Introduction
In 2015, there were 45,780 new cases of cancer of
the oral cavity and pharynx diagnosed with 90% of them attributed
to oral squamous cell carcinoma (OSCC) and 8,650 people succumbed
to this disease, since 69.4% of these new cases were diagnosed at
late stage III and IV of the disease (1). Therapeutic treatment of OSCC is based
on the stage of the disease and includes, either separately or in
combination, surgery, radiation and chemotherapeutic agents such as
cisplatin (2–4). However, despite many novel
chemotherapeutic agents and molecularly targeted therapies, the
essential molecular mechanisms underlying the pathogenesis of OSCC
and chemotherapy resistance have yet to be fully elucidated, which
is important especially to late-stage patients, since the 5-year
survival rates have not significantly improved in decades (5,6).
The initiation of oral cancer cells in primary tumor
invasion-metastasis cascade is enabled by epithelial-mesenchymal
transition (EMT), a complex and reversible process which leads to
loss of epithelial markers, such as E-cadherin and gain of
mesenchymal markers, such as N-cadherin, fibronectin and vimentin
with increased migration and invasive capabilities (7). EMT is initiated and regulated by
various different cytokines and growth factors during tumor
progression (8). The gain of EMT
markers has been associated with the resistance of ovarian
carcinoma epithelial cell lines to paclitaxel (9). Although emerging evidence indicates
that OSCCs which undergo EMT are responsible for
radio-chemoresistance, the underlying mechanism remains unclear
(10–12). Therefore, considering the oncogenic
potential of EMT, it is essential to investigate the possible role
of EMT in drug resistance to commonly used OSCC chemotherapeutic
agents such as cisplatin, in order to acquire promising
improvements.
The presence of recurrent copy number aberrations of
3q is one of the most frequent chromosomal aberrations in OSCC and
is related to a more substantial risk of metastasis (11,13).
To date, eukaryotic translation initiation factor 5A-2 (eIF5A2)
located on 3q26 is the only known substrate of deoxyhypusine
synthase (DHPS), an enzyme catalyzing hypusination that contributes
to the progression of tumor malignancy and poor prognosis (14,15).
The overexpression of eIF5A2 has been demonstrated to be related to
cancer metastasis in hepatocellular and colorectal carcinomas and
to poor prognosis in bladder and ovarian cancers (16–18).
Several studies have found that N1-guanyl-1,7-diaminoheptane (GC7),
a novel DHPS inhibitor, suppresses tumor cell proliferation by
inhibiting eIF5A2 (18–22). These findings indicated that
aberrant eIF5A2 expression was a response to the malignant behavior
of cancer cells. However, the potential oncogenic role and the
underlying molecular mechanisms of eIF5A2 in OSCC have not been
elucidated.
In addition, EMT progression was significantly
impacted by eIF5A2 through different molecular pathways in many
solid tumors (19,20,22,23).
In the present study, we examined the antitumor effect of
cisplatin-based treatment combined with GC7 in OSCC cells. We also
investigated the possible underlying molecular mechanisms through
which the activation of eIF5A2 was involved in the enhanced
cisplatin sensitivity in OSCC cells and found that the inactivation
of eIF5A2 induced by GC7 increased cisplatin chemosensitivity in
mesenchymal phenotype OSCC cells via inhibition of the signal
transducer and activator of transcription 3 (STAT3) signaling
pathway feedback activation.
Materials and methods
Cell culture and reagents
The human OSCC cell lines, Cal27, HN4, HN30 and
Tca8113, were obtained from the Chinese Oral Tissue Culture and
Collection Center (Shanghai, China) and were maintained as
monolayers in RPMI-1640 medium (Gibco, Carlsbad, CA, USA) with 10%
fetal bovine serum (FBS; HyClone Laboratories, Logan, UT, USA), 1%
penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA) in 5%
CO2 at 37°C. Cisplatin was obtained from Sigma-Aldrich
and stock solutions were prepared in dimethyl sulphoxide (DMSO).
GC7 was obtained from Calbiochem (Merck KGaA, Darmstadt, Germany).
Unless otherwise indicated, all other chemicals were the purest
grade available and were obtained from Sigma-Aldrich.
CCK-8 cell viability assay and EdU
incorporation assay
Cell viability was analyzed with the Cell Counting
Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) in accordance
with the manufacturers protocols. Briefly, OSCC cells
(3×103 cells/well) were seeded onto 96-well culture
plates, allowed to attach for 12 h and serum-starved overnight to
synchronize the cells. Subsequently, the culture medium was
replaced with complete medium containing cisplatin or cisplatin
combined with GC7 at indicated doses for 48 h. Subsequnelty, CCK-8
solution (10 µl/well) was added and the cells were incubated at
37°C for 3 h, and then absorbance was assessed at 450 nm using an
MRX II microplate reader (Dynex Technologies Inc., Chantilly, VA,
USA). The cell viability was calculated as a percentage of
untreated control cells. Assessement of the inhibition rate of cell
proliferation was performed using a Click-iT EdU Imaging kit
(Invitrogen Life Technologies, Carlsbad, CA, USA) following the
manufacturers protocol. Each experiment was performed in triplicate
and repeated three times.
eIF5A2 siRNA transfection
OSCC cells were transfected with eIF5A2 siRNA or
negative control siRNA (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) using Lipofectamine 2000 (Invitrogen Life Technologies)
according to the manufacturers instructions. The transfection
medium (Opti-MEM) was replaced with complete medium 6 h after
transfection, and then the cells were incubated for 24 h, before
all subsequent experiments were performed. Each experiment was
performed in triplicate and repeated three times.
Apoptosis analysis
For the evaluation of apoptosis in HN30 cells
induced by cisplatin (IC30 and IC50) alone or
combined with 5 µM GC7 for 48 and 24 h, respectively, the Annexin
V-FITC PI Apoptosis Detection kit (BD Pharmingen; BD Biosciences,
San Diego, CA, USA) was used according to the manufacturers
protocols. The apoptosis cells were identified using an FC 500 flow
cytometer (Beckman Coulter, Inc., Los Angeles, CA, USA) at 488 nm.
Each experiment was performed in triplicate and repeated three
times.
Western blot analysis
OSCC cells were collected and whole cellular
extracts were lysed in 50 µl cell of lysis buffer (Cell Signaling
Technology, Beverly, MA, USA) containing protease inhibitors
(BioVision, Milpitas, CA, USA) according to the manufacturers
instructions. The protein concentration was quantified using the
BCA Protein kit (Thermo Fisher Scientific, Rockford, IL, USA).
Equal amounts of proteins were separated by 10% SDS-PAGE and
transferred to PVDF membranes (Millipore, Billerica, MA, USA),
blocked with TBS/T containing 5% bovine serum albumin (BSA) for 2 h
on ice, and then incubated with primary antibodies against
E-cadherin (mouse monoclonal, ab1416), vimentin (mouse monoclonal,
ab8978), eIF5A2 (mouse polyclonal, ab168122), p53 (mouse
monoclonal, ab26), STAT3 (rabbit monoclonal, ab68153), p-STAT3
(rabbit monoclonal, ab76315), c-Myc (rabbit monoclonal, ab32072) or
GAPDH (mouse monoclonal, ab8245) (1:1,000 diluted in TBS/T; Abcam,
Cambridge, MA, USA) at 4°C overnight, washed three times with TBS/T
and then incubated with the appropriate HRP-conjugated secondary
antibodies (rabbit anti-mouse, ab6728; goat anti-rabbit, ab6721)
(dilution 1:2,000; Abcam) for 1 h at room temperature. The protein
bands were developed using chemiluminescence (GE Healthcare Life
Sciences, Piscataway, NJ, USA) and visualized using autoradiography
(Kodak, Rochester, NY, USA). The quantification of proteins was
performed by estimation of the protein band densities using
Image-Pro Plus 6.0 software (Media Cybernetics Inc., Bethesda, MD,
USA) and the protein levels were standardized to GAPDH.
Establishment and treatment of tumor
xenografts in vivo
All animal studies were carried out in compliance
with the Guide for the Care and Use of Laboratory Animals of
Zhejiang University (Zhejiang, China). All applicable
international, national, and/or institutional guidelines for the
care and use of animals were followed. Male athymic BALB/c nude
mice (Shanghai Experiment Animal Centre, Shangai, China), 5–6 weeks
old and weighing 15–20 g, were used in the present study. OSCC
Tca8113 cells (1×106) suspended in 0.1 ml phosphate
buffered saline (PBS) were injected subcutaneously (s.c.) into the
left flank of each mouse and tumor growth was monitored every other
day. Tumor length (L) and width (W) were assessed with a sliding
caliper and tumor volume was determined with the standard formula
(LxW2)/2 (24,25). Drug treatment was initiated when the
tumor volume reached 30–75 mm3. Twenty nude mice
injected with OSCC cells were randomly divided into four groups:
three experimental groups including GC7 (1 mg/kg), cisplatin (2
mg/kg) and GC7 (1 mg/kg) + cisplatin (2 mg/kg), as well as one
vehicle-treated control group (equal volume of diluents). The drugs
were administered intraperitoneally (i.p.) every 2 days for 2
weeks. Subsequently, the mice were euthanized by cervical
dislocation, and then the tumors were carefully dissected from each
mouse for tumor weight assessement. The relative tumor inhibitory
rate was calculated as follows: (mean tumor weight of the control
group - mean tumor weight of the experimental group)/(mean tumor
weight of the control group) × 100%.
Statistical analysis
The results are presented as the mean ± standard
deviation (SD). Data were statistically analyzed using the SPSS
17.0 software (SPSS Inc., Chicago, IL, USA). The effects of the
combined treatment were compared using two-way ANOVA, followed by
Bonferronis post hoc test. Analyses of the comparisons of two
groups were carried out using Students t-tests. P<0.05 was
considered to indicate a statistically significant difference.
Results
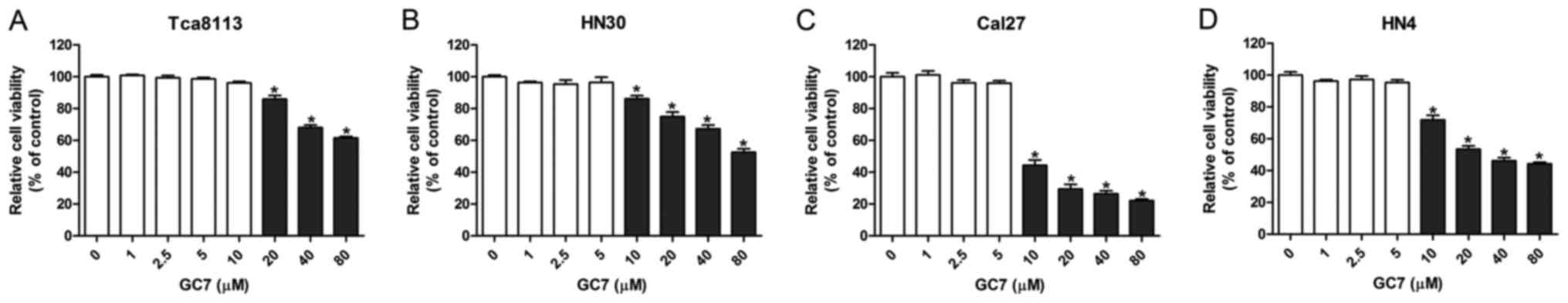
Low concentrations of GC7 exhibit
slight cytotoxicity against OSCC cells
By inhibiting the hypusination of eIF5A2 by DHPS,
GC7 specifically prevents the activation of eIF5A2. However, the
cytotoxicity of GC7 against OSCC cells has rarely been reported. To
determine the appropriate concentration of GC7 for
co-administration with cisplatin, CCK-8 cell viability assays were
performed in order to test the cytotoxicity of GC7 in Tca8113,
Cal27, HN30 and HN4 OSCC cell lines. The results revealed that GC7
had almost no effect on Cal27, HN30 and HN4 cell viability at a
concentration between 0 and 5 µM, however, GC7 significantly
inhibited cell viability at a concentration exceeding 10 µM
(Fig. 1B-D). Similarly, the cell
viability of Tca8113 cells was not affected when the GC7
concentration was <10 µM, whereas it was significantly inhibited
at a GC7 concentration exceeding 20 µM (Fig. 1A). Our data indicated that low
concentrations of GC7 exerted little cytotoxicity against OSCC
cells. Several studies have reported that low concentrations of GC7
could inhibit the hypusination of eIF5A2 effectively in some tumor
cells (26,27). Subsequently, 5 µM GC7, which exerted
little cytotoxicity against OSCC cells but could inhibit the
efficiency of eIF5A2 activation, was used for the following
co-treatments with cisplatin.
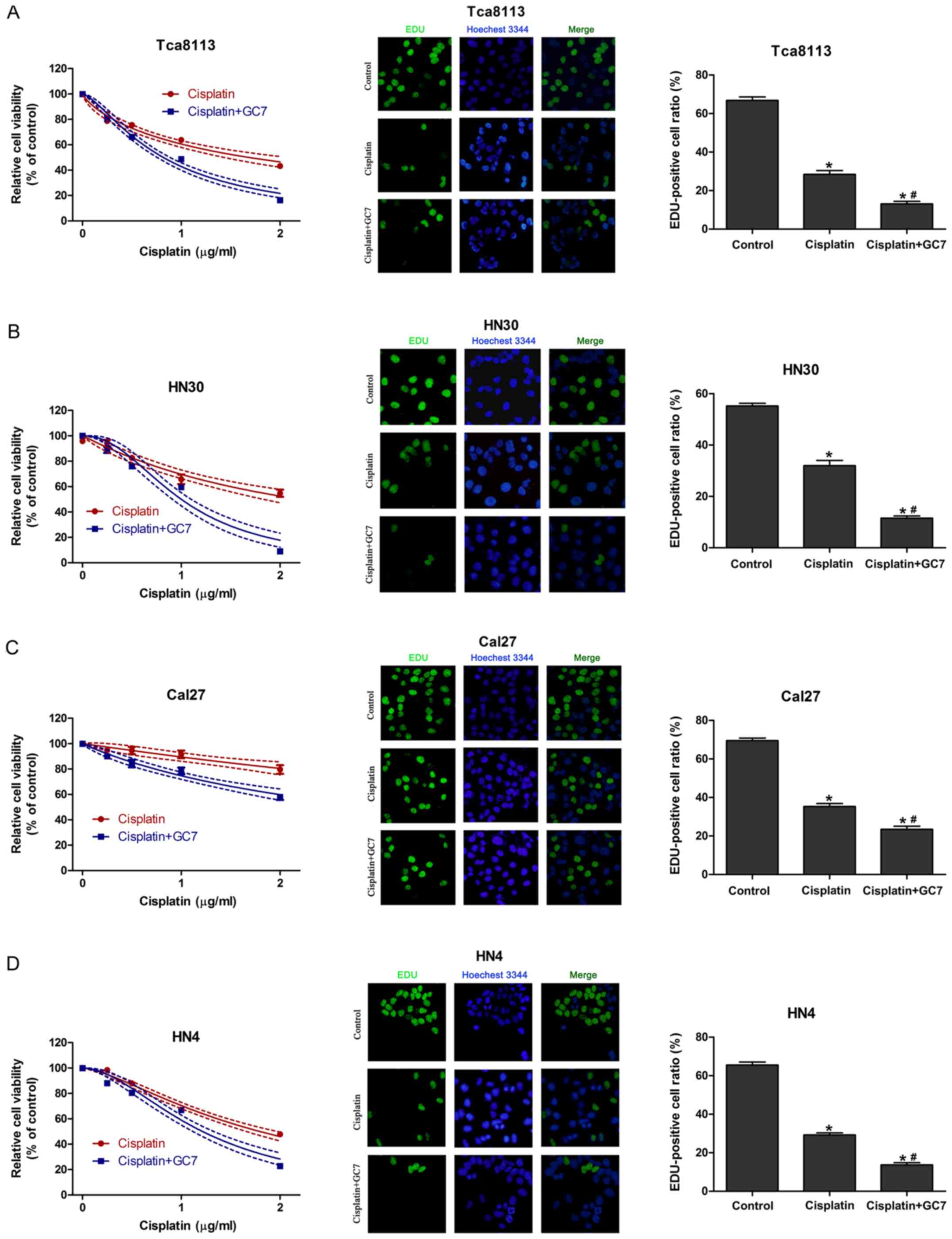
GC7 enhances the sensitivity of
cisplatin in OSCC cells through inhibition of cell proliferation
without induction of apoptosis
To assess the synergistic cytotoxic effect of
cisplatin plus GC7, we used a CCK-8 assay to assess cell viability
and EdU incorporation assay to test the inhibition of proliferation
of OSCC cells treated with cisplatin alone or cisplatin plus GC7
for 48 h. The results revealed that increasing the concentration
doses of cisplatin reduced cell viability in all cell lines
(Fig. 2). Tca8113, HN4 and HN30
cells exhibited a higher sensitivity to cisplatin than Cal27 cells.
The IC50 values for cisplatin alone at 48 h in Tca8113,
HN30, HN4 and Cal27 cells were 1.685 (1.535–1.835), 2.201
(1.971–2.431), 1.797 (1.722–1.872) and 7.729 µg/ml (4.162–11.296
µg/ml), respectively (Table I).
When cisplatin was combined with 5 µM GC7, cisplatin sensitivity
significantly increased and exerted a stronger anti-proliferative
effect in all four cell lines compared to cisplatin alone and the
IC50 values at 48 h in Tca8113, HN30, HN4 and Cal27
cells were decreased to 0.818 (0.787–0.850, P<0.05), 1.000
(0.955–1.046, P<0.05), 1.215 (1.164–1.266, P<0.05) and 2.994
µg/ml (2.611–3.37 µg/ml, P<0.05), respectively (Fig. 2 and Table I). Therefore, GC7 significantly
sensitized OSCC cells to cisplatin.
 | Table I.IC50 values of cisplatin
in OSCC cell lines with or without GC7 treatment. |
Table I.
IC50 values of cisplatin
in OSCC cell lines with or without GC7 treatment.
|
| IC50
(µg/ml)a |
|---|
|
|
|
|---|
| OSCC cell
lines | Cisplatin | Cisplatin +
GC7 |
|---|
| Cal27 | 7.729
(4.162–11.296) | 2.994
(2.611–3.377)b |
| HN4 | 1.797
(1.722–1.872) | 1.215
(1.164–1.266)b |
| HN30 | 2.201
(1.971–2.431) | 1.000
(0.955–1.046)b |
| Tca8113 | 1.685
(1.535–1.835) | 0.818
(0.787–0.850)b |
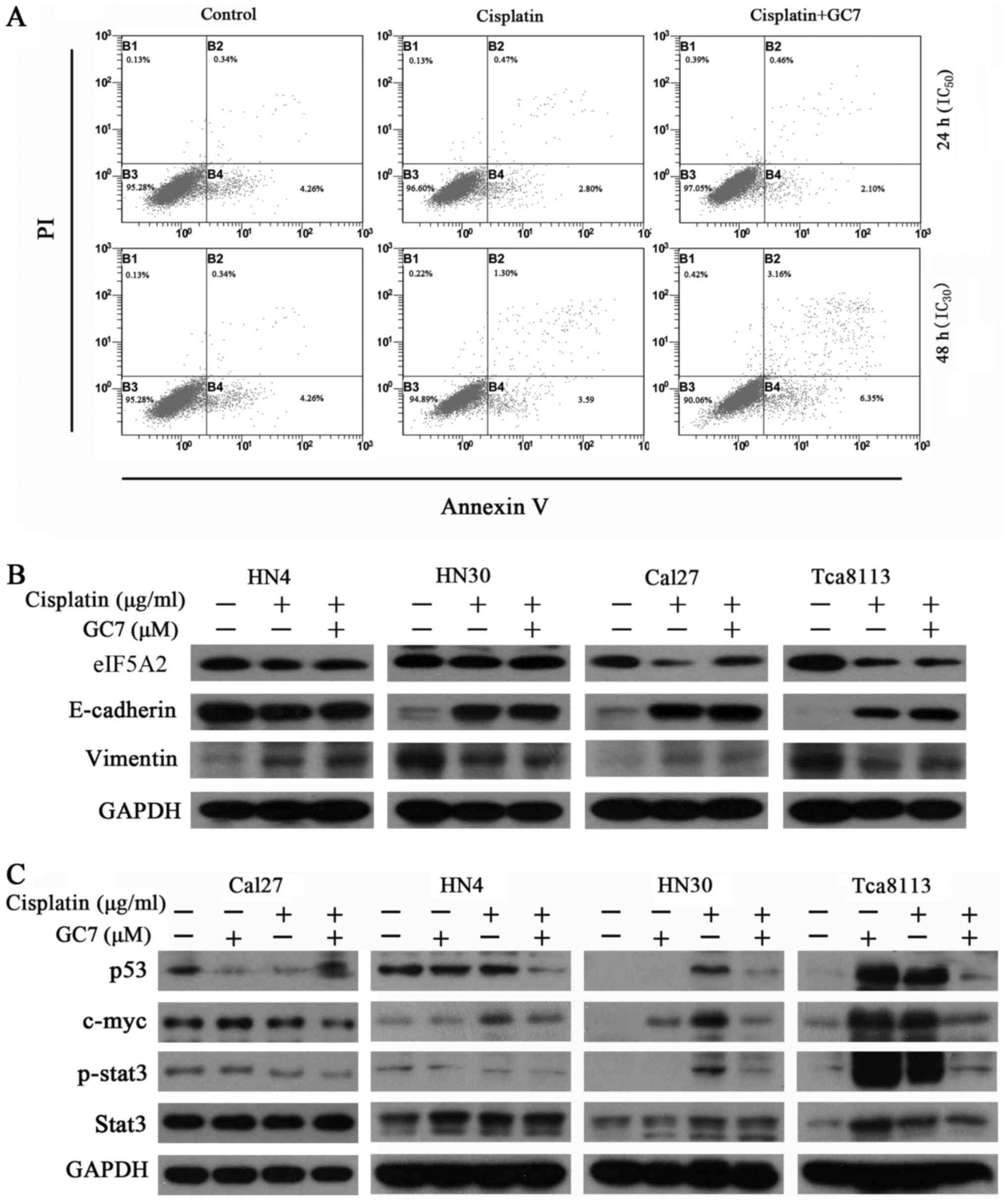
The induction of apoptosis in HN30 cells was
analyzed by flow cytometry following administration of cisplatin
(IC50 and IC30 values) alone or in
combination with 5 µM GC7 for 24 and 48 h, respectively. Notably,
co-treatment with GC7 had no evident effect on apoptosis induction
compared to cisplatin alone (Fig.
3A, P>0.05).
GC7 sensitizes the cytotoxicity of
cisplatin in mesenchymal phenotype OSCC cells through STAT3 pathway
inhibition and eIF5A2 inactivation
In order to determine whether the phenotype of OSCC
cells contributed to their differing chemosensitivity to combined
therapy, we assessed the expression of epithelial/mesenchymal
markers in OSCC cells. The results revealed that the
E-cadherin/vimentin ratio was clearly higher in the HN4 and Cal27
cells with an epithelial phenotype than in the HN30 and Tca8113
cells, which have a mesenchymal phenotype (Fig. 3B). Notably, mesenchymal OSCC cells
exhibited more sensitivity to cisplatin than epithelial OSCC cells.
Cisplatin alone reversed EMT in mesenchymal OSCC cells through
upregulation of E-cadherin and downregulation of vimentin. Although
cisplatin alone increased the expression of E-cadherin, it also
slightly increased the expression of vimentin in epithelial OSCC
cells. There were no significant changes in the expression of
E-cadherin and vimentin in both epithelial and mesenchymal OSCC
cells treated with cisplatin alone as compared to the combined
treatment (Fig. 3B). Therefore, the
ability of GC7 to enhance the cytotoxicity of cisplatin did not
occur in relation to phenotype transition in OSCC cells.
STAT3, one of the seven members of the STAT protein
family that regulates cell cycle progression and cell growth,
exhibits constitutive activity in various human malignancies,
including breast, prostate, head and neck, lung, colon, liver and
pancreatic cancers, and lymphoma, leukemia and multiple myeloma
(1–3). Notably, we revealed that the ability
of GC7 to enhance the cytotoxicity of cisplatin in mesenchymal
phenotype OSCC cells may be mediated through STAT3 pathway
inhibition and the eIF5A2 inactivation. As shown in Fig. 3B and C, c-Myc and p-STAT3 were
upregulated in OSCC cells treated with cisplatin or GC7 alone
compared to the control, whereas the combined treatment could
significantly reverse this upregulation in mesenchymal phenotype
Tca8113 and HN30 cells. Similar results were not observed in
epithelial phenotype Cal27 and HN4 cells. Notably, we found that
the expression of eIF5A2 was decreased via combined treatment in
Tca8113 cells.
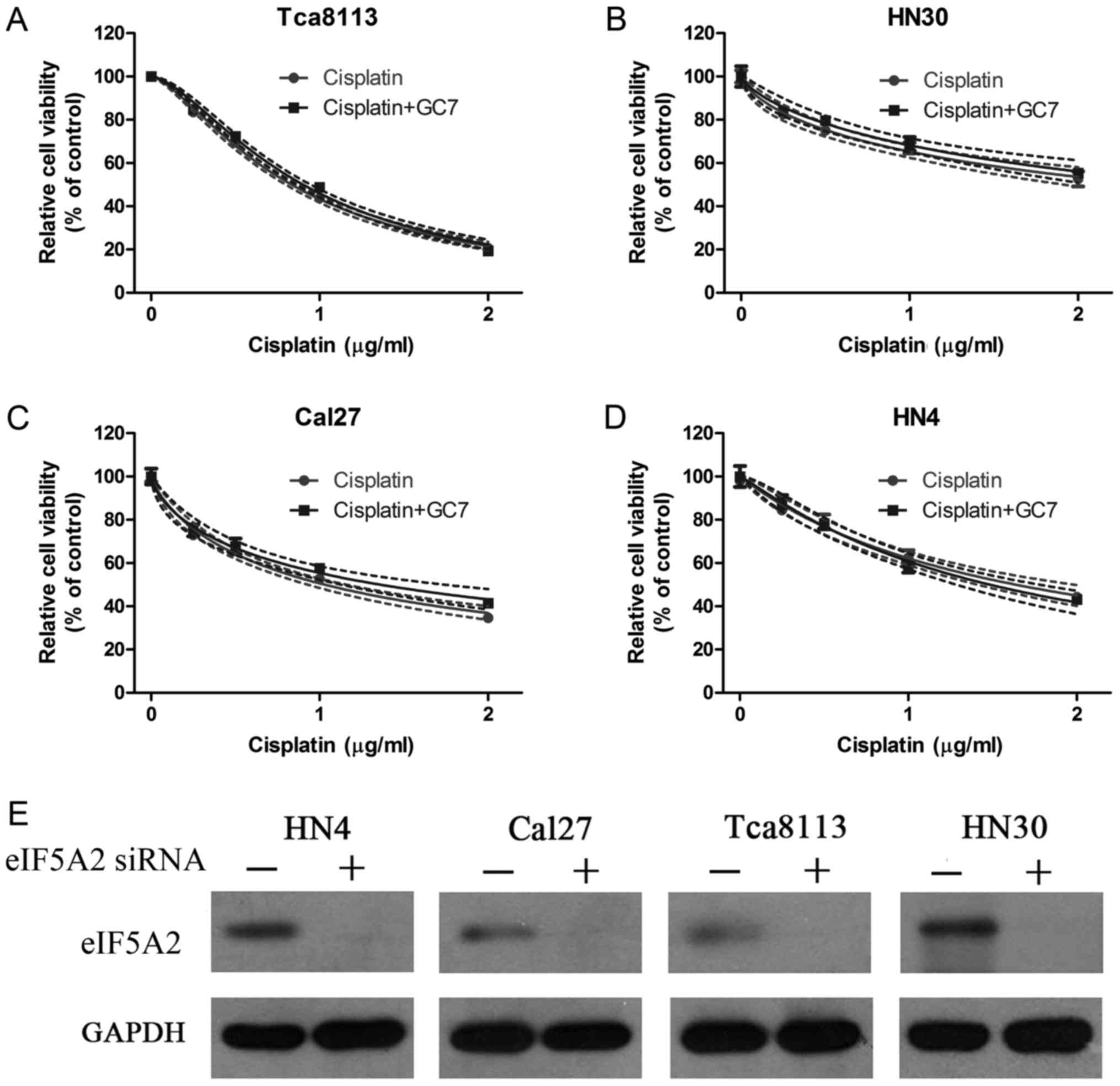
To further ascertain the mechanism by which GC7
enhanced the cytotoxicity of cisplatin in OSCC cells, eIF5A2 siRNA
was transfected into HN4, Cal27, HN30 and Tca8113 cells to inhibit
the eIF5A2 expression and found that the expression of eIF5A2 was
significantly inhibited in all four OSCC cell lines (Fig. 4E). CCK-8 cell viability assays were
then carried out in these transfected OSCC cells treated with
cisplatin alone or combined with GC7 for 48 h. The results
indicated that increasing the concentration doses of cisplatin
reduced cell viability in all cell lines (Fig. 4A-D). Although the sensitivity to
cisplatin alone increased significantly in Cal27 cells after the
eIF5A2 siRNA transfection the IC50 value at 48 h was
decreased from 7.729 (4.162–11.296) to 1.030 (0.975–1.085 µg/ml)
(Tables I and II). The Tca8113 cells were the most
sensitive to cisplatin alone after eIF5A2 siRNA transfection and
the IC50 value at 48 h was 0.833 (0.816–0.851 µg/ml)
(Table II). However, when
cisplatin treatment was combined with GC7 after eIF5A2 siRNA
transfection, no significant increase of cisplatin sensitivity was
observed in all four OSCC cell lines and the IC50 values
at 48 h in Tca8113, HN30, HN4 and Cal27 cells were 0.901
(0.880–0.922), 2.771 (2.293–3.249), 1.483 (1.368–1.598) and 1.359
µg/ml (1.220–1.498 µg/ml), respectively (Table II and Fig. 4A-D).
| Table II.IC50 values of cisplatin
in eIF5A2-siRNA transfected OSCC cell lines with or without GC7
treatment. |
Table II.
IC50 values of cisplatin
in eIF5A2-siRNA transfected OSCC cell lines with or without GC7
treatment.
|
| IC50
(µg/ml)a |
|---|
|
|
|
|---|
| OSCC cell
lines | Cisplatin | Cisplatin +
GC7 |
|---|
| Cal27 | 1.030
(0.975–1.085) | 1.359
(1.220–1.498) |
| HN4 | 1.637
(1.504–1.770) | 1.483
(1.368–1.598) |
| HN30 | 2.452
(2.096–2.808) | 2.771
(2.293–3.249) |
| Tca8113 | 0.833
(0.816–0.851) | 0.901
(0.880–0.922) |
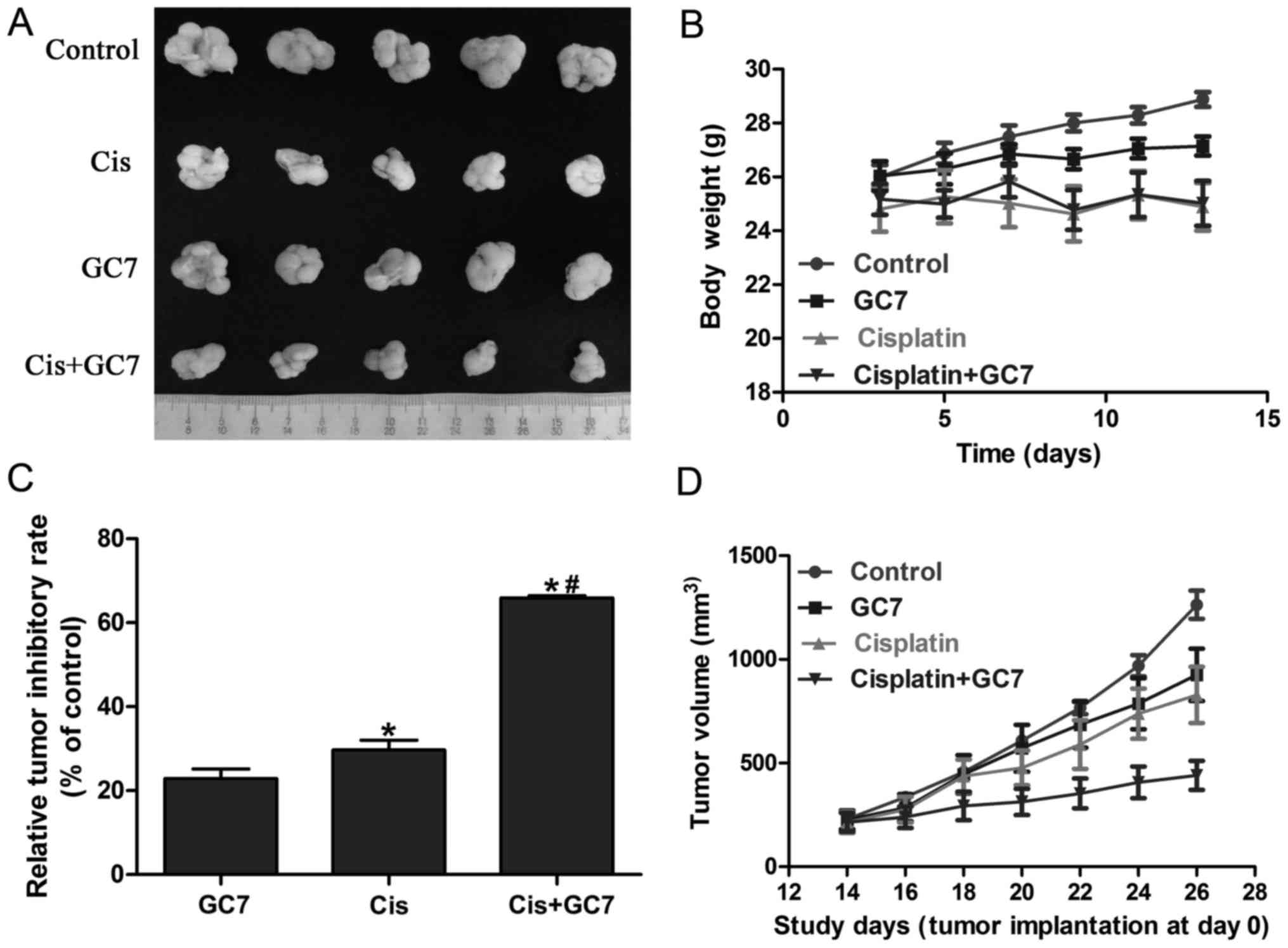
The antitumor effect of cisplatin is
boosted by GC7 without evident toxic side effect increase in the
Tca8113 cells in vivo
Based on the significant enhancement of cytotoxicity
of cisplatin by GC7 in OSCC cells in vitro and the essential
role of STAT3 pathway inhibition via eIF5A2 inactivation, we
further examined the antitumor effect of the cisplatin/GC7
combination in Tca8113 cells in vivo. As shown in Fig. 5A-C, GC7 alone exhibited a
significant antitumor inhibitory rate compared to the control which
indicated the crucial role of eIF5A2 activation in OSCC
tumorigenesis. Cisplatin alone revealed lower tumor volume. In
addition, cisplatin alone resulted into relatively more body weight
loss compared to GC7 alone, which may indicate a higher toxic side
effect (Fig. 5B and D). The
differences of body weight between the cisplatin and cisplatin/GC7
groups were marginal. Furthermore, tumor volume and weight were
significantly decreased in the cisplatin/GC7 group as compared to
the cisplatin group alone (Fig. 5B and
D). Our results strongly indicated that GC7 may enhance the
antitumor effect of cisplatin in vivo without causing severe
systemic side-effects (body weight change).
Discussion
The most significant clinical factor responsible for
the vast majority of OSCC deaths is not only metastasis to cervical
lymph nodes, but also the impact of primary tumors. Despite the
progress in surgery and radiation therapy especially in adjuvant or
neo-adjuvant chemotherapy, the 5-year survival rate for patients
with advanced or recurrent OSCC has not significantly improved over
the past 30 years (1). Therefore,
an improved, more promising therapeutic intervention to improve the
prognosis of OSCC patients is essential. Recently, combined
treatment based on chemotherapy drugs or molecularly-targeted
agents revealed promising synergistic antitumor effects and
relieved the toxic side-effects usually caused by chemotherapy
(4,21,28,29).
The overexpression of eIF5A2 has been reported to be associated
with invasion and metastasis in various human malignances,
including colon, bladder, ovarian and hepatocellular cancer
(14,15,17).
In addition, eIF5A2 has been shown to induce EMT in colorectal and
hepatocellular cancer (22,23,26).
In the present study, we first demonstrated that GC7, an inhibitor
of the eIF5A2 activation, exerted better antitumor effects in the
OSCC cells in vitro and in vivo combined with
cisplatin-based therapy, the most frequently used chemotherapy
drugs in OSCC patients, and then identified the potential molecular
mechanism mediating the synergistic antitumor effects.
As the substrate of DHPS, eIF5A2 isolated from 3q26
using chromosome microdissection and hybrid selection, is essential
for the proliferation of mammalian cells and has recently been
considered as a novel candidate oncogene highly associated to tumor
metastasis and invasion (30–33).
Despite the fact that the properties of malignancy of various human
cancers are closely related with the expression of eIF5A2, the
expression and function of eIF5A2 in OSCC is not fully elucidated.
In the present study, we observed a high level of eIF5A2 expression
in HN4, Cal27, HN30 and Tca8113 cells, while the inactivation of
eIF5A2 by GC7 or the knockdown of eIF5A2 expression by siRNA
transfection revealed a significant inhibition of proliferation of
the OSCC cells in vitro and in vivo. Thus, we came to
the conclusion that eIF5A2 may also play a key role in the
properties of malignancy of the OSCC cells which may be a potential
therapeutic target.
EMT has been demonstrated to be associated with
tumorigenicity, growth and invasiveness in various human tumors
including OSCC and it has been suggested that EMT may be involved
in drug resistance in OSCC (7,8,12,34,35).
In the present study, we found that mesenchymal Tca8113 and HN30
cells were more sensitive to cisplatin compared to epithelial Cal27
and HN4 cells. Compared to the untreated control cells, cisplatin
downregulated vimentin and upregulated E-cadherin in the Tca8113
and HN30 cells, which indicated that the cells underwent EMT
reversion in response to cisplatin. Furthermore, cisplatin
downregulated E-cadherin and upregulated vimentin in the relatively
less sensitive Cal27 and HN4 cells which indicated that the cells
underwent EMT. However, when treated with the combination of
cisplatin and GC7, a novel inhibitor of DHPS which is required for
the activation of eIF5A2, the expression of E-cadherin and vimentin
did not change, while the sensitivity to cisplatin was
significantly inceased compared to cisplatin treatment alone in the
OSCC cell lines. This study revealed that GC7 enhanced the
cytotoxicity of cisplatin in OSCC cells through the inactivation of
eIF5A2 without EMT involvement. To further determine the role of
eIF5A2 in the increased GC7-induced cisplatin sensitivity, the OSCC
cells were transfected with eIF5A2 siRNA. Consistent with our
hypothesis, the synergistic effect of cisplatin and GC7 was
eliminated by eIF5A2 siRNA transfection.
STAT3 is one of the seven members of the STAT
protein family with SH2 (Src Homology-2) domains that act as signal
messengers and transcription factors and mediate the actions of
many cytokines and growth factors (11). The constitutive activity of STAT3
has been associated with a wide variety of human malignancies,
including breast, prostate, head and neck, lung, colon, liver and
pancreatic cancers, and multiple myeloma (36–39).
The in vitro data in the present study revealed that the
mesenchymal phenotype Tca8113 and HN30 cells were more sensitive to
cisplatin compared to the epithelial Cal27 and HN4 cells. To
ascertain the molecular mechanisms involved in the combination of
GC7 with cisplatin in different phenotype OSCC cells, we observed
that cisplatin significantly upregulated p-STAT3 and c-Myc which
was abolished when co-treated with GC7 in mesenchymal phenotype
Tca8113 and HN30 cells (40).
Similar results were not observed in epithelial phenotype Cal27 and
HN4 cells, which indicated that GC7 may enhance cisplatin sentivity
via other molecular mechanisms in epithelial phenotype Cal27 and
HN4 cells. Therefore, our data revealed that the STAT3 signaling
pathway may be involved in chemoresistance to cisplatin in more
aggressive mesenchymal OSCC cells and that inhibition of eIF5A2
activity could eliminate this effect. The specific mechanisms
involved in STAT3 pathway regulation and eIF5A2 activity are worth
further investigation in mesenchymal phenotype OSCC cells.
Our aim for the future is not only to increase the
efficiency of cisplatin in more aggressive OSCC cells, but more
importantly, to reduce the adverse side-effects associated with the
administration of high doses of cisplatin. The results obtained
with OSCC cells in vitro prompted us to evaluate the
efficacy of GC7 in increasing cisplatin sensitivity in vivo.
In combination with GC7, it is interesting to note that a
significant inhibition of tumor growth was found without causing
distinct body weight reduction compared with cisplatin alone.
Accordingly, we considered that the potent effect of cisplatin
combined with GC7 may be more specific in blocking OSCC cell growth
than other normal cells, which could represent a remarkable key
point in developing efficient and safe adjuvant treatment of OSCC.
Further in vitro and in vivo studies are required to
investigate the underlying molecular mechanisms involved in the
increase of cisplatin sensitivity via GC7-mediated inactivation of
eIF5A2 in OSCC cells, especially with a more aggressive mesenchymal
phenotype.
In conclusion, the present study revealed that
combined treatment with GC7 enhanced the cytotoxicity of cisplatin
in OSCC cells through inhibition of eIF5A2 activation. In addition,
this is the first evidence that combined treatment with GC7
presents a significant antineoplastic effect in more aggressive
mesenchymal phenotype OSCC cells in vitro and in vivo
by preventing cisplatin- or GC7-induced STAT3 signaling pathway
activation. Therefore, the combination therapy with GC7 may
contribute to a better effect with a lower recurrence rate and
adverse side-effects after cisplatin chemotherapy. Collectively,
these findings not only provided strong evidence to consider eIF5A
as a novel target in OSCC, but also offered a new strategy to
improve the treatment of patients with advanced or recurrent OSCC
in clinical practice.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (grant no. 81302353).
References
|
1
|
Cancer Stat Facts SEER: Oral Cavity and
Pharynx Cancer. National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/statfacts/html/oralcav.html
|
|
2
|
Rohde S, Kovács AF, Turowski B, Yan B,
Zanella F and Berkefeld J: Intra-arterial high-dose chemotherapy
with cisplatin as part of a palliative treatment concept in oral
cancer. AJNR Am J Neuroradiol. 26:1804–1809. 2005.PubMed/NCBI
|
|
3
|
Kovács AF: Chemoembolization using
cisplatin crystals as neoadjuvant treatment of oral cancer. Cancer
Biother Radiopharm. 20:267–279. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Andreadis C, Vahtsevanos K, Sidiras T,
Thomaidis I, Antoniadis K and Mouratidou D: 5-Fluorouracil and
cisplatin in the treatment of advanced oral cancer. Oral Oncol.
39:380–385. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
da Silva SD, Hier M, Mlynarek A, Kowalski
LP and Alaoui-Jamali MA: Recurrent oral cancer: Current and
emerging therapeutic approaches. Front Pharmacol. 3:1492012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mehrotra R, Ibrahim R, Eckardt A, Driemel
O and Singh M: Novel strategies in therapy of head and neck cancer.
Curr Cancer Drug Targets. 11:465–478. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Smith A, Teknos TN and Pan Q: Epithelial
to mesenchymal transition in head and neck squamous cell carcinoma.
Oral Oncol. 49:287–292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chang JY, Wright JM and Svoboda KK: Signal
transduction pathways involved in epithelial-mesenchymal transition
in oral cancer compared with other cancers. Cells Tissues Organs.
185:40–47. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.PubMed/NCBI
|
|
10
|
Lewin B, Siu A, Baker C, Dang D, Schnitt
R, Eisapooran P and Ramos DM: Expression of Fyn kinase modulates
EMT in oral cancer cells. Anticancer Res. 30:2591–2596.
2010.PubMed/NCBI
|
|
11
|
Sasahira T, Kirita T and Kuniyasu H:
Update of molecular pathobiology in oral cancer: A review. Int J
Clin Oncol. 19:431–436. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Attramadal CG, Kumar S, Boysen ME, Dhakal
HP, Nesland JM and Bryne M: Tumor budding, EMT and cancer stem
cells in T1-2/N0 oral squamous cell carcinomas. Anticancer Res.
35:6111–6120. 2015.PubMed/NCBI
|
|
13
|
Zhang Z, Filho MS and Nör JE: The biology
of head and neck cancer stem cells. Oral Oncol. 48:1–9. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mathews MB and Hershey JWB: The
translation factor eIF5A and human cancer. Biochim Biophys Acta.
1849:836–844. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Clement PMJ, Henderson CA, Jenkins ZA,
Smit-McBride Z, Wolff EC, Hershey JW, Park MH and Johansson HE:
Identification and characterization of eukaryotic initiation factor
5A-2. Eur J Biochem. 270:4254–4263. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xing ZH, Wei JH, Cheang TY, Wang ZR, Zhou
X, Wang SS, Chen W, Wang SM, Luo JH and Xu AW: Bifunctional
pH-sensitive Zn(II)-curcumin nanoparticles/siRNA effectively
inhibit growth of human bladder cancer cells in vitro and in vivo.
J Mater Chem B Mater Biol Med. 2:2714–2724. 2014. View Article : Google Scholar
|
|
17
|
Wang FW, Guan XY and Xie D: Roles of
eukaryotic initiation factor 5A2 in human cancer. Int J Biol Sci.
9:1013–1020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang DJ, Dong SS, Ma NF, Xie D, Chen L, Fu
L, Lau SH, Li Y, Li Y and Guan XY: Overexpression of eukaryotic
initiation factor 5A2 enhances cell motility and promotes tumor
metastasis in hepatocellular carcinoma. Hepatology. 51:1255–1263.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Y, Liu R, Fu P, Du F, Hong Y, Yao M,
Zhang X and Zheng S: N1-Guanyl-1,7-diaminoheptane sensitizes
estrogen receptor negative breast cancer cells to doxorubicin by
preventing epithelial-mesenchymal transition through inhibition of
eukaryotic translation initiation factor 5A2 activation. Cell
Physiol Biochem. 36:2494–2503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J, Yu H, Shen M, Wei W, Xia L and
Zhao P: N1-guanyl-1,7-diaminoheptane sensitizes bladder cancer
cells to doxorubicin by preventing epithelial-mesenchymal
transition through inhibition of eukaryotic translation initiation
factor 5A2 activation. Cancer Sci. 105:219–227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu G, Yu H, Shi X, Sun L, Zhou Q, Zheng D,
Shi H, Li N, Zhang X and Shao G: Cisplatin sensitivity is enhanced
in non-small cell lung cancer cells by regulating
epithelial-mesenchymal transition through inhibition of eukaryotic
translation initiation factor 5A2. BMC Pulm Med. 14:1742014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lou B, Fan J, Wang K, Chen W, Zhou X,
Zhang J, Lin S, Lv F and Chen Y: N1-guanyl-1,7-diaminoheptane (GC7)
enhances the therapeutic efficacy of doxorubicin by inhibiting
activation of eukaryotic translation initiation factor 5A2 (eIF5A2)
and preventing the epithelial-mesenchymal transition in
hepatocellular carcinoma cells. Exp Cell Res. 319:2708–2717. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bao Y, Lu Y, Wang X, Feng W, Sun X, Guo H,
Tang C, Zhang X, Shi Q and Yu H: Eukaryotic translation initiation
factor 5A2 (eIF5A2) regulates chemoresistance in colorectal cancer
through epithelial mesenchymal transition. Cancer Cell Int.
15:1092015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tomayko MM and Reynolds CP: Determination
of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother
Pharmacol. 24:148–154. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dykes DJ, Harrison SD Jr, Mayo JG and
Griswold DP Jr: Excision assay for initial evaluation of antitumor
drug activity in mice bearing human tumor xenografts. J Natl Cancer
Inst. 84:528–530. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee NP, Tsang FH, Shek FH, Mao M, Dai H,
Zhang C, Dong S, Guan XY, Poon RT and Luk JM: Prognostic
significance and therapeutic potential of eukaryotic translation
initiation factor 5A (eIF5A) in hepatocellular carcinoma. Int J
Cancer. 127:968–976. 2010.PubMed/NCBI
|
|
27
|
Nakanishi S and Cleveland JL: Targeting
the polyamine-hypusine circuit for the prevention and treatment of
cancer. Amino Acids. 48:2353–2362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang L, Mosel AJ, Oakley GG and Peng A:
Deficient DNA damage signaling leads to chemoresistance to
cisplatin in oral cancer. Mol Cancer Ther. 11:2401–2409. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu ZW, Zhong LP, Ji T, Zhang P, Chen WT
and Zhang CP: MicroRNAs contribute to the chemoresistance of
cisplatin in tongue squamous cell carcinoma