Introduction
Lung cancer is the leading cause of cancer mortality
worldwide (1), and non-small cell
lung cancer (NSCLC) is the most common type of lung cancer.
Approximately 85% of lung cancers are NSCLC, and the subtypes of
NSCLC include squamous cell carcinoma, adenocarcinoma and large
cell carcinoma. When diagnosed, most NSCLC patients are at an
advanced and inoperable stage and therefore have limited
therapeutic options (2). Moreover,
the metastatic phenotype is a major cause of lung cancer mortality.
Therefore, earlier detection and prevention of metastasis is very
important to prevent the progression of NSCLC (3).
The homeobox transcription factor CUTL1, also known
as CutX1 or CCAAT displacement protein (cux/CDP), plays a key role
in the control of normal embryonic development, differentiation and
cell growth (4). CUTL1 is known as
a transcriptional activator or as a transcriptional repressor
(5,6). CUTL1 is associated with cellular
proliferation and cell cycle progression (7,8), and
it functions as an oncogene because its increased expression
promotes the growth and migration of cancer cells (9–11).
Epithelial-mesenchymal transition (EMT) is a key process that
facilitates cell invasion and metastasis and is characterized by a
convergent loss or relocalization of epithelial markers such as
E-cadherin and β-catenin and gain of mesenchymal markers such as
N-cadherin and vimentin (12).
Accumulating evidence suggests that EMT is important for the
initial step of tumor metastasis (13,14);
however, whether CUTL1 participates in EMT is currently
unknown.
Materials and methods
Ethics statement
Patient information and samples were obtained with
written informed consent. Each patient in the study gave written
informed consent to publish the case details. The study was
approved by the ethics committee of Harbin Medical University
Cancer Hospital.
Cell lines, cell culture and
transfection
The human NSCLC lines (H292, H358, H322, A549,
Calu1, Calu6 and H23) and human embryonic kidney cell line 293T
were purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and maintained in Dulbeccos Modified Eagles
medium (DMEM) supplemented with 10% fetal bovine serum (FBS;
Invitrogen, Carlsbad, CA, USA) containing 100 units/ml penicillin
and 100 units/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA)
at 37°C with 5% CO2. A549 and H322 cells were
transfected using X-tremeGENE (Roche Applied Science, Indianapolis,
IN, USA) and HEK293T cells were transfected using Lipofectamine
2000 (Invitrogen) according to the manufacturers directions. Human
CUTL1 siRNA (sc-35051; Santa Cruz Biotechnology, Santa Cruz, CA,
USA) was transfected into cells for 24 h using Lipofectamine
RNAiMAX reagent (Invitrogen). TβR-I inhibitor SB431542 (10 µM) was
from R&D Systems (Minneapolis, MN, USA).
Western blot analysis
Total cellular proteins were extracted using lysis
buffer and 40 µg of total proteins was used for western blot
analysis. The polyvinylidene difluoride membranes were probed with
antibodies against CUTL1 (1:1,000 dilution)(Novus Biologicals,
Littleton, CO, USA); E-cadherin (1:1,000 dilution), N-cadherin
(1:1,000 dilution), Snail (1:1,000 dilution), p-Smad3 (1:1,000
dilution) (Cell Signaling Technology, Danvers, ΜΑ, USA); or GAPDH
(1:1,000 dilution). The blots were subsequently developed using the
enhanced chemiluminescence method (Millipore, Billerica, MA, USA).
The GAPDH signal was used as a control.
Transcription reporter assay
Cells were treated with or without 2 ng/ml TGF-β1 20
h after transfection. The cells were then harvested and analyzed
with the Dual-luciferase reporter assay system (Promega, Madison,
WI, USA). All assays were performed in triplicate, and all values
were normalized for transfection efficiency against Renilla
luciferase activities.
Real-time RT-PCR (qRT-PCR)
Total RNAs were obtained using the TRIzol reagent
(Invitrogen). Reverse transcription of the RNA was performed using
the ImProm-II reverse transcription system (Promega) according to
the manufacturers instructions. Quantitative reverse transcriptase
(qRT)-PCR was performed using an ABI PRISM 7500 sequence detector
system (Applied Biosystems, Foster City, CA, USA) with
gene-specific primers.
Immunofluorescence
A549 and Calu1 cells were cultured in a 12-well
plate and transfected with siCUTL1. The cells were then fixed with
4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and
incubated with primary antibody against E-cadherin or N-cadherin
for 1 h at 37°C. The cells were then incubated with Alexa Fluor 488
for 30 min at 37°C. The fluorescent images were captured using a
confocal laser-scanning microscope.
