
and Martelli AM: Targeting phosphatidylinositol 3-kinase signaling
in acute myelogenous leukemia. Expert Opin Ther Targets.
17:921–936. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Callera F, Lopes CO, Rosa ES and Mulin CC:
Lack of antileukemic activity of rapamycin in elderly patients with
acute myeloid leukemia evolving from a myelodysplastic syndrome.
Leuk Res. 32:1633–1634. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zou H, Li L, Carcedo I Garcia, Xu ZP,
Monteiro M and Gu W: Synergistic inhibition of colon cancer cell
growth with nanoemulsion-loaded paclitaxel and PI3K/mTOR dual
inhibitor BEZ235 through apoptosis. Int J Nanomed. 11:1947–1958.
2016.
|
|
39
|
Park HS, Hong SK, Oh MM, Yoon CY, Jeong
SJ, Byun SS, Cheon J, Lee SE and Moon G: Synergistic antitumor
effect of NVP-BEZ235 and sunitinib on docetaxel-resistant human
castration-resistant prostate cancer cells. Anticancer Res.
34:3457–3468. 2014.PubMed/NCBI
|