Introduction
In China, there are ~0.4779 million new cases of
oesophageal carcinoma (EC) each year (1). The most prevalent histologic type of
EC is esophageal squamous cell carcinoma (ESCC) (2). Radiotherapy (RT) is an important
strategy for treating EC, and it markedly improves survival rates.
Although there have been considerable advances in EC therapy, poor
prognosis is inevitable due to the rapidly proliferating tumour
cells (3). Therefore,
identification of the key molecules involved in ESCC may provide
new therapeutic targets and further improve survival rates. It has
been shown that multiple genetic alterations are closely associated
with cell growth, metastasis and DNA damage repair in ESCC
(4–5).
Epigenetic regulator polycomb group (PcG) genes are
related to cell proliferation, invasion and migration.
B-cell-specific Moloney murine leukaemia virus integration site-1
(BMI-1), the core component of PcG, is dysregulated in various
types of cancers (6,7). BMI-1 is an important indicator for
predicting cancer invasion (8),
since it promotes cell viability, causes apoptosis resistance and
enhances transfer capabilities according to gene chip analysis
(9). Unfortunately, overexpression
of BMI-1 is related to treatment failure in many malignancies, such
as breast and prostatic cancer, and hepatocellular carcinoma
(10–12). BMI-1 is known as a PcG protein and
is required for H2AX ubiquitination-associated transcriptional
silencing (13). Ubiquitylation of
H2AX is likely activated by ATM kinase and causes H2AX
phosphorylation at serine 139, followed by the recruitment of
downstream genes, such as MDC1 and 53BP1, to the impaired sites and
induction of the DNA damage response (DDR). MDC1 and 53BP1 are
closely correlated with cell cycle arrest caused by the DDR.
Inhibition of these molecules can delay the DDR and promote cell
death (14,15). However, how BMI-1 is involved in
radiosensitivity, particularly in the regulatory mechanisms of the
DDR, is still unknown.
To identify the novel role of BMI-1 in the DDR of
ESCC, a proteomic and molecular biology analysis was conducted to
determine the function of this gene. To date, there have been few
studies regarding the mechanism through which BMI-1 promotes cell
viability in EC after RT, particularly studies including clinical
and in vitro and in vivo data. There are also few
studies regarding the regulatory effects of BMI-1 on the
radiosensitivity of EC via the PI3K/Akt signalling pathway. Given
the vital function of BMI-1 in DNA damage-induced DDR via the
PI3K/Akt pathway, we hypothesized that BMI-1 knockdown may result
in DDR defects and further increase radiosensitization by
inactivating the PI3K/Akt pathway. In this article, different cell
lines were used to verify this hypothesis in vitro and in
vivo, as well as to further explore the regulatory mechanisms
of BMI-1.
Materials and methods
Tissue specimens and
immunohistochemical analysis
Sixty ESCC and matched adjacent non-tumour tissues
were collected at the Department of Thoracic Surgery, Hebei
Hospital of the Fourth Affiliated Medical University (Hebei,
China), from January 2010 to December 2010. Patient consent was
obtained for the collection of specimens, and all study protocols
were approved by the Ethics Committee for Clinical Research of the
Fourth affiliated Medical University. Immunohistochemistry
protocols were performed as previously described (16). Briefly, slides were incubated with
an anti-BMI-1 monoclonal antibody (ab126783, 1:100; Abcam,
Cambridge, MA, USA), and then with a horseradish
peroxidase-conjugated anti-mouse secondary antibody (Dako,
Glostrup, Denmark). Phosphate-buffered saline (PBS) was used
instead of the anti-BMI-1 antibody for the negative control
sections. Immunostaining results were independently evaluated by
two pathologists. At least 4–5 random high-power fields
(magnification, ×200) from each section were observed. Positive
samples were cases with >10% of cells staining for BMI-1
(17).
Cell lines and cell culture
The human normal oesophageal cell line HEEC and
various human ESCC cell lines, including ECA109, KYSE30, EC9706 and
TE13, were used for screening. These cells were obtained from the
Research Center of the Fourth Hospital of Hebei Medical University
(Shijiazhuang, China). To note, authentification of the TE13 cell
line was verified by our laboratory. They were cultivated at 37°C
and 5% CO2 in RPMI-1640 medium (Gibco, Gaithersburg, MD,
USA) supplemented with 10% foetal bovine serum (FBS) (Invitrogen,
Carlsbad, CA, USA), penicillin (100 U/ml), and streptomycin (0.1
mg/ml).
X-ray irradiation
Post-irradiation (IR) was conducted using a 6-MV
Siemens linear accelerator (Siemens, Concord, CA, USA) at a dose
rate of 5 Gy/min. After that, cells were cultivated in an incubator
before harvesting.
shRNA transfection
For the shRNA analyses, the sequences of shRNA
against BMI-1 were: BMI-1 shRNA1, 5′-UCCUCAUCCACAGUUUCCUCACAUU-3′
(sense); BMI-1 shRNA2, 5′-GGGUCAUCAGCAACUUCUUCUGGUU-3′ (sense); and
BMI-1 shRNA3, 5′-GCUUAUCCAUUGAAUUCUUUGACCA-3′ (sense); and the
negative control scrambled shRNA (NC-shRNA) sequence was:
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense); these sequences were designed
and purchased from Invitrogen. Lipofectamine RNAiMAX (Invitrogen)
was used to transfect the cells. Briefly, cells
(5×105/well) were cultured in 6-well plates until they
reached 50% confluency, and were then transiently transfected with
either BMI-1-shRNA or NC-shRNA (100 nM). Cells were collected at 24
h after transfection with shRNA, followed by the selection of
stable clones with puromycin and irradiation.
Western blotting
The cultured tumour cells or tissues were lysed in
500 µl of lysis buffer. The lysates were separated by sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
were transferred onto polyvinylidene difluoride (PVDF) membranes.
The PVDF membranes were incubated overnight with specific dilutions
of the primary antibodies at 4°C. The antibodies included rabbit
anti-human mAbs against BMI-1 (ab126783, 1:10,000), γH2AX (ab26350,
1:1,000), MDC1 (ab114143, 1:800), 53BP1 (ab36823, 1:10,000), P16
(ab108349, 1:2,000), cyclin D2 (ab81359, 1:500), CDK4 (ab137675;
1:2,000), MCL-1 (ab32087, 1:3,000), and Bax (ab32503, 1:5,000)
(Abcam) and anti-H2AK119ub (AB10029, 1:1,000) (Millipore,
Billerica, MA, USA), rabbit anti-Akt (4685S, 1:1,000), anti-pAkt
polyclonal (4058S, 1:1,000) (Cell Signaling Technology, Inc.,
Beverly, MA, USA) and β-actin (AP0060, 1:10,000) (Bioworld, Dublin,
OH, USA) antibodies. The PVDF membranes were incubated with
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibodies, followed by visualization using the Odyssey infrared
imaging system. The levels of these proteins were calculated as the
ratio of the intensity of the specified protein to that of
β-actin.
Cell viability assay
MTT assays were performed to examine the effect of
BMI-1 shRNA on cell viability (Sigma-Aldrich Chemical Co., St.
Louis, MO, USA). Briefly, the transfected cells were cultured into
a 96-well plate, followed by irradiation for 24 h. After incubation
for 24, 48 and 72 h at 37°C, 5 µl of MTT reagent (5 mg/ml in PBS)
was added to the cells, followed by incubation for 4 h. After that,
the cells were incubated with 150 µl of dimethyl sulfoxide (DMSO)
for 15–20 min. Absorbance at 492 nm was measured. This experiment
was repeated 3 times. Cells were preincubated for 24 h with
LY294002 and IGF-1 from Cell Signaling Technology (Beverly, MA,
USA).
Colony formation assay
A standard colony formation assay was performed to
generate cell survival curves (18). Graded single doses of IR (0–8 Gy)
were used to irradiate tumour cells, followed by cultivation in
RPMI-1640 supplemented with 10% FBS for 2 weeks. Then, the cells
were fixed and stained with crystal violet (0.6%). The survival
fraction was calculated according to a previous study (19). A single-hit multitarget formula was
performed to analyse the data using GraphPad Prism 5 software: S =
1 - (1 - e−D/D0)N, where D0 is the
dose on the straight-line portion of the survival curve required to
decrease the survival to 37%. The Dq, the intercept of the
extrapolated high dose, was calculated. N, the extrapolation
number, was the measure of the width of the shoulder of the
survival curve. SF after irradiation at 2 Gy (SF2) and
the sensitization enhancement ratio (SER) were obtained from the
above parameters.
Flow cytometry (FCM)
To assess the effects of BMI-1 shRNA on cell cycle
progression and apoptosis, FCM was performed. The transfected cells
were treated with IR for 24 h, and then harvested to analyse the
cell cycle distribution. Cells were fixed with 70% ethanol at 4°C
overnight, followed by washing with PBS and resuspension in
staining solution for 30 min at room temperature in the dark. To
measure cell apoptosis under different treatment conditions, cells
were harvested and incubated with Annexin V and 7-AAD stains (BD
Pharmingen™).
Animals and tumour xenograft
assay
Stably transfected tumour cells were inoculated into
the left hind paw of 4- to 6-week-old BALB/c nude mice. A dosage of
15 Gy was used to irradiate the mice for 21 days after the
injection. To protect the upper part of the mice from irradiation,
mice were placed in custom-made holders called a collimator
container when irradiated. Tumour dimensions and volumes
(mm3) were measured and calculated with callipers once
every week. In the end, the nude mice were sacrificed by cervical
dislocation in the fifth week after exposure to irradiation and the
tumors were harvested. Proteins were extracted from the tumor
tissues and detected by western blotting (for Akt, pAkt). The other
tumor tissues were fixed in formalin to obtain sections for the
TUNEL assay and immunohistochemistry [for BMI-1 (ab126783, 1:100),
Ki67 (27309–1-AP, 1:2,000) (ProteinTech Group, Inc., Chicago, IL,
USA)]. The experimental protocols were evaluated and approved by
the Animal Care and Use Committee of the Fourth Hospital of Hebei
Medical University.
Terminal deoxynucleotidyl transferase
(TdT)-mediated dUTP nick end labeling (TUNEL) assay for cell
apoptosis
The DeadEnd™ System was employed to assay cell
apoptosis. For detection of apoptosis in tissue sections,
paraffin-embedded tissue sections were deparaffinized and
permeabilized with proteinase K, and then coverslips were immersed
in 4% paraformaldehyde for 25 min, 0.2% Triton X-100 in PBS for 5
min, and 100 µl equilibration buffer at room temperature for 5 min,
followed by addition of 100 µl TdT reaction mix (98 µl
equilibration buffer, 1 µl biotinylated nucleotide mix and 1 µl
rTdT Eenzyme) and incubation at 37°C for 60 min. The coverslips
were then immersed in 2X SSC for 15 min, 0.3% hydrogen peroxide for
3 min, and 100 µl streptavidin-HRP for 30 min, after which 100 µl
DAB was added until a light brown background developed. The nuclei
of apoptosis cells were stained dark brown. Quantitative analysis
was performed blindly by counting the number of TUNEL-positive
cells in 10 microscopic fields, as previously described. Apoptosis
rate (%) = (apoptosis cells/total cells) (%).
Statistical analysis
SPSS software package version 13.0 (SPSS, Inc.,
Chicago, IL, USA) was used to conduct statistical analyses. All
data are presented as the mean ± standard deviation (SD) of at
least 3 independent experiments and were analysed by ANOVA.
P-values of <0.05 and 0.01 were considered to indicate a
statistically significant result.
Results
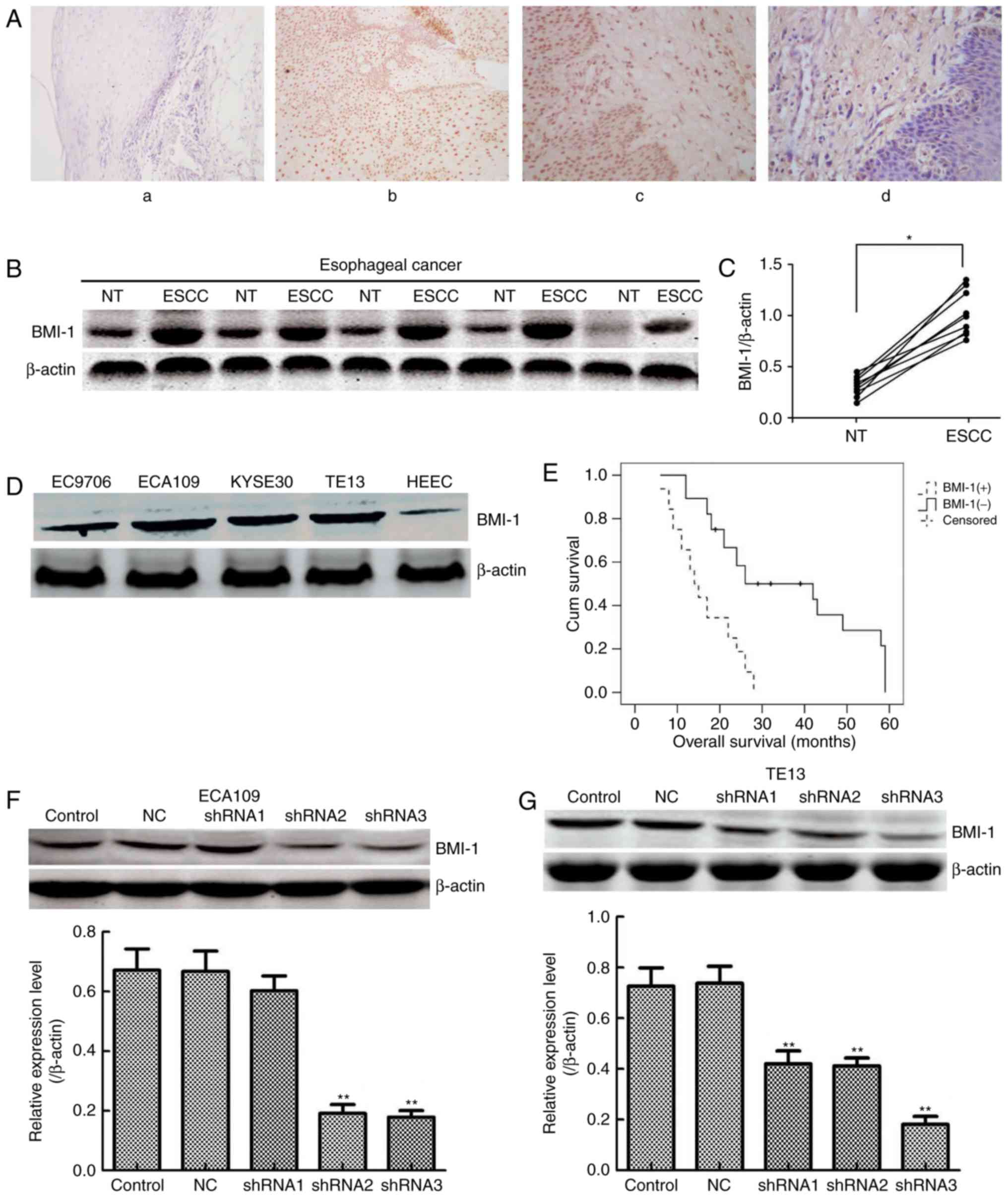
BMI-1 is highly expressed in ESCC
cells and specimens
To evaluate BMI-1 expression, immunohistochemistry
assays were performed on 60 ESCC and matched adjacent normal
oesophageal tissues. In agreement with a previous study (20), BMI-1 was mainly expressed in the
cell nuclei, but occasionally in the neoplastic epithelial
cytoplasm and plasmalemma (Fig.
1A); its expression was observed in 32 of the 60 cases of
oesophageal cancer (53.33%) (Table
I). No staining or only weak staining was noted in normal
epithelial cells. In addition, the expression of BMI-1 was measured
in 30 pairs of ESCC and matched adjacent non-tumour tissues by
western blotting (Fig. 1B). The
expression of BMI-1 in tumour tissues was obviously higher than
that of the corresponding normal oesophageal tissues (Fig. 1C; P<0.05), indicating that high
expression of BMI-1 may result in the development and progression
of ESCC.
 | Table I.Correlation between the expression of
BMI-1 and clinicopathological variables of the patients with
ESCC. |
Table I.
Correlation between the expression of
BMI-1 and clinicopathological variables of the patients with
ESCC.
|
| BMI-1
expression |
|
|---|
|
|
|
|
|---|
| Variables | Positive | Negative | P-value |
|---|
| Age, years |
|
| 0.448 |
|
>60 | 18 | 13 |
|
|
≤60 | 14 | 15 |
|
| Gender |
|
| 0.755 |
|
Male | 17 | 16 |
|
|
Female | 15 | 12 |
|
| Tumour size
(cm) |
|
| 0.022 |
| ≤5 | 10 | 17 |
|
|
>5 | 22 | 11 |
|
| Histological
grade |
|
| 0.466 |
| Good or
moderate | 13 | 14 |
|
|
Poor | 19 | 14 |
|
| TNM stage |
|
| 0.021 |
|
I–II | 12 | 16 |
|
|
III–IV | 20 | 12 |
|
| Lymph node
metastasis |
|
| 0.011 |
|
Positive | 23 | 11 |
|
|
Negative | 9 | 17 |
|
Furthermore, 4 ESCC cell lines (EC9706, ECA109,
KYSE30 and TE13) and the immortal oesophageal epithelial cell line
HEEC were selected for measuring the expression of BMI-1 (Fig. 1D). The expression of BMI-1 was high
in ESCC cells, particularly ECA109 and TE13 cells. Collectively,
the results of the western blot assays were in accordance with
those of the IHC analyses, which supported that BMI-1 contributes
to oesophageal carcinogenesis as an oncogene in oesophageal
cancer.
BMI-1 overexpression is related to the
progression and poor prognosis of ESCC
To explore the clinical significance of BMI-1 during
ESCC development, the correlation between the expression of BMI-1
and the clinicopathological features of patients with ESCC were
analysed. As shown in Table I,
nuclear accumulation of BMI-1 in ESCC was markedly related to
tumour size, clinical stage and lymph node metastasis (P<0.05),
displaying a correlation between BMI-1 expression and cell
proliferation and metastasis. However, no obvious correlation was
observed between BMI-1 and patient age, sex or histological grade
(P>0.05). Moreover, Kaplan-Meier survival analysis results
indicated that patients harbouring low BMI-1 expression had a
markedly longer overall survival time than that of patients with
high levels of BMI-1 (P<0.001, log-rank test; Fig. 1E). Collectively, the above results
demonstrated that BMI-1 may be used to evaluate the prognosis of
patients with ESCC.
BMI-1 knockdown inhibits cell
viability and promotes the radiosensitivity of ESCC cells after IR
in vitro
Either NC shRNA or shRNA targeting the BMI-1 gene
was used to treat ECA109 and TE13 cells to reveal the important
role of BMI-1. As shown in Fig. 1F and
G, shRNA3 BMI-1 significantly inhibited the expression of BMI-1
in both cell types (P<0.01). Therefore, ECA109-BMI-1 shRNA3 and
TE13-BMI-1 shRNA3 cells were further characterized.
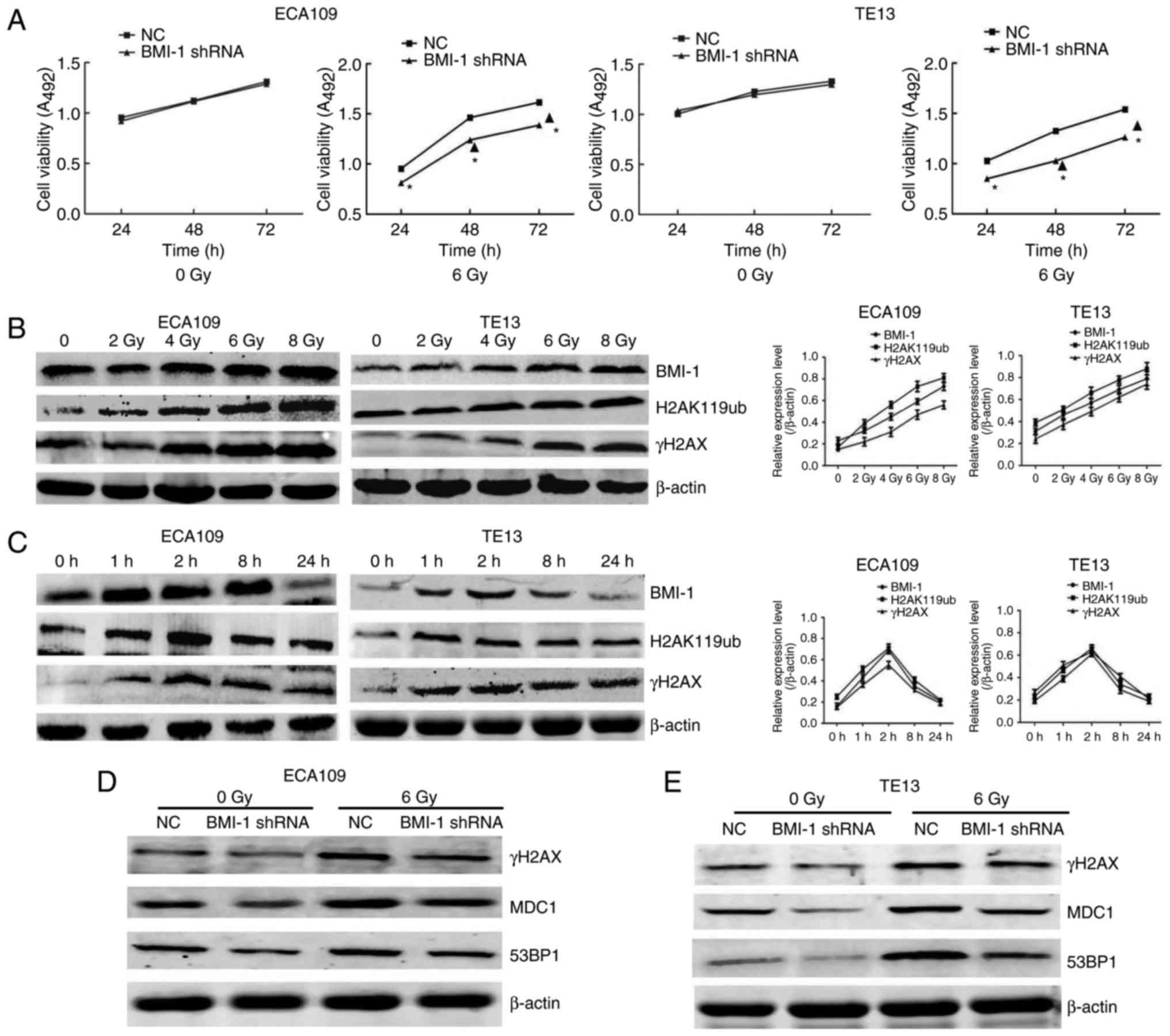
To investigate the effect of BMI-1 knockdown on the
growth of cells, MTT assays were employed to detect cell viability.
As shown in Fig. 2A, cell viability
was not markedly different in both groups of ECA109 and TE13 cells
at the indicated time points before IR. In contrast, cell viability
was obviously increased after IR, particularly at 48 and 72 h
(P<0.05), but BMI-1 knockdown markedly suppressed cell viability
compared to the NC group (P<0.05). Additionally,
radiosensitivity in both groups after IR was also detected. As
shown in our previous study, the BMI-1 shRNA cells had greater
radiosensitivity than that of the NC group (21). Collectively, our data suggested that
IR combined with BMI-1 knockdown significantly inhibited cell
viability and increased radiosensitivity in vitro.
BMI-1 regulates DNA damage
repair-related genes in a DNA damage-induced manner
In the present study, IR promoted the expression of
BMI-1, H2AK119ub and γH2AX at the protein level in a dose-dependent
manner (Fig. 2B). Furthermore, the
changes to these proteins were consistent in both cell types. The
expression levels all reached their highest level at 1 and 2 h, and
then gradually decreased (Fig. 2C).
By 24 h, their levels were restored to unirradiated levels. These
data led us to propose that there was a relationship between BMI-1
and H2AK119ub and γH2AX, which was in accordance with previous data
that indicated that the interaction between BMI-1 and H2AK119ub and
γH2AX increased after IR as determined by co-immunoprecipitation
(Co-IP) assay (21). In addition,
we also evaluated the expression of γH2AX and its downstream genes,
such as MDC1 and 53BP1, in the NC and BMI-1 shRNA groups after IR
or not. Our results demonstrated that the levels of these indexes
were not obviously altered before IR. Although IR induced the
expression of γH2AX, MDC1 and 53BP1 in both groups, their protein
levels were obviously lower in the BMI-1 shRNA group than those of
the NC group in ECA109 (Fig. 2D)
and TE13 cells (Fig. 2E) after IR.
Collectively, our data suggested that BMI-1 regulates the
expression of proteins associated with DNA damage repair, including
γH2AX, MDC1 and 53BP1.
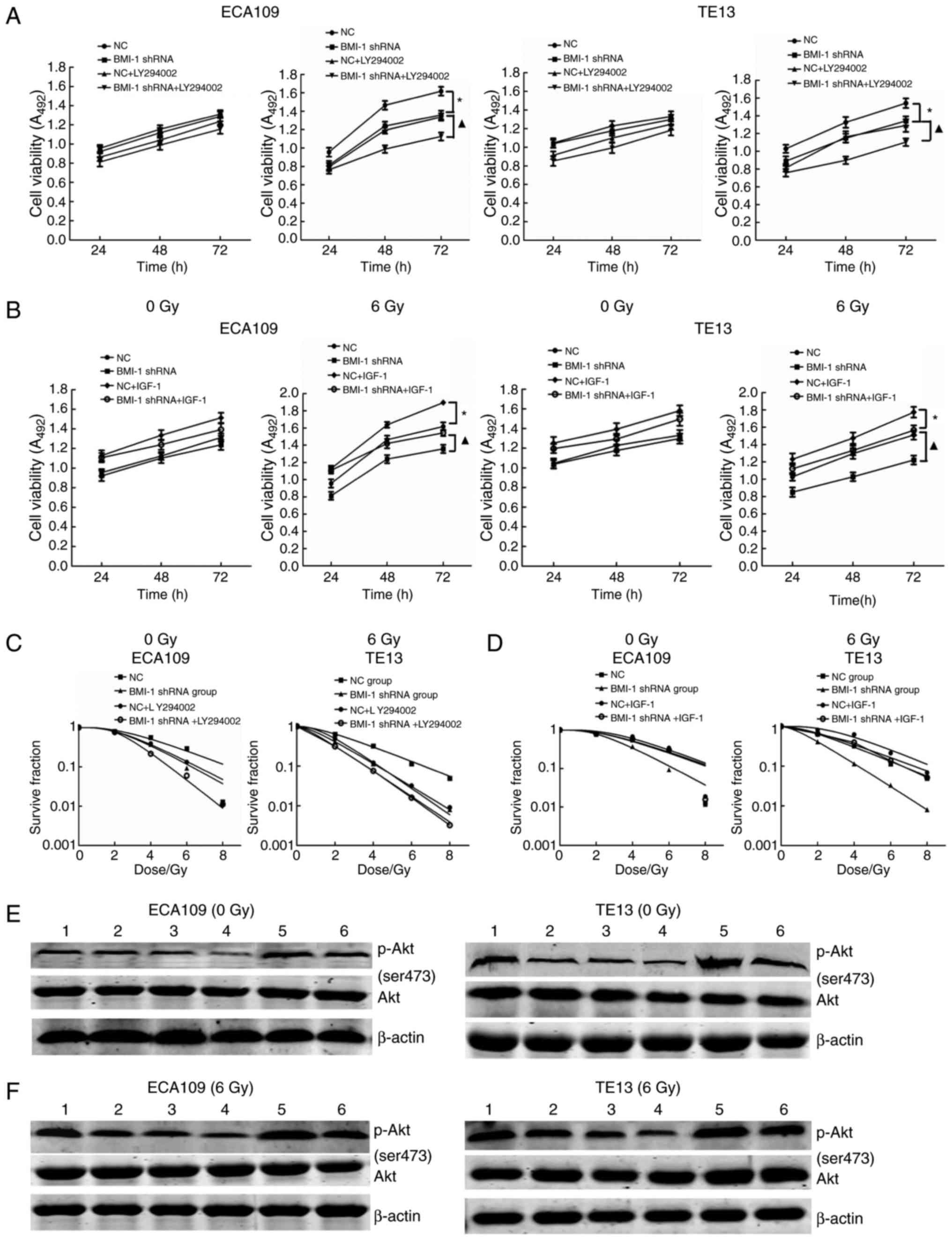
BMI-1 silencing inhibits cell
viability and improves radiosensitivity in cells by inhibiting the
PI3K/Akt pathway
LY-294002 is a specific inhibitor of PI3K and can
significantly inhibit the expression of p-Akt. After the addition
of 20 mmol/l LY-294002, cell viability and the expression of p-Akt
were decreased, and the radiosensitivity of the cells was
increased, but was not significantly different from that of the
BMI-1 shRNA and NC + LY-294002 groups with or without IR (all
P>0.05); cell viability and the expression of p-Akt were
significantly decreased, and the radiosensitivity of the cells was
improved in the BMI-1 shRNA + LY-294002 group after IR compared
with the BMI-1 shRNA group (P<0.05) (Fig. 3A, C, E and F).
IGF-1, as an agonist of PI3K, can activate PI3K and
increase the expression of p-AKT. When 3 ng/ml IGF-1 was added to
the cell culture medium, the effects of BMI-1 knockdown on cell
viability and radiosensitivity were reversed. Cell viability and
the expression of p-Akt were increased, and radiosensitivity was
decreased but was not significantly different compared to that of
the BMI-1 shRNA + IGF-1 and NC groups before and after IR (all
P>0.05); cell viability and the expression of p-Akt were
significantly increased, while the radiosensitivity of the cells
was obviously decreased in the BMI-1 shRNA + IGF-1 group after IR
compared with that of the BMI-1 shRNA group (P<0.05) (Fig. 3B, D, E and F).
BMI-1 knockdown-mediated mechanisms of
radiosensitization
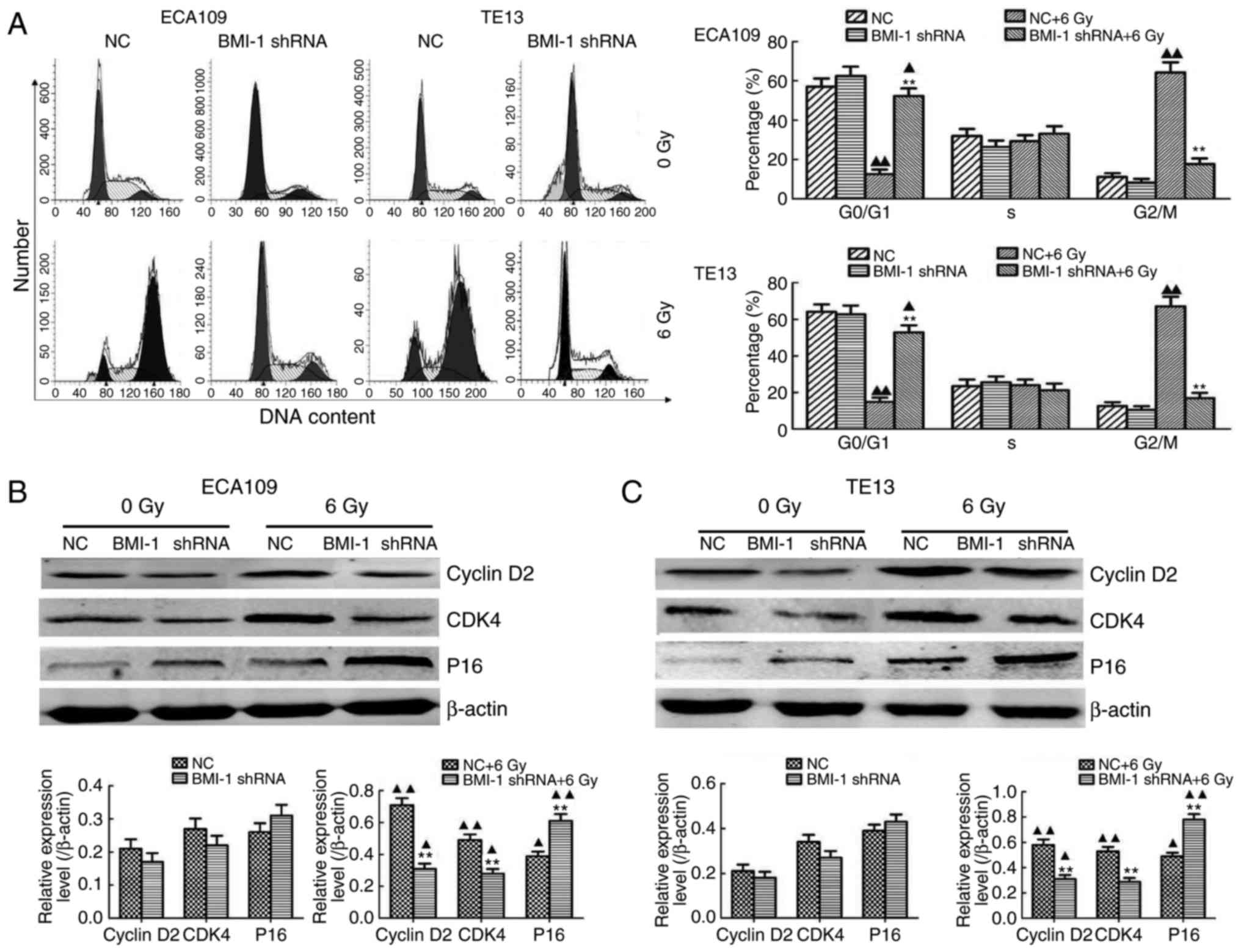
FCM was performed to explore the mechanisms involved
in BMI-1 knockdown-mediated radiosensitization, and we demonstrated
that IR obviously increased the proportion of cells in the G2/M
phase of the cell cycle in both cell types in vitro;
however, BMI-1 knockdown obviously decreased the proportion of
cells in the G2/M phase (Fig. 4A)
when considerable time was allowed for the damage repair of tumour
cells. This repair may decrease the killing effect of IR, thus
inducing radioresistance. However, the DDR was not significantly
different in the both groups without IR. Collectively, our results
showed that IR obviously induced cell cycle arrest at the G2/M
phase, while BMI-1 knockdown decreased the proportion of cells in
the G2/M phase to a certain degree, reducing the opportunity for
tumour cells to repair, thereby improving radiosensitization.
Moreover, the regulatory effect of BMI-1 on cell cycle-related
proteins was detected by western blotting. Notably, their
expression was detected before IR but was not obviously altered
between the BMI-1 shRNA and NC groups. However, IR induced their
expression in both groups. Although the expression of cyclin D2 and
cyclin-dependent kinase 4 (CDK4) at the protein level was markedly
inhibited, the expression of P16 was significantly increased in the
BMI-1 shRNA group, compared to that of the NC group (P<0.05)
(Fig. 4B and C).
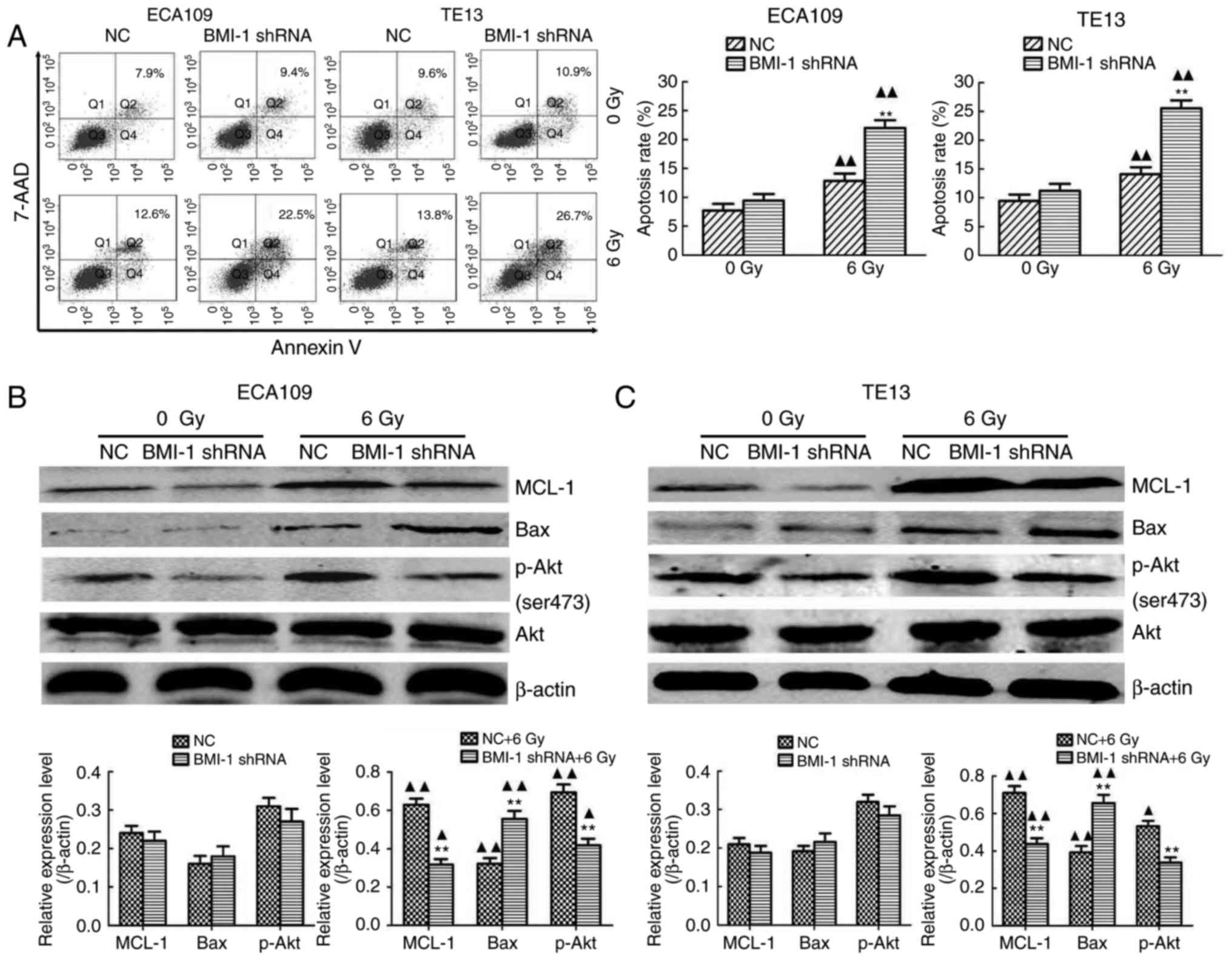
Additionally, the apoptosis rate of both cell types
in the BMI-1 shRNA group was slightly higher, compared to that of
the NC group, but was not significantly different between the BMI-1
shRNA and NC groups before IR (all P>0.05). IR induced a
dramatic increase in apoptosis, and BMI-1 knockdown further
promoted cell apoptosis after IR (P<0.01) (Fig. 5A). Moreover, IR induced the protein
expression of MCL-1 and Bax. The expression of MCL-1 was
downregulated, and the expression of Bax was upregulated after
BMI-1 knockdown (P<0.01). However, their expression was not
markedly altered before IR (Fig. 5B and
C).
BMI-1 knockdown inhibits the
activation of the PI3K/Akt pathway
As shown in Fig. 5B and
C, BMI-1 silencing did not increase the total amount of Akt in
either cell type before or after IR (P>0.05). The
phosphorylation of Akt is a characteristic of PI3K activation.
Compared with the NC group, the expression of p-AKT in ECA109 and
TE13 cells in the BMI-1 shRNA group was slightly reduced, but was
not significantly different before IR (P>0.05). IR induced the
expression of phosphorylated Akt in each group (P<0.05) and
BMI-1 knockdown markedly inhibited its expression (P<0.01).
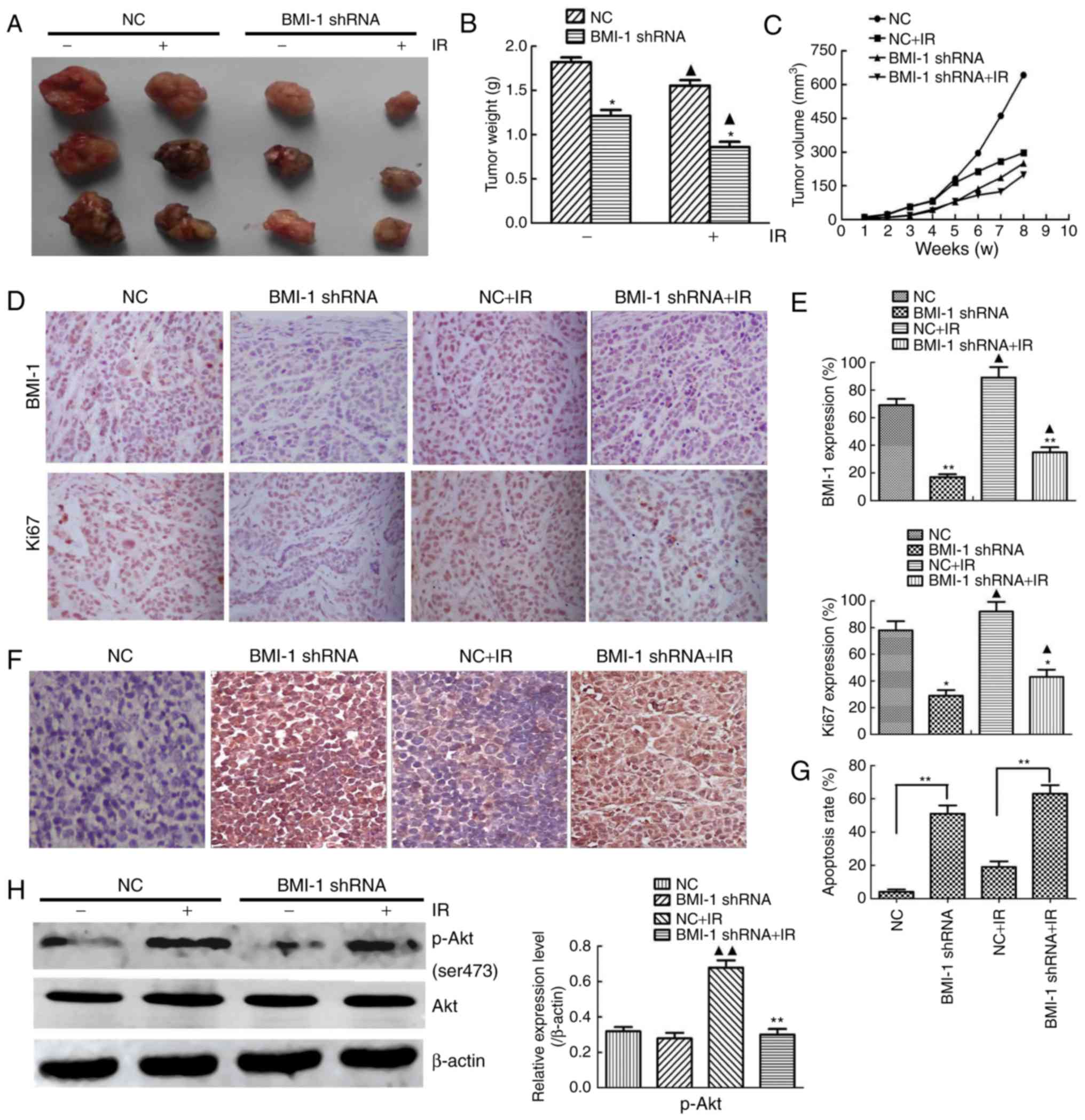
BMI-1 knockdown suppresses the tumour
formation of oesophageal cancer cells in vivo
TE13 cells in the BMI-1 knockdown and NC groups were
inoculated into nude mice to determine the function of BMI-1 in
tumour formation in vivo. Although the NC and BMI-1 shRNA
cells induced palpable tumours by 7 weeks after injection, the
BMI-1 shRNA cells developed smaller tumours than those derived from
the NC cells (Fig. 6A). BMI-1
knockdown did not affect cell viability in vitro before IR,
but it markedly slowed tumour growth in vivo. Furthermore,
IR caused smaller tumours, and the weights and volumes of tumours
from the BMI-1 shRNA group were obviously lower compared to those
of the other group after IR, indicating that BMI-1 knockdown may
improve the radiosensitivity of oesophageal cancer in vivo
(Fig. 6B and C).
The protein expression of BMI-1 and Ki67 in the
tumour tissues was analysed by immunohistochemistry (Fig. 6D). IR induced the expression of Ki67
and BMI-1 in the tumour tissues, and the tumour tissues formed by
the BMI-1 shRNA cells exhibited lower protein expression of Ki67
and BMI-1 than those formed by the NC cells before and after IR
in vivo, which was different from the results in
vitro (Fig. 6E), demonstrating
that BMI-1 promoted tumour formation and the development of
oesophageal cancer by accelerating the growth of cells in
vivo.
To examine whether BMI-1 leads to cell apoptosis
in vivo, we performed TUNEL assays in tumour tissues. These
data demonstrated significantly higher apoptosis rates in the BMI-1
shRNA group before IR compared to those in the NC group (Fig. 6F and G), and this tendency was more
obvious after IR (P<0.01). As shown in Fig. 6H, the expression of p-Akt was
downregulated in xenografts treated with shRNA and IR. The
expression of p-Akt markedly increased in the NC group after IR
compared with the corresponding unirradiated groups (P<0.01),
but it was not markedly different in the BMI-1 shRNA group,
regardless of IR (P>0.05).
Discussion
It has been shown that BMI-1 is closely related to
tumour radiosensitivity (22); it
is involved in DNA damage repair, aberrant activation of multiple
signalling pathways (23) and
autophagy, all of which are involved in the complicated process of
developing radioresistance. Given the ability of BMI-1 to regulate
multiple oncogenic processes, including the response to RT, we
considered investigation of the role of BMI-1 a promising avenue
for furthering our understanding of radiation resistance.
Therefore, any attempt to improve our understanding of the
molecular mechanisms regarding radiosensitivity was the goal of the
investigators.
In the present study, BMI-1 overexpression was
observed in both ESCC-derived cell lines and ESCC tissues. Cell
viability in both groups without IR was not significantly
different, but cell viability in the BMI-1 shRNA group was slightly
lower. However, the viability of ESCC cells in the BMI-1 shRNA
group was significantly inhibited, and the radiosensitivity of the
tumour cells was markedly improved after IR in vitro. This
tendency was more obvious after BMI-1 silencing, indicating that IR
combined with shRNA effectively gave rise to the suppression of
cell viability and enhanced the radiosensitivity of the cells. Our
data is in agreement with previous studies, which showed that BMI-1
knockdown inhibited the viability of cancer cells and further
decreased their radioresistance and chemoresistance (7,24,25). A
previous study demonstrated that BMI-1 was related closely to the
ubiquitination and phosphorylation of H2AX (26). In agreement with the above results,
our previous results from a co-IP assay revealed that the
interaction between BMI-1 and H2AK119ub and γH2AX was significantly
promoted in ESCC cells after IR, although there was no obvious
interaction before IR, showing that IR induces the correlation
between BMI-1 and the ubiquitylation and phosphorylation of H2AX
(H2AK119ub and γH2AX) (21). To
further explore their correlation, we measured their expression at
different doses and times and found that IR induced similar dose-
and time-dependent effects on the protein levels of BMI-1,
H2AK119ub and γH2AX. Furthermore, BMI-1 knockdown resulted in
decreased expression of important indexes of the DDR, such as
γH2AX, MDC1 and 53BP1, confirming that BMI-1 contributed to the DDR
through inducing the phosphorylation of H2AX (γH2AX) and its
downstream targets, such as MDC1 and 53BP1, which play a vital
regulatory role in the cell cycle.
PI3K/AKT, a signal transduction pathway, was most
closely correlated with cell viability. Abnormal activation of the
PI3K/AKT signalling pathway induced abnormal cell viability and
differentiation and promoted the growth of tumour cells (27). Phosphorylation of Akt generated
p-Akt, increased the expression of p-mTOR and p-70S6K through
further activating the mTOR pathway, and ultimately increased cell
viability (28). The present study
found that BMI-1 knockdown can inhibit cell viability and reduce
the expression of p-AKT. The changes were more obvious after IR.
Then, we used LY-294002, a specific inhibitor of PI3K, to pre-treat
ECA109 and TE13 cells and found that LY-294002 inhibited the
expression of p-AKT and cell viability and improved
radiosensitivity. Additionally, IGF-1, an agonist of PI3K, was also
used to pre-treat ECA109 and TE13 cells. We found that IGF-1
significantly increased the expression of p-AKT and reversed the
effects of BMI-1 knockdown on cell viability and radiosensitivity.
This tendency was more obvious after IR. The results finally showed
that BMI-1 knockdown inhibited the growth of cells and improved
radiosensitivity by suppressing the PI3K/Akt pathway.
Additionally, IR increased the proportion of cells
in the G2/M phase, but BMI-1 knockdown significantly eliminated
this phenomenon after IR. However, without IR, the cell cycle
distribution was not significantly different in either group. These
results are in accordance with previous studies (29,30).
To clarify whether BMI-1-mediated cell cycle arrest was involved in
regulating cell cycle-related proteins, we investigated the
expression levels of P16, cyclin D2 and CDK4 and found that BMI-1
knockdown resulted in a decrease in cyclin D2 and CDK4 expression
and an increase in P16 expression. P16, a CDK inhibitor, negatively
regulates cell cycle progression by binding various cyclin-CDK
complexes and suppressing their viability (31). It has also been demonstrated that
γH2AX, a marker of DDR, is closely associated with P16 and P53
(32,33). It has been reported that DNA damage
induces the expression of P53 and P16, accompanied by increases in
the proportion of cells in the G1 phase (34). In the presents study, BMI-1
knockdown downregulated the protein expression of cyclin D2 and
CDK, upregulated the expression of P16, and altered the cell cycle
distribution, suggesting that the effects of BMI-1 on tumour growth
are correlated with increased cell viability via the regulation of
some cyclins and P16, further regulating the cell cycle.
In addition to the cell cycle, the cellular response
to DNA damage includes apoptosis. BMI-1 knockdown caused the
suppression of cell viability and the induction of cell apoptosis,
indicating that it may have a significant therapeutic effect on
oesophageal carcinoma (35). Our
data showed that IR markedly induced cell apoptosis and that BMI-1
knockdown facilitated this trend. However, cell apoptosis was not
significantly different in either group without IR, but it was
slightly higher in the BMI-1 shRNA group, thereby indicating the
effect of BMI-1 knockdown on radiosensitization. Moreover, our
results indicated that the effect of BMI-1 knockdown on promoting
cell apoptosis after IR was accompanied by the downregulation of
MCL-1 and the upregulation of Bax, which are related to cell
apoptosis after DNA damage. The results of the present study are in
agreement with previous studies (36,37).
In addition to its effects on cell viability and radiosensitivity,
the PI3K signalling pathway is a common mechanism involved in
tumour cell metastasis and apoptosis (27,38,39).
p-Akt is the core component of the PI3K signalling pathway, and its
activation stimulates the growth of tumour cells (40). Therefore, full inactivation of p-Akt
could be the key to enhancing radiosensitivity and promoting
apoptosis in tumour cells. We found that high expression of BMI-1
after IR activated the PI3K/Akt pathway and promoted the expression
of p-Akt, accompanied by the upregulation of MCL-1 and
downregulation of Bax, which were associated with radiosensitivity
and apoptosis. In addition, BMI-1 knockdown suppressed the effect
of PI3K/Akt on cell apoptosis, further suggesting that the PI3K/Akt
signalling pathway is involved in BMI-1-mediated
radioresistance.
Given the role of BMI-1 in oesophageal carcinoma
in vitro, we explored whether the same effect appeared in
vivo. It was shown that BMI-1 knockdown obviously suppressed
the growth of tumours, including the weights and volumes, in nude
mice after IR. The Ki67 index has been shown to be upregulated in
many tumours, and its high expression is also significantly
associated with decreased survival (41). The results demonstrated that IR
induced the expression of BMI-1 and Ki67 in tumour tissues, but
BMI-1 knockdown significantly decreased Ki67-positive cells in the
tumours of nude mice. Additionally, BMI-1 knockdown also caused
decreased expression of Ki67 before IR, which was different from
the results in vitro, indicating that BMI-1 knockdown also
inhibits cell viability in vivo. The reason for this may be
that there were still certain non-specific immune functions in the
nude mice, but no specific immune functions, in addition to the
more complex microenvironment in vivo. Our TUNEL assay
results revealed that IR induced apoptosis in nude mice, but the
apoptosis rate was obviously increased after BMI-1 knockdown,
indicating that inhibition of BMI-1 knockdown in oesophageal
carcinoma is also mediated by the induction of apoptosis and thus
increases radiosensitivity in vivo. We found that BMI-1
knockdown also downregulated the expression of p-Akt only in
vivo and did not decrease the total amount of Akt, indicating
that BMI-1 may contribute to radioresistance at least partly by
activating the PI3K/Akt signalling pathway.
In summary, the present study demonstrated that
BMI-1 was overexpressed in ESCC cells and was related to poor
prognosis in ESCC patients. Moreover, BMI-1 knockdown decreased
cyclin D2 and CDK4 expression, enhanced P16 expression, eliminated
IR-induced G2/M cell cycle arrest, inhibited cell viability,
improved radiosensitivity and induced apoptosis by inactivating the
PI3K/Akt signalling pathway. To the best of our knowledge, we
systematically demonstrated overall and systematically, that BMI-1
may be an important target gene for oesophageal carcinoma
therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81372416), the Natural
Science Foundation of China of Hebei Province (no. H2017206170),
and the Medical Research Institute of Hebei Province (nos. 20160183
and 20170154).
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ma M, Zhao LM, Yang XX, Shan YN, Cui WX,
Chen L and Shan BE: p-Hydroxylcinnamaldehyde induces the
differentiation of oesophageal carcinoma cells via the
cAMP-RhoA-MAPK signalling pathway. Sci Rep. 6:313152016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bertolini G, Roz L, Perego P, Tortoreto M,
Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S,
et al: Highly tumorigenic lung cancer CD133+ cells display
stem-like features and are spared by cisplatin treatment. Proc Natl
Acad Sci USA. 106:pp. 16281–16286. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao JX and Xie XL: Regulation of gene
expression in laryngeal carcinama by microRNAs. Int J Pathol Clin
Med. 32:222–225. 2012.
|
|
5
|
Nacerddine K, Beaudry JB, Ginjala V,
Westerman B, Mattiroli F, Song JY, van der Poel H, Ponz OB,
Pritchard C, Cornelissen-Steijger P, et al: Akt-mediated
phosphorylation of Bmi1 modulates its oncogenic potential, E3
ligase activity, and DNA damage repair activity in mouse prostate
cancer. J Clin Invest. 122:1920–1932. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruan ZP, Xu R, Lv Y, Tian T, Wang WJ, Guo
H and Nan KJ: Bmi1 knockdown inhibits hepatocarcinogenesis. Int J
Oncol. 42:261–268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song W, Tao K, Li H, Jin C, Song Z, Li J,
Shi H, Li X, Dang Z and Dou K: Bmi-1 is related to proliferation,
survival and poor prognosis in pancreatic cancer. Cancer Sci.
101:1754–1760. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bosch A, Panoutsopoulou K, Corominas JM,
Gimeno R, Moreno-Bueno G, Martín-Caballero J, Morales S, Lobato T,
Martínez-Romero C, Farias EF, et al: The Polycomb group protein
RING1B is overexpressed in ductal breast carcinoma and is required
to sustain FAK steady state levels in breast cancer epithelial
cells. Oncotarget. 5:2065–2076. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Su WJ, Fang JS, Cheng F, Liu C, Zhou F and
Zhang J: RNF2/Ring1b negatively regulates p53 expression in
selective cancer cell types to promote tumor development. Proc Natl
Acad Sci USA. 110:pp. 1720–1725. 2013; View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang L, Liu JL, Yu L, Liu XX, Wu HM, Lei
FY, Wu S and Wang X: Downregulated miR-495 [corrected] inhibits the
G1-S phase transition by targeting Bmi-1 in breast cancer.
Medicine. 94:e7182015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin M, Zhang T, Liu C, Badeaux MA, Liu B,
Liu R, Jeter C, Chen X, Vlassov AV and Tang DG: miRNA-128
suppresses prostate cancer by inhibiting BMI-1 to inhibit
tumor-initiating cells. Cancer Res. 74:4183–4195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang F, Lv LZ, Cai QC and Jiang Y:
Potential roles of EZH2, Bmi-1 and miR-203 in cell proliferation
and invasion in hepatocellular carcinoma cell line Hep3B. World J
Gastroenterol. 21:13268–13276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Wang L, Erdjument-Bromage H, Vidal
M, Tempst P, Jones RS and Zhang Y: Role of histone H2A
ubiquitination in Polycomb silencing. Nature. 431:873–878. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ward IM, Minn K, van Deursen J and Chen J:
p53 Binding protein 53BP1 is required for DNA damage responses and
tumor suppression in mice. Mol Cell Biol. 23:2556–2563. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moudry P, Lukas C, Macurek L, Neumann B,
Heriche JK, Pepperkok R, Ellenberg J, Hodny Z, Lukas J and Bartek
J: Nucleoporin NUP153 guards genome integrity by promoting nuclear
import of 53BP1. Cell Death Differ. 19:798–807. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li DQ, Qiu M, Nie XM, Gui R and Huang MZ:
Oxidored-nitro domain-containing protein 1 expression is associated
with the progression of hepatocellular carcinoma. Oncol Lett.
11:3003–3008. 2016.PubMed/NCBI
|
|
17
|
Song LB, Zeng MS, Liao WT, Zhang L, Mo HY,
Liu WL, Shao JY, Wu QL, Li MZ, Xia YF, et al: Bmi-1 is a novel
molecular marker of nasopharyngeal carcinoma progression and
immortalizes primary human nasopharyngeal epithelial cells. Cancer
Res. 66:6225–6232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang XX, Ma M, Sang MX, Wang XX, Song H,
Liu ZK and Zhu SC: Radiosensitization of esophageal carcinoma cells
by knockdown of RNF2 expression. Int J Oncol. 48:1985–1996. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie G, Zhan J, Tian Y, Liu Y, Chen Z, Ren
C, Sun Q, Lian J, Chen L, Ruan J, et al: Mammosphere cells from
high-passage MCF7 cell line show variable loss of tumorigenicity
and radioresistance. Cancer Lett. 316:53–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Allegra E, Puzzo L, Zuccalà V, Trapasso S,
Vasquez E, Garozzo A and Caltabiano R: Nuclear BMI-1 expression in
laryngeal carcinoma correlates with lymph node pathological status.
World J Surg Oncol. 10:206–211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang XX, Sang MX, Zhu SC, Liu ZK and Ma M:
Radiosensitization of esophageal carcinoma cells by the silencing
of BMI-1. Oncol Rep. 35:3669–3678. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dong Q, Sharma S, Liu H, Chen L, Gu B, Sun
X and Wang G: HDAC inhibitors reverse acquired radio resistance of
KYSE-150R esophageal carcinoma cells by modulating Bmi-1
expression. Toxicol Lett. 224:121–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang MC, Jiao M, Wu T, Jing L, Cui J, Guo
H, Tian T, Ruan ZP, Wei YC, Jiang LL, et al: Polycomb complex
protein BMI-1 promotes invasion and metastasis of pancreatic cancer
stem cells by activating PI3K/AKT signaling, an ex vivo, in vitro,
and in vivo study. Oncotarget. 7:9586–9599. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang W, Zhu D, Cui X, Su J, Liu H, Han J,
Zhao F and Xie W: Knockdown BMI1 expression inhibits proliferation
and invasion in human bladder cancer T24 cells. Mol Cell Biochem.
382:283–291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma M, Zhao L, Sun G, Zhang C, Liu L, Du Y,
Yang X and Shan B: Mda-7/IL-24 enhances sensitivity of B cell
lymphoma to chemotherapy drugs. Oncol Rep. 35:3122–3130. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ginjala V, Nacerddine K, Kulkarni A, Oza
J, Hill SJ, Yao M, Citterio E, van Lohuizen M and Ganesan S: BMI1
is recruited to DNA breaks and contributes to DNA damage-induced
H2A ubiquitination and repair. Mol Cell Biol. 31:1972–1982. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang XJ, Yu HY, Cai YJ and Ke M: Lycium
barbarum polysaccharides inhibit proliferation and migration of
bladder cancer cell lines BIU87 by suppressing Pi3K/AKT pathway.
Oncotarget. 8:5936–5942. 2017.PubMed/NCBI
|
|
28
|
Fotouhi Ghiam A, Taeb S, Huang X, Huang V,
Ray J, Scarcello S, Hoey C, Jahangiri S, Fokas E, Loblaw A, et al:
Long non-coding RNA urothelial carcinoma associated 1 (UCA1)
mediates radiation response in prostate cancer. Oncotarget.
8:4668–4689. 2017.PubMed/NCBI
|
|
29
|
Liu WL, Guo XZ, Zhang LJ, Wang JY, Zhang
G, Guan S, Chen YM, Kong QL, Xu LH, Li MZ, et al: Prognostic
relevance of Bmi-1 expression and autoantibodies in esophageal
squamous cell carcinoma. BMC Cancer. 10:4672010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Min L, Dong-Xiang S, Xiao-Tong G, Ting G
and Xiao-Dong C: Clinicopathological and prognostic significance of
Bmi-1 expression in human cervical cancer. Acta Obstet Gynecol
Scand. 90:737–745. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boquoi A, Arora S, Chen T, Litwin S, Koh J
and Enders GH: Reversible cell cycle inhibition and premature aging
features imposed by conditional expression of p16Ink4a. Aging Cell.
14:139–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blanco D, Vicent S, Fraga MF,
Fernandez-Garcia I, Freire J, Lujambio A, Esteller M,
Ortiz-de-Solorzano C, Pio R, Lecanda F, et al: Molecular analysis
of a multistep lung cancer model induced by chronic inflammation
reveals epigenetic regulation of p16 and activation of the DNA
damage response pathway. Neoplasia. 9:840–852. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wen W, Peng C, Kim MO, Ho Jeong C, Zhu F,
Yao K, Zykova T, Ma W, Carper A, Langfald A, et al: Knockdown of
RNF2 induces apoptosis by regulating MDM2 and p53 stability.
Oncogene. 33:421–428. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shapiro GI, Edwards CD, Ewen ME and
Rollins BJ: p16INK4A participates in a G1 arrest checkpoint in
response to DNA damage. Mol Cell Biol. 18:378–387. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yao XB, Wang XX, Liu H, Zhang SQ and Zhu
HL: Silencing Bmi-1 expression by RNA interference suppresses the
growth of laryngeal carcinoma cells. Int J Mol Med. 31:1262–1272.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Siddique HR, Parray A, Tarapore RS, Wang
L, Mukhtar H, Karnes RJ, Deng Y, Konety BR and Saleem M: BMI1
polycomb group protein acts as a master switch for growth and death
of tumor cells: Regulates TCF4-transcriptional factor-induced BCL2
signaling. PLoS One. 8:e606642013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu D, Wan X, Huang H, Chen X, Liang W,
Zhao F, Lin T, Han J and Xie W: Knockdown of Bmi1 inhibits the
stemness properties and tumorigenicity of human bladder cancer stem
cell-like side population cells. Oncol Rep. 31:727–736. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bussink J, van der Kogel AJ and Kaanders
JH: Activation of the PI3-K/AKT pathway and implications for
radioresistance mechanisms in head and neck cancer. Lancet Oncol.
9:288–296. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Deng R, Tang J, Ma JG, Chen SP, Xia LP,
Zhou WJ, Li DD, Feng GK, Zeng YX and Zhu XF: PKB/Akt promotes DSB
repair in cancer cells through upregulating Mre11 expression
following ionizing radiation. Oncogene. 30:944–955. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Y, Zheng L, Ding Y, Li Q, Wang R,
Liu T, Sun Q, Yang H, Peng S, Wang W, et al: miR-20a induces cell
radioresistance by activating the PTEN/PI3K/Akt signaling pathway
in hepatocellular carcinoma. Int J Radiat Oncol Biol Phys.
92:1132–1140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ji Y, Zheng MF, Ye SG, Chen J and Chen Y:
PTEN and Ki67 expression is associated with clinicopathologic
features of non-small cell lung cancer. J Biomed Res. 28:462–467.
2014.PubMed/NCBI
|