Immunohistochemistry
Tissue specimens were embedded in paraffin. The
sections were then deparaffinized in xylene and rehydrated in an
ethanol gradient. After antigen retrieval, the sections were
treated with 3% H2O2 for 10 min followed by
5% bovine serum albumin (BSA) for 30 min. Then, the sections were
incubated with primary antibodies against CUTL1 (1:200 dilution),
E-cadherin (1:100 dilution), N-cadherin (1:100 dilution) or Snail
(1:100 dilution) overnight at 4°C. Visualization of antibody
binding was performed using DAB staining. The nuclei were stained
with hematoxylin, and the immunostaining results were independently
assessed by two pathologists. The percentage of the positive cancer
cells: 0, no positive cells; 1, <10% positive cells; 2, 10–50%
positive cancer cells; 3, 51–75% positive cancer cells; and 4 ≥75%
positive cancer cells. The staining intensity: 1, no staining; 2,
weak staining; 3, moderate staining; and 4, strong staining. The
final score was equal to the area score and the intensity score. A
final staining score ≥4 was considered overexpression, and score
<4 as not overexpressed.
Patients
Lung cancer specimens (n=57) were collected from
patients with NSCLC at Harbin Medical University Cancer Hospital
from 2009 to 2012. The tissues were stored at −80°C until use. All
samples were from patients who had not undergone preoperative
radiotherapy or chemotherapy.
The pathological staging of the 57 tumors was
performed according to the tumor node metastasis (TNM) staging
system.
Cell migration and invasion assay
Migration assays were performed with 8-µm filters
(BD Biosciences, San Jose, CA, USA). Each well was loaded with
~1×105 cells. After incubation for 20 h, the cells
passing through the filter into the bottom wells were fixed in
formalin and stained with crystal violet. The cells in 10 randomly
selected fields (x200) from each well were counted. Transwell
invasion assays were performed under the same conditions as the
Transwell migration assays, but before the invasion assays, the
polycarbonate filter was coated with Matrigel.
Statistical analyses
All statistical analyses were performed using SPSS
17.0 software. The χ2 test was used to analyze the
expression of CUTL1 in NSCLC tissues and their adjacent normal
tissues. Other statistical analyses were performed by Student's
t-test. The data are shown as the mean ± SD from 3 independent
assays. A statistically significant difference was considered at
P<0.05.
Results
The CUTL1 expression levels are higher
in clinical NSCLC tissues
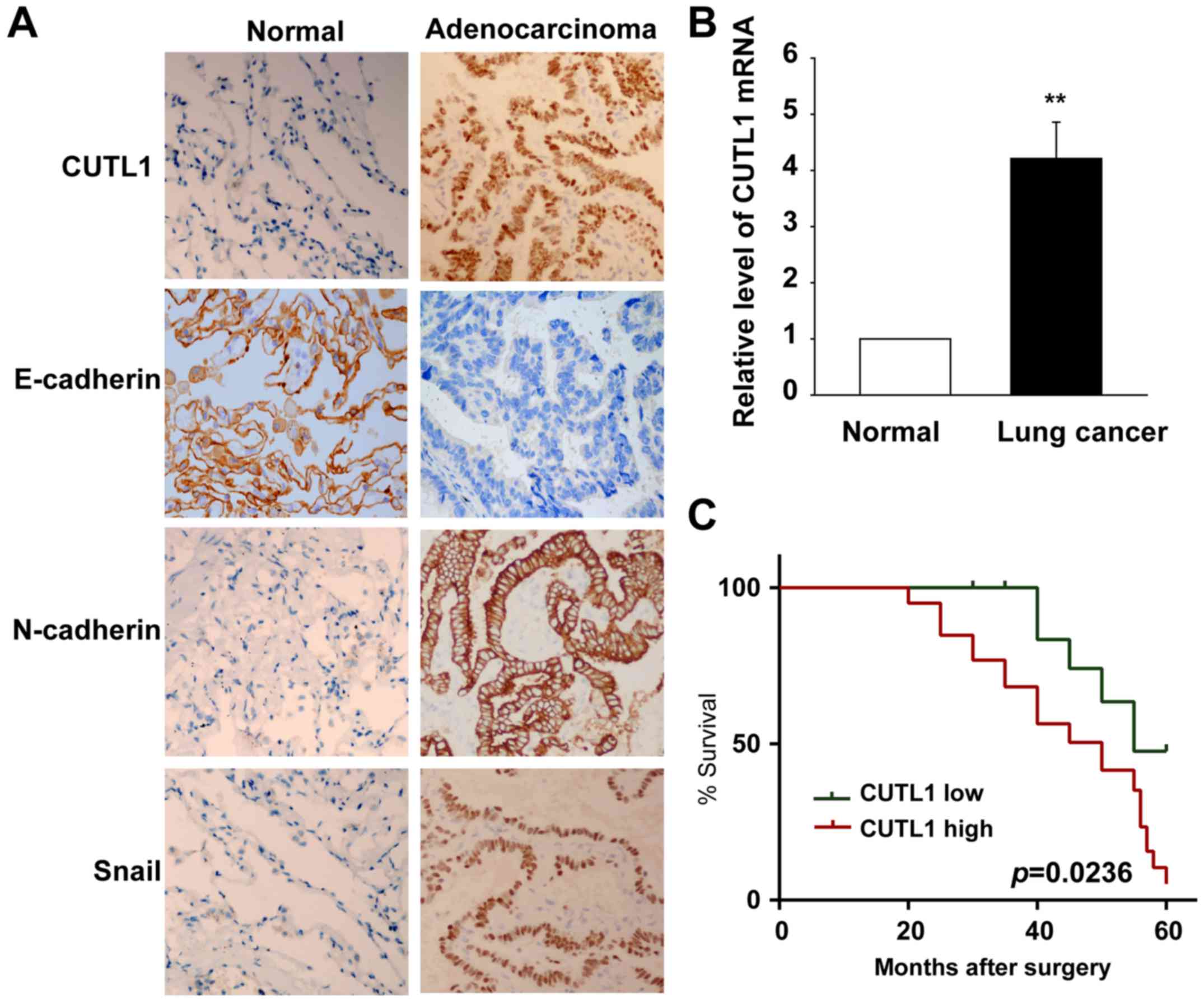
To explore the roles of CUTL1 in lung cancer, we
first tested the CUTL1 expression in NSCLC tissues and their
corresponding normal tissues by IHC assays. The results are
summarized in Table I. Specimens
overexpressing CUTL1 accounted for 70.18% of the cancer samples,
and most of these samples exhibited intermediate to strong
staining. However, specimens overexpressing CUTL1 accounted for
only 30.77% of the adjacent normal lung tissues and most of the
samples exhibited weak to moderate staining. The expression of
CUTL1 was significantly higher in the NSCLC specimens than in the
normal lung tissues (Fig. 1A).
CUTL1 expression was mainly localized in the nucleus of the NSCLC
tumor cells (Fig. 1A). To study the
roles of CUTL1 in metastasis, we investigated the role of CUTL1 in
EMT. First, E-cadherin, N-cadherin and Snail were detected in the
lung cancer tissues and in their corresponding normal lung tissues
using IHC assays (Fig. 1A). The
expression of E-cadherin decreased in the cancer tissues compared
to their adjacent normal tissues, but the expression of N-cadherin
and Snail increased in the cancer tissues compared with their
adjacent normal tissues (Fig. 1A).
The levels of CUTL1 in 8 lung cancer tissues and their
corresponding normal lung tissues were further detected using
real-time PCR analyses. The results showed that CUTL1 mRNA was
increased in the lung cancer tissues (Fig. 1B). The Kaplan-Meier survival curves
showed that patients with CUTL1 higher expression were at a notably
greater risk of an earlier death than those with CUTL1 lower
expression (P=0.0236) (Fig.
1C).
 | Table I.Expression of CUTL1 in NSCLC tissues
and their adjacent normal tissues. |
Table I.
Expression of CUTL1 in NSCLC tissues
and their adjacent normal tissues.
| Variables | n | CUTL1 Overexpression,
n (%)a | P-value |
|---|
| Cancer tissue | 57 | 40 (70.18 | <0.001 |
| Adjacent tissue | 39 | 12 (30.77) |
|
High CUTL1 expression in NSCLC is
associated with its mesenchymal-like phenotype
To establish the relationship between CUTL1
expression and the NSCLC cell phenotype, a panel of human NSCLC
cell lines was screened for CUTL1 expression. CUTL1 was highly
expressed in cell lines that were characterized as mesenchymal-like
cell lines (Fig. 2A). In contrast,
epithelial-like cells almost completely lacked CUTL1 (Fig. 2A).
The cellular migratory behavior of the
A549 cells were studied using migration and invasion assays
Compared to the control group, fewer A549 cells
passed through the Matrigel after depletion of CUTL1 (Fig. 2B). Consistently, there were fewer
A549 cells that migrated when CUTL1 was downregulated (Fig. 2C), but the number of migrated H322
cells was significantly increased after the overexpression of CUTL1
(Fig. 2D and E). In A549 cells,
CUTL1 knockdown resulted in a morphological switch from a
migratory, mesenchymal-like phenotype to a resting, epithelium-like
phenotype (Fig. 2F). These data
suggest that CUTL1 is involved in an EMT-like switch and
facilitates cell migration in vitro.
Effects of CUTL1 on EMT in NSCLC cell
lines
To further confirm the effects of CUTL1 on EMT, we
detected the expression of CUTL1, E-cadherin, N-cadherin and Snail
using western blot analysis. The expression of CUTL1, N-cadherin
and Snail increased, whereas the expression of E-cadherin decreased
in H322 and H292 cells transfected with the pcDNA3-HA-CUTL1 plasmid
(Fig. 3A). Moreover, the A549 and
Calu1 cells exhibited lower levels of CUTL1, N-cadherin and Snail
proteins and higher levels of the E-cadherin protein when
transfected with CUTL1 siRNA (Fig.
3B). Finally, we determined the expression of E-cadherin and
N-cadherin by immunofluorescence staining. As shown in Fig. 3C and D, treatment with siCUTL1
significantly increased E-cadherin expression and decreased
N-cadherin expression in A549 and Calu1 cells. These data suggest
that CUTL1 may induce EMT in NSCLC cell lines.
CUTL1 induces EMT through the TGF-β
signaling pathway
It is well known that the TG-Fβ signaling pathway
plays a key role in EMT (15–17).
To test whether CUTL1 regulation of EMT is associated with the
TGF-β signaling pathway, we overexpressed CUTL1 in H322 cells and
then treated them with the TβR-I inhibitor SB431542. The morphology
of the control groups treated with or without SB431542 remained
unchanged (Fig. 4A). In contrast,
the mesenchymal-like morphology of CUTL1-overexpressing H322 cells
was reverted back to epithelial-like morphology upon treatment with
SB431542 (Fig. 4A). Western blot
analysis further showed that SB431542 treatment restored E-cadherin
expression and abolished N-cadherin expression in
CUTL1-overexpressing H322 and H292 cells (Fig. 4B). Moreover, TGF-β-mediated
E-cadherin and N-cadherin expression at the protein level was
inhibited in A549 and Calu1 cells following CUTL1 depletion. These
data suggest that CUTL1-induced EMT depends on the TGF-β signaling
pathway.
CUTL1 promotes TβR-I expression to
positively regulate the TGF-β signaling pathway
To identify the TGF-β signaling pathway component
that is critical for CUTL-mediated EMT, we analyzed the mRNA levels
of TβR-I, TβR-II and TβR-III in H322 cells ectopically expressing
CUTL. Among the TβRs, the amount of TβR-I increased >7-fold in
H322 cells upon ectopic CUTL1 expression, but no significant change
was detected for TβR-II or TβR-III (Fig. 5A). CUTL1 knockdown in A549 cells
impaired Smad3 phosphorylation in response to TGF-β exposure
(Fig. 5B). In addition, knockdown
of CUTL1 in A549 cells resulted in reduced activity of
TGF-β/Smad3-driven SBE4-luciferase transcriptional reporter
activity (Fig. 5C). These results
indicate that CUTL promotes TβR-I expression to positively regulate
the TGF-β signaling pathway in lung cancer cell lines.
Discussion
The homeobox transcription factor CUTL1 is
evolutionarily highly conserved and known to regulate cell growth
and differentiation in mammals (4,5). CUTL1
has been shown to be an important mediator of tumor cell motility
and invasiveness in many types of tumors. The molecular mechanism
for promoting migration by CUTL1 has been reported. For instance,
CUTL1 accelerates tumor cell migration via decreasing
proteasome-mediated Src degradation (9), and CUTL1 uses WNT5A as an important
target to promote invasiveness and tumor progression in pancreatic
cancer (11). However, it is
unknown whether CUTL1 plays a key role in the EMT process (18,19).
In the present study, we provide evidence that CUTL1 induces EMT in
NSCLC. CUTL1 may facilitate tumor progression, and targeting its
expression will be a promising strategy for cancer therapy
(6). In this study, we detected the
expression of CUTL1 in NSCLC tissues. The results showed that CUTL1
was overexpressed in human NSCLC. Additionally, the effects of
CUTL1 on the migration and invasion of lung cancer cells were
studied, and we found that CUTL1 could enhance NSCLC cellular
migration and invasion. These data suggest that CUTL1 is an
oncogene and provide a strategy for cancer treatment.
EMT plays a pivotal role in cancer metastasis.
During this process, tumor cells derived from epithelial cells lose
their epithelial features and acquire mesenchymal features to
escape from the primary tissue and invade the surrounding stroma
(12,20,21).
E-cadherin is a cell adhesion molecule involved in maintaining the
epithelial phenotype. The loss of E-cadherin often results in a
loss of epithelial morphology (22). Notably, the decreased expression of
E-cadherin is often accompanied by upregulation of the expression
of N-cadherin, which promotes tumor cell invasiveness (23). Snail is another important EMT marker
that is well known to enhance tumor invasion (24). In this study, CUTL1 was shown to
decrease the expression of E-cadherin and increase the expression
of N-cadherin and Snail. These results suggest that CUTL1 is able
to induce EMT. Moreover, the migration and invasion assays further
confirm that CUTL1 promotes cell migration and invasion in
vitro.
CUTL1 (CCAAT displacement protein 1) belongs to the
homeodomain transcription factor family. Because the transcription
factor CUTL1 contains the DNA-binding domain, we speculated that
CUTL1 interacts with TβR-I promoter through its DNA-binding site,
which facilitates CUTL promoting TβR-I expression. We show that
CUTL1 directly facilitates TβR-I expression by activating TβR-I
gene transcription, indicating that activation of the TGF-β
signaling network may be related to CUTL1-induced EMT. This is
clearly supported by the observation that CUTL1-induced EMT in
epithelial-like lung cancer cells was reversed by the TβR-I
inhibitor SB431542. It is well-known that TGF-β functions as a
characterized inducer of EMT during cancer progression and
metastasis (25). TGF-β signaling
is activated through a heteromeric complex of type I and type II
transmembrane serine/threonine kinase receptors. Then, the receptor
complex results in the activation of Smad2 and Smad3. Next,
phosphorylated Smad2/3 and Smad4 translocate to the nucleus, where
they cooperate with transcription factors such as Snail to inhibit
the expression of epithelial markers and promote the expression of
mesenchymal markers at the mRNA level (26,27).
Tumor cells induced by TGF-β become invasive
concomitant with the loss of epithelial characteristics (28). TGF-β-induced EMT leads to cadherin
switching, i.e., the loss of E-cadherin and the increased presence
of N-cadherin, which results in cells becoming more motile and
invasive (29,30). Tumor cells often secrete active
TGF-β; therefore, the measurement of the serum concentration of
TGF-β has been a complementary diagnostic method in lung cancer
detection (31). Our experiments
showed that CUTL1 induced EMT in NSCLC. All of these studies
indicate that CUTL1 is a potential target for anti-lung cancer
therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (81172818; 81172215).
References
|
1
|
Shtivelman E, Hensing T, Simon GR, Dennis
PA, Otterson GA, Bueno R and Salgia R: Molecular pathways and
therapeutic targets in lung cancer. Oncotarget. 5:1392–1433. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reck M, Heigener DF, Mok T, Soria JC and
Rabe KF: Management of non-small-cell lung cancer: Recent
developments. Lancet. 382:709–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hsiao SH, Chung CL, Chou YT, Lee HL, Lin
SE and Liu HE: Identification of subgroup patients with stage
IIIB/IV non-small cell lung cancer at higher risk for brain
metastases. Lung Cancer. 82:319–323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nepveu A: Role of the multifunctional
CDP/Cut/Cux homeodomain transcription factor in regulating
differentiation, cell growth and development. Gene. 270:1–15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sansregret L and Nepveu A: The multiple
roles of CUX1: Insights from mouse models and cell-based assays.
Gene. 412:84–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu KC, Lin BS, Zhao M, Wang KY and Lan
XP: Cutl1: A potential target for cancer therapy. Cell Signal.
25:349–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Truscott M, Raynal L, Premdas P, Goulet B,
Leduy L, Bérubé G and Nepveu A: CDP/Cux stimulates transcription
from the DNA polymerase alpha gene promoter. Mol Cell Biol.
23:3013–3028. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Gurp MF, Pratap J, Luong M, Javed A,
Hoffmann H, Giordano A, Stein JL, Neufeld EJ, Lian JB, Stein GS, et
al: The CCAAT displacement protein/cut homeodomain protein
represses osteocalcin gene transcription and forms complexes with
the retinoblastoma protein-related protein p107 and cyclin A.
Cancer Res. 59:5980–5988. 1999.PubMed/NCBI
|
|
9
|
Aleksic T, Bechtel M, Krndija D, von
Wichert G, Knobel B, Giehl K, Gress TM and Michl P: CUTL1 promotes
tumor cell migration by decreasing proteasome-mediated Src
degradation. Oncogene. 26:5939–5949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fan X, Wang H, Zhou J, Wang S, Zhang X, Li
T, Nie Y and Liu B: The transcription factor CUTL1 is associated
with proliferation and prognosis in malignant melanoma. Melanoma
Res. 24:198–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ripka S, König A, Buchholz M, Wagner M,
Sipos B, Klöppel G, Downward J, Gress T and Michl P: WNT5A - target
of CUTL1 and potent modulator of tumor cell migration and invasion
in pancreatic cancer. Carcinogenesis. 28:1178–1187. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng H and Kang Y: Multilayer control of
the EMT master regulators. Oncogene. 33:1755–1763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Foroni C, Broggini M, Generali D and Damia
G: Epithelial-mesenchymal transition and breast cancer: Role,
molecular mechanisms and clinical impact. Cancer Treat Rev.
38:689–697. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fabregat I, Fernando J, Mainez J and
Sancho P: TGF-beta signaling in cancer treatment. Curr Pharm Des.
20:2934–2947. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hong S, Noh H, Teng Y, Shao J, Rehmani H,
Ding HF, Dong Z, Su SB, Shi H, Kim J, et al: SHOX2 is a direct
miR-375 target and a novel epithelial-to-mesenchymal transition
inducer in breast cancer cells. Neoplasia. 16:279–90.e1, 5. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michl P and Downward J: CUTL1: A key
mediator of TGFbeta-induced tumor invasion. Cell Cycle. 5:132–134.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Michl P, Ramjaun AR, Pardo OE, Warne PH,
Wagner M, Poulsom R, DArrigo C, Ryder K, Menke A, Gress T, et al:
CUTL1 is a target of TGF(beta) signaling that enhances cancer cell
motility and invasiveness. Cancer Cell. 7:521–532. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tiwari N, Gheldof A, Tatari M and
Christofori G: EMT as the ultimate survival mechanism of cancer
cells. Semin Cancer Biol. 22:194–207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nagathihalli NS, Massion PP, Gonzalez AL,
Lu P and Datta PK: Smoking induces epithelial-to-mesenchymal
transition in non-small cell lung cancer through HDAC-mediated
downregulation of E-cadherin. Mol Cancer Ther. 11:2362–2372. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang X, Liu G, Kang Y, Dong Z, Qian Q and
Ma X: N-cadherin expression is associated with acquisition of EMT
phenotype and with enhanced invasion in erlotinib-resistant lung
cancer cell lines. PLoS One. 8:e576922013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng P, Meng HM, Gao WZ, Chen L, Liu XH,
Xiao ZQ, Liu YX, Sui HM, Zhou J, Liu YH, et al: Snail as a key
regulator of PRL-3 gene in colorectal cancer. Cancer Biol Ther.
12:742–749. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feng XH and Derynck R: Specificity and
versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev
Biol. 21:659–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cavallaro U and Christofori G: Cell
adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev
Cancer. 4:118–132. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shirakihara T, Saitoh M and Miyazono K:
Differential regulation of epithelial and mesenchymal markers by
deltaEF1 proteins in epithelial mesenchymal transition induced by
TGF-beta. Mol Biol Cell. 18:3533–3544. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
González-Santiago AE, Mendoza-Topete LA,
Sánchez-Llamas F, Troyo-Sanromán R and Gurrola-Díaz CM: TGF-β1
serum concentration as a complementary diagnostic biomarker of lung
cancer: Establishment of a cut-point value. J Clin Lab Anal.
25:238–243. 2011. View Article : Google Scholar : PubMed/NCBI
|