Introduction
Glioblastoma is the most common type of primary
brain tumor in adults. This highly malignant tumor creates a
serious social and economic burden and is associated with high
mortality and morbidity (1).
Although multimodal treatment consisting of surgery, radiation
therapy and chemotherapy has been used, glioblastoma still exhibits
a poor prognosis, with a less than 12-month survival period
(2). In addition, less than 5% of
patients with glioblastoma survive more than 5 years after
diagnosis (3). Thus, more effective
therapeutic strategies are imperative.
Class I phosphatidylinositol 3-kinases (PI3Ks) play
critical roles in a variety of cellular processes, such as
differentiation, metabolism, migration and survival (4). The PI3K family is subdivided into 3
classes, and class I PI3K is further divided into 4 members (p110α,
p110β, p110γ and p110δ) (5).
Previous studies have revealed that p110β can be activated by
growth factor receptors and G protein-coupled receptors, and its
overexpression is capable of transforming cells (6). In addition, the known p110-β-selective
inhibitor TGX-221 blocked activation of PKB/Akt in PTEN-deficient
cells (7,8). For example, p110β expression was
significantly increased in malignant prostate tissues compared with
that in their surrounding non-malignant counterparts, and its mRNA
levels were correlated with disease progression in prostate cancer
patients (9). Compared with the
solvent control, TGX-221 significantly decreased xenograft tumor
growth in nude mice (10).
Furthermore, this result was supported by other groups using
transgenic mouse models (11,12)
and cell culture models (13).
Previous studies have revealed that PTEN restoration
and PIK3CB knockdown synergistically suppressed glioblastoma growth
in vitro and in xenografts (7). However, whether TGX-221 inhibits
proliferation and induces apoptosis of glioblastoma cells has not
been well studied. Thus, we treated U87 and U251 cells with TGX-221
to determine the effect of TGX-221 on glioblastoma cells.
Materials and methods
Cell culture
The human glioblastoma cell lines U251 and U87 were
acquired from the State Key Laboratory of Molecular Biology,
Institute of Biochemistry and Cell Biology, Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
The cells were cultured in Dulbeccos modified Eagles medium (DMEM)
containing 10% fetal bovine serum and incubated at 37°C in a
humidified atmosphere containing 5% carbon dioxide. DMEM was
acquired from GINOM Co., Ltd. (Guangzhou, China). TGX-221 was
purchased from Selleckchem (Houston, TX, USA) and dissolved in
dimethyl sulfoxide (DMSO), which was a product purchased from
Sigma-Aldrich (St. Louis, MO, USA).
CCK-8 assay
Cell viability was assessed using Cell Counting
Kit-8 (CCK-8) according to the manufacturer's instructions. CCK-8
was purchased from Dojindo China Co., Ltd. (Shanghai, China).
Approximately 8×103 cells were seeded in a volume of 100
µl of DMEM into each well of a 96-well plate. TGX-221 was added to
the medium and evaluated at different concentrations at a single
time-point or at different time-points at a single concentration.
In addition, 100 µl of fresh medium containing 5 µl of CCK-8
solution was added into each well and incubated at 37°C for 30 min.
The absorbance at 450 nm was measured using a spectrophotometric
plate reader. Each group was assessed in triplicate.
5-Ethynyl-2′-deoxyuridine (EdU)
staining
The Cell-Light EdU DNA Cell Proliferation kit was
purchased from RiboBio Co., Ltd. (Guangzhou, China) and used
according to the manufacturer's instructions. Approximately
8×103 cells were seeded in a volume of 100 µl of DMEM
into each well of a 96-well plate. The medium was mixed with
TGX-221 at different concentrations, and the cells were evaluated
48 h after exposure to TGX-221. The cells were treated with 50
µmol/l EdU for 12 h at 37°C. After fixation with 4%
paraformaldehyde for 15 min, the cells were treated with 0.5%
Triton X-100 for 20 min and rinsed with phosphate-buffered saline
(PBS) 3 times. Next, the cells were incubated with 100 µl of 1X
Apollo® reaction cocktail for 30 min, and the cell
nuclei were stained for 30 min with 5 µg/ml Hoechst 33342.
Fluorescence images of the EdU and Hoechst in the cells were
captured using a fluorescence microscope (Olympus, Tokyo, Japan).
The number of EdU- and DAPI-positive cells was quantified using
ImageJ software, and the EdU-labeling index was calculated as the
ratio of the number of EdU-positive cells to the number of
DAPI-positive cells.
Flow cytometry for cell apoptosis and
cell cycle distribution analysis
The effects of TGX-221 on apoptosis and cell cycle
distribution were determined using the Annexin V-FITC/propidium
iodide (PI) apoptosis and cell cycle kit independently, according
to the manufacturer's instructions from MultiSciences Biotech
(Hangzhou, China). The cells were examined after 48 h following
exposure to TGX-221 at different concentrations. At the end of the
treatment period, 3×105 or more cells were trypsinized,
collected by centrifugation at 1,000 rpm for 5 min and rinsed with
cold PBS. Next, the corresponding dyes and solution were added and
incubated according to the manufacturer's instructions. Cell
apoptosis and cell cycle distribution were analyzed using a flow
cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA), and the data
were analyzed using FlowJo software (version 7.6).
Migration and invasion assays
Migration and invasion assays were performed using a
Transwell chamber with an 8.0-µm pore polycarbonate membrane. The
cells were seeded into the top chambers containing the membranes,
which were either coated or not with Matrigel for migration and
invasion assays, respectively. Then, the chambers were placed into
a 24-well plate, and medium containing 10% fetal bovine serum was
added. The cells were fixed and stained with crystal violet, which
penetrated the underside surfaces of the membranes. Subsequently,
the cells were quantified under a microscope. The assays were
performed 3 times.
Western blot analysis
Cell proteins were extracted with RIPA lysis buffer
and assessed using the standard BCA method (Beyotime Institute of
Biotechnology, Jiangsu, China). Equal amounts of protein were
separated using SDS-PAGE and electroblotted onto polyvinylidene
difluoride membranes (Millipore Corp., Bedford, MA, USA). The
membranes were blocked in TBS containing 0.1% Tween-20 and 5%
powdered milk, and incubated at 4°C overnight with primary
antibodies against cleaved caspase-3, caspase-3, Bcl-2, Lc-3b, MMP9
and β-actin. Then, the membranes were incubated in the secondary
antibody Alex Fluor 680/790-labeled goat anti-rabbit IgG (LI-COR
Biosciences, Lincoln, NE, USA) for 1 h. Subsequently, the blots
were visualized using the LI-COR Odyssey Infrared Imaging
System.
Statistical analyses
All experimental results are expressed as the mean ±
SD. A Student's t-test was performed to determine the significance
between two mean values. The results were considered significant at
P-values of <0.05.
Results
TGX-221 inhibits glioblastoma cell
proliferation
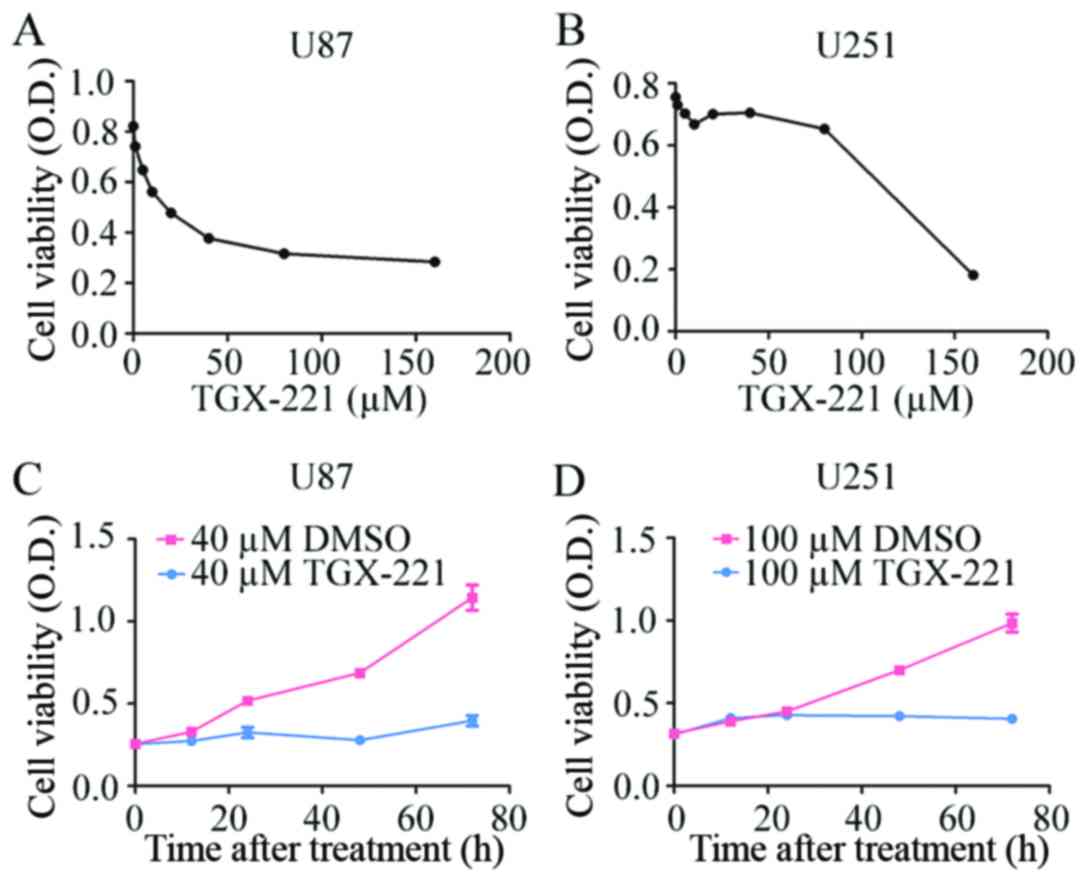
To confirm the effect of TGX-221 on glioblastoma
cell proliferation, we performed the CCK-8 assay using different
concentrations of TGX-221 in U87 and U251 cells. As shown in
Fig. 1A and B, TGX-221
significantly inhibited the viability of U87 and U251 cells in a
dose-dependent manner. The IC50 values of TGX-221 in U87
and U251 cells were ~40 and 100 µM, respectively. We then performed
the CCK-8 assay at different time-points with the IC50
values of TGX-221. Glioblastoma cell proliferation was inhibited
significantly by TGX-221 in a time-dependent manner (Fig. 1C and D).
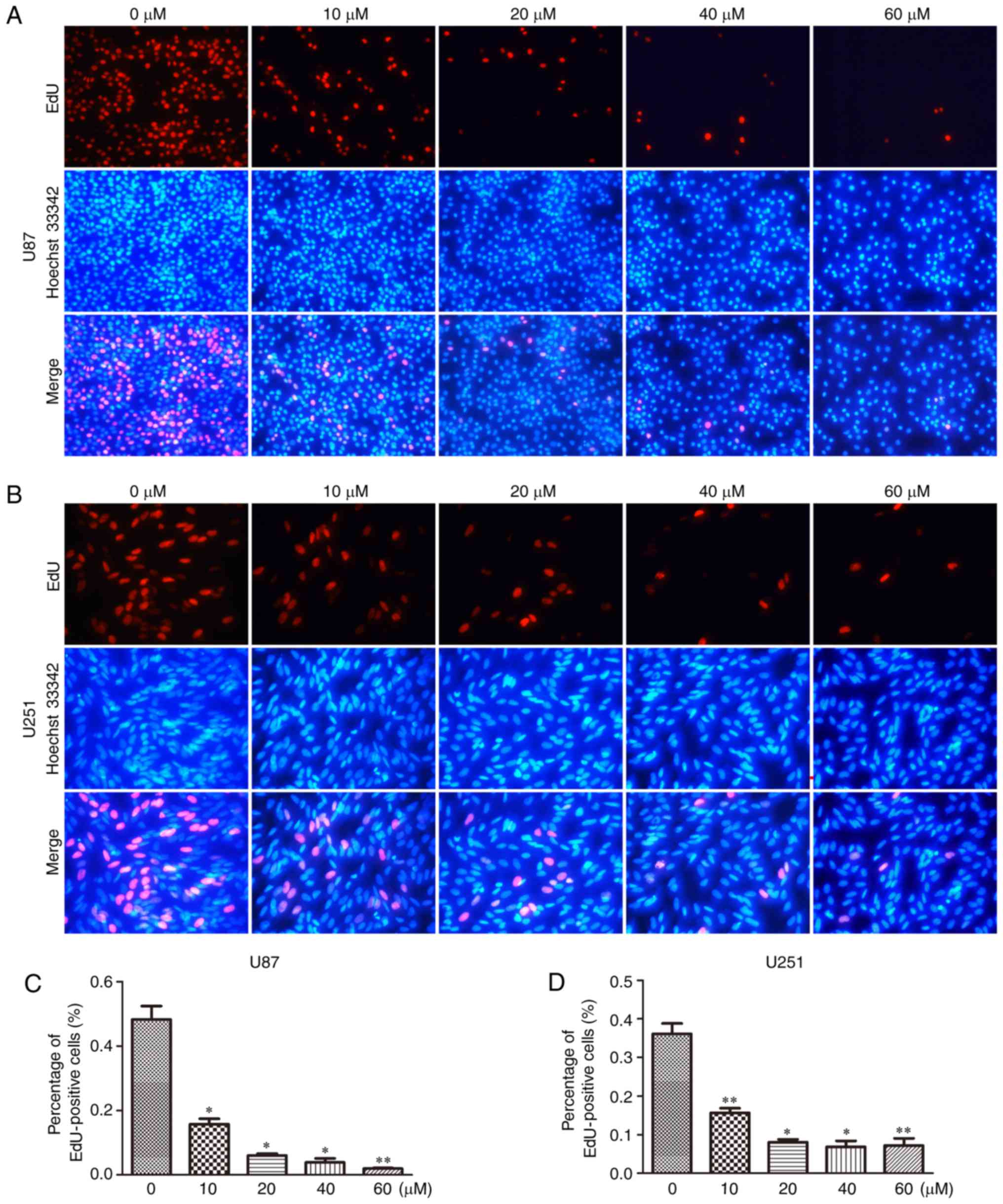
To further confirm the inhibitory effect of TGX-221
on cell proliferation in glioblastoma cells, we performed the EdU
assay in both U87 and U251 cells (Fig.
2A and B). A significant inhibition of cell proliferation was
observed in both U87 and U251 cells in a dose-dependent manner.
With an increase in TGX-221 concentration, the number of cell
nuclei with thymidine analog incorporation was decreased. The total
percentage of stained nuclei in cells treated with TGX-221 was
lower than that in the cells treated with DMSO (Fig. 2C and D).
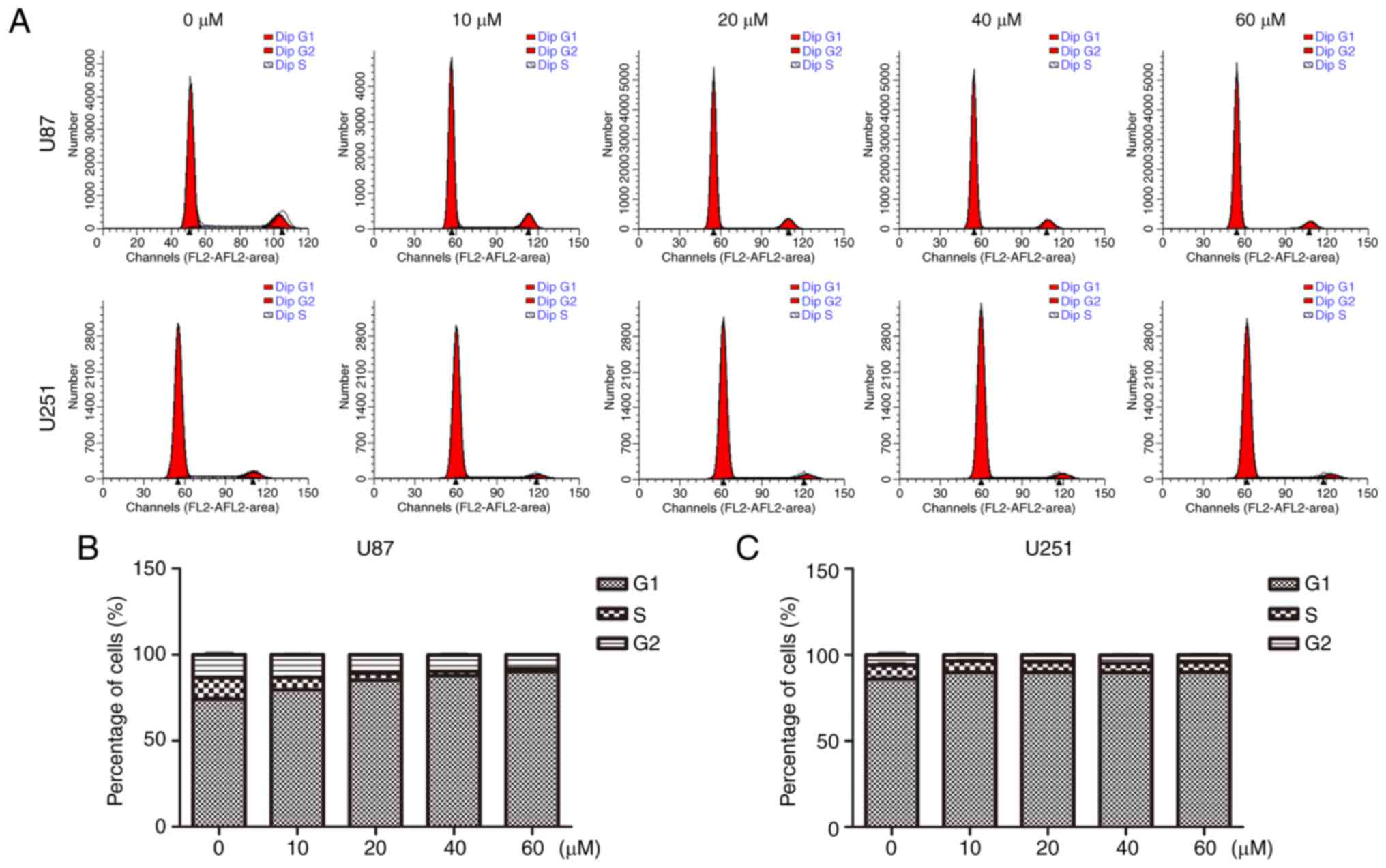
In addition, we performed flow cytometry to analyze
the cell cycle distribution. As shown in Fig. 3, U87 and U251 cells were cultured
with TGX-221 for 48 h. The percentage of cells in the G1 phase was
increased compared with that in the control groups. The percentage
of cells in the S and G2 phases was decreased, which suggested that
TGX-221 inhibited glioblastoma cell proliferation. Importantly, the
percentage of S and G2 phases decreased with an increase in TGX-221
concentration in glioblastoma cells.
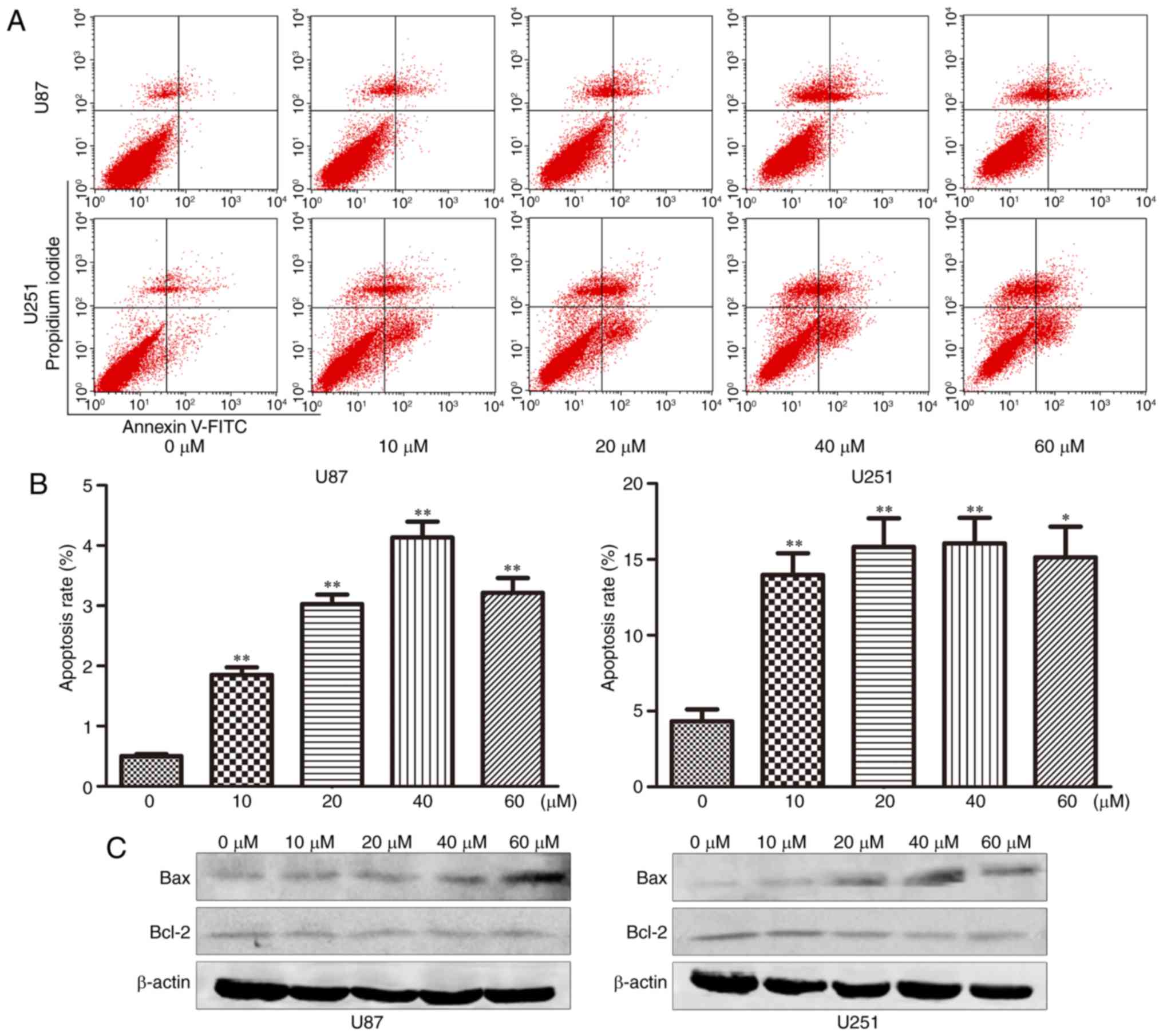
TGX-221 induces apoptosis in
glioblastoma cells
The effect of TGX-221 on cell apoptosis was
investigated using flow cytometry. The apoptosis rates at 48 h
after treatment at different concentrations (0,10, 20, 40 and 60
µM) are shown in Fig. 4A and B. We
found that the apoptosis rate increased significantly with
increasing TGX-221 concentrations.
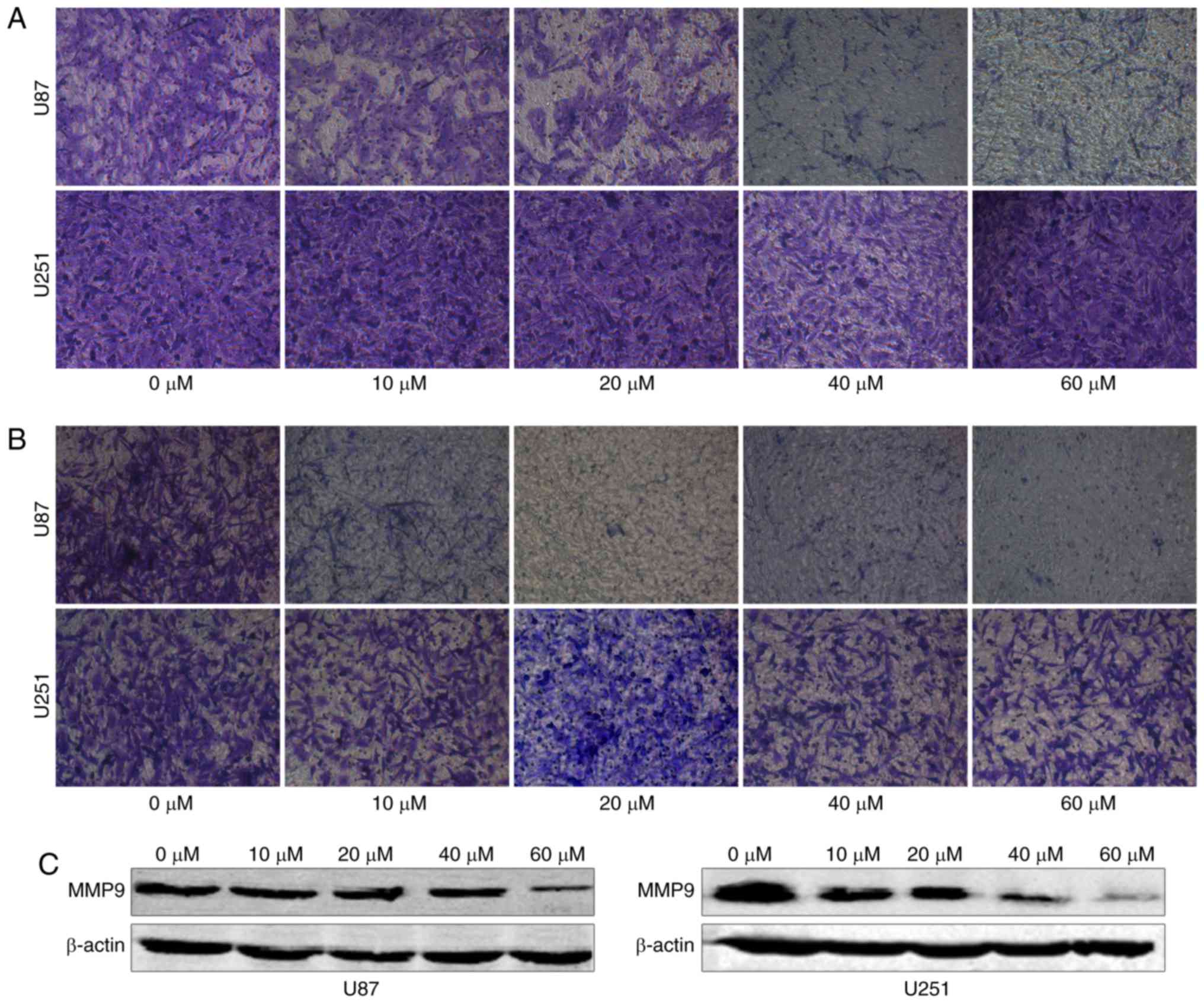
TGX-221 inhibits glioblastoma cell
migration and invasion
To examine whether TGX-221 inhibits the migration
and invasion of glioblastoma cells, we performed a migration and
invasion assay in U87 and U251 cells at different concentrations
(0, 10, 20, 40 and 60 µM). We found that TGX-221 inhibited
glioblastoma cell migration and invasion (Fig. 5A and B). These results were further
confirmed using western blot analyses. Furthermore, we demonstrated
that MMP9 gradually decreased with increasing concentrations of
TGX-221 (Fig. 5C).
Discussion
The PI3K family can be divided into 3 classes
according to their homology and function (14). Class I PI3Ks consists of two groups
(A and B), and previous research has shown that only Class IA
enzymes are expressed in human types of cancer. Class IA is a
heterodimeric protein consisting of a p110-kDa catalytic subunit
and a p85-kDa regulatory subunit. The p85 regulatory subunit
inhibits the catalytic activity of the p110 subunit in quiescent
cells (15). Previous studies have
demonstrated that activating point mutations or amplifications in
the PIK3CA gene are present in many types of human cancer (16–21).
Moreover, these findings revealed that an aberration of PIK3CA
affects the occurrence and development of human types of cancer.
Furthermore, PIK3CB has been demonstrated to play an important role
in PI3K/AKT signaling in glioblastomas (7). Recent studies examining mutant PIK3CA
also revealed that p110α was the most effective therapeutic target
in many human tumors. However, PTEN-deficient types of cancer
appear to be dependent on PIK3CB, but not PIK3CA. Several studies
have confirmed these findings in many human tumor cells, including
prostate, glioma, breast and endometrial cancer cells (22–25).
In a previous study, we used the combined treatment of PTEN
restoration and PIK3CB-siRNA and demonstrated that it was an
effective gene therapy approach for PTEN-deficient glioblastomas
(7).
We examined the role of the PI3K p110β isoform in
signaling pathways and found that TGX-221 inhibited p110β based on
a detailed structure and function analysis of LY294002. TGX-221
exhibited a >1,000-fold selectivity for PI3K p110β over a broad
range of protein kinases. Similar to LY294002, the concentration of
ATP affected the inhibitory effects of TGX-221 (26). Furthermore, TGX-221 consists of a
chiral center with an aniline moiety, and uses racemic material to
exert its functions (27). TGX-221
has been successfully used to inhibit p110β activity in some human
tumors. Recent studies also demonstrated that TGX-221 effectively
blocked tumor growth in prostate cancer xenograft mouse (10), transgenic mouse (11,12)
and cell culture models (13). In
the present study, we investigated U87 and U251 cells treated with
TGX-221 to examine the effect of TGX-221 in glioblastoma cells. We
hypothesized that TGX-221 inhibited proliferation and induced
apoptosis in human glioblastoma cells based on the findings
obtained in previous studies (10–13).
Our results indicated that TGX-221 inhibited
proliferation, migration and invasion, and induced apoptosis. In
addition, we found that U87 cells were more sensitive to TGX-221
than U251 cells. A previous study revealed that PIK3CB knockdown
suppressed glioblastoma growth with PTEN restoration in
vitro and in xenografts (7).
Thus, TGX-221 could be more effective in U87 cells potentially due
to their lack of PTEN expression.
Previous studies have provided some clues regarding
the mechanism of TGX-221 (27).
First, TGX-221 is an inhibitor of p110β, which participates in the
PI3K/Akt signaling pathway (5).
Thus, we proposed that the effect of TGX-221 may potentially
involve the PI3K/Akt signaling pathway. Akt regulates cell
apoptosis and survival (4,28), and Akt may exert its effects via an
NF-κB signaling pathway to affect cell survival. The mechanistic
effects observed were similar to our results. Thus, TGX-221 may
induce apoptosis and inhibit proliferation in glioblastoma cells
via the PI3K/Akt signaling pathway. Many studies have illustrated
that P110β plays a role in thrombosis and stenosis reduction
(29–31). The effect of TGX-221 in thrombosis
and stenosis potentially occurs via the regulation of ERK
phosphorylation (31). ERK can
affect cell proliferation via the MAPK signaling pathway. Thus,
TGX-221 may also affect cell proliferation via the MAPK signaling
pathway. Previous studies have proposed that p110β plays an
important role in insulin signaling (32–34).
Moreover, some studies have also demonstrated that p110β can be
activated by GPCRs (35–36). Furthermore, p110β exhibited
different requirements for Ras activation (6). On the basis of these studies, we found
that p110β affected cell apoptosis, the cell cycle, cell
proliferation and cell survival via several pathways. Thus, TGX-221
may inhibit p110β to affect the biological behaviors of
glioblastoma cells.
Although we obtained some results that can explain
the effects observed with TGX-221, the present study has several
limitations. First, we only demonstrated the effect of TGX-221, but
we did not examine its underlying mechanism. Furthermore, we only
performed our studies using U87 and U251 cells, and did not examine
the effect of TGX-221 in vivo. Finally, we did not have
sufficient clinical evidence to demonstrate our results. However,
our results are credible and aligned with our expectations.
Collectively, our study illustrated that TGX-221 can
inhibit proliferation and induce apoptosis in human glioblastoma
cells, which may represent a promising strategy for the treatment
of glioblastoma.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (nos. 81372683 and 81572489)
(to Q.-X.C.), and (no. 81502175) (to B.-H.L.).
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davis FG, McCarthy BJ, Freels S, Kupelian
V and Bondy ML: The conditional probability of survival of patients
with primary malignant brain tumors: Surveillance, epidemiology,
and end results (SEER) data. Cancer. 85:485–491. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ostrom QT, Gittleman H, Farah P, Ondracek
A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 15
Suppl 2:ii1–ii56. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vanhaesebroeck B, Stephens L and Hawkins
P: PI3K signalling: The path to discovery and understanding. Nat
Rev Mol Cell Biol. 13:195–203. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kang S, Denley A, Vanhaesebroeck B and
Vogt PK: Oncogenic transformation induced by the p110beta, -gamma,
and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl
Acad Sci USA. 103:pp. 1289–1294. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen H, Mei L, Zhou L, Shen X, Guo C,
Zheng Y, Zhu H, Zhu Y and Huang L: PTEN restoration and PIK3CB
knockdown synergistically suppress glioblastoma growth in vitro and
in xenografts. J Neurooncol. 104:155–167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Edgar KA, Wallin JJ, Berry M, Lee LB,
Prior WW, Sampath D, Friedman LS and Belvin M: Isoform-specific
phosphoinositide 3-kinase inhibitors exert distinct effects in
solid tumors. Cancer Res. 70:1164–1172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu Q, Youn H, Tang J, Tawfik O, Dennis K,
Terranova PF, Du J, Raynal P, Thrasher JB and Li B:
Phosphoinositide 3-OH kinase p85alpha and p110beta are essential
for androgen receptor transactivation and tumor progression in
prostate cancers. Oncogene. 27:4569–4579. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen R, Zhao Y, Huang Y, Yang Q, Zeng X,
Jiang W, Liu J, Thrasher JB, Forrest ML and Li B: Nanomicellar
TGX221 blocks xenograft tumor growth of prostate cancer in nude
mice. Prostate. 75:593–602. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee SH, Poulogiannis G, Pyne S, Jia S, Zou
L, Signoretti S, Loda M, Cantley LC and Roberts TM: A
constitutively activated form of the p110beta isoform of PI3-kinase
induces prostatic intraepithelial neoplasia in mice. Proc Natl Acad
Sci USA. 107:pp. 11002–11007. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee
SH, Zhang J, Signoretti S, Loda M, Roberts TM, et al: Essential
roles of PI(3)K-p110beta in cell growth, metabolism and
tumorigenesis. Nature. 454:776–779. 2008.PubMed/NCBI
|
|
13
|
Jiang X, Chen S, Asara JM and Balk SP:
Phosphoinositide 3-kinase pathway activation in phosphate and
tensin homolog (PTEN)-deficient prostate cancer cells is
independent of receptor tyrosine kinases and mediated by the
p110beta and p110delta catalytic subunits. J Biol Chem.
285:14980–14989. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fruman DA, Meyers RE and Cantley LC:
Phosphoinositide kinases. Annu Rev Biochem. 67:481–507. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wee S, Lengauer C and Wiederschain D:
Class IA phosphoinositide 3-kinase isoforms and human
tumorigenesis: Implications for cancer drug discovery and
development. Curr Opin Oncol. 20:77–82. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shayesteh L, Lu Y, Kuo WL, Baldocchi R,
Godfrey T, Collins C, Pinkel D, Powell B, Mills GB and Gray JW:
PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet.
21:99–102. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC,
Whang-Peng J, Liu JM, Yang DM, Yang WK and Shen CY: PIK3CA as an
oncogene in cervical cancer. Oncogene. 19:2739–2744. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Samuels Y, Wang Z, Bardelli A, Silliman N,
Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al:
High frequency of mutations of the PIK3CA gene in human cancers.
Science. 304:5542004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Campbell IG, Russell SE, Choong DY,
Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB and
Phillips WA: Mutation of the PIK3CA gene in ovarian and breast
cancer. Cancer Res. 64:7678–7681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu G, Mambo E, Guo Z, Hu S, Huang X,
Gollin SM, Trink B, Ladenson PW, Sidransky D and Xing M: Uncommon
mutation, but common amplifications, of the PIK3CA gene in thyroid
tumors. J Clin Endocrinol Metab. 90:4688–4693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Phillips WA, Russell SE, Ciavarella ML,
Choong DY, Montgomery KG, Smith K, Pearson RB, Thomas RJ and
Campbell IG: Mutation analysis of PIK3CA and PIK3CB in esophageal
cancer and Barrett's esophagus. Int J Cancer. 118:2644–2646. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pu P, Kang C, Zhang Z, Liu X and Jiang H:
Downregulation of PIK3CB by siRNA suppresses malignant glioma cell
growth in vitro and in vivo. Technol Cancer Res Treat. 5:271–280.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
An HJ, Cho NH, Yang HS, Kwak KB, Kim NK,
Oh DY, Lee SW, Kim HO and Koh JJ: Targeted RNA interference of
phosphatidylinositol 3-kinase p110-beta induces apoptosis and
proliferation arrest in endometrial carcinoma cells. J Pathol.
212:161–169. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oda K, Okada J, Timmerman L,
Rodriguez-Viciana P, Stokoe D, Shoji K, Taketani Y, Kuramoto H,
Knight ZA, Shokat KM, et al: PIK3CA cooperates with other
phosphatidylinositol 3′-kinase pathway mutations to effect
oncogenic transformation. Cancer Res. 68:8127–8136. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wee S, Wiederschain D, Maira SM, Loo A,
Miller C, deBeaumont R, Stegmeier F, Yao YM and Lengauer C:
PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA.
105:pp. 13057–13062. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jackson SP, Schoenwaelder SM, Goncalves I,
Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM,
Yuan Y, et al: PI 3-kinase p110β: A new target for antithrombotic
therapy. Nat Med. 11:507–514. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin H, Erhard K, Hardwicke MA, Luengo JI,
Mack JF, McSurdy-Freed J, Plant R, Raha K, Rominger CM, Sanchez RM,
et al: Synthesis and structure-activity relationships of
imidazo[1,2-a]pyrimidin-5(1H)-ones as a novel series of beta
isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg
Med Chem Lett. 22:2230–2234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: Rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sturgeon SA, Jones C, Angus JA and Wright
CE: Advantages of a selective beta-isoform phosphoinositide
3-kinase antagonist, an anti-thrombotic agent devoid of other
cardiovascular actions in the rat. Eur J Pharmacol. 587:209–215.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bird JE, Smith PL, Bostwick JS, Shipkova P
and Schumacher WA: Bleeding response induced by anti-thrombotic
doses of a phosphoinositide 3-kinase (PI3K)-β inhibitor in mice.
Thromb Res. 127:560–564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garcia A, Kim S, Bhavaraju K,
Schoenwaelder SM and Kunapuli SP: Role of phosphoinositide 3-kinase
β in platelet aggregation and thromboxane A2 generation
mediated by Gi signalling pathways. Biochem J.
429:369–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roche S, Downward J, Raynal P and
Courtneidge SA: A function for phosphatidylinositol 3-kinase beta
(p85alpha-p110beta) in fibroblasts during mitogenesis: Requirement
for insulin- and lysophosphatidic acid-mediated signal
transduction. Mol Cell Biol. 18:7119–7129. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hooshmand-Rad R, Hájková L, Klint P,
Karlsson R, Vanhaesebroeck B, Claesson-Welsh L and Heldin CH: The
PI 3-kinase isoforms p110(alpha) and p110(beta) have differential
roles in PDGF- and insulin-mediated signaling. J Cell Sci. 113(Pt
2): 1–214. 2000.
|
|
34
|
Asano T, Kanda A, Katagiri H, Nawano M,
Ogihara T, Inukai K, Anai M, Fukushima Y, Yazaki Y, Kikuchi M, et
al: p110beta is up-regulated during differentiation of 3T3-L1 cells
and contributes to the highly insulin-responsive glucose transport
activity. J Biol Chem. 275:17671–17676. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kubo H, Hazeki K, Takasuga S and Hazeki O:
Specific role for p85/p110beta in GTP-binding-protein-mediated
activation of Akt. Biochem J. 392:607–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kurosu H, Maehama T, Okada T, Yamamoto T,
Hoshino S, Fukui Y, Ui M, Hazeki O and Katada T: Heterodimeric
phosphoinositide 3-kinase consisting of p85 and p110beta is
synergistically activated by the betagamma subunits of G proteins
and phosphotyrosyl peptide. J Biol Chem. 272:24252–24256. 1997.
View Article : Google Scholar : PubMed/NCBI
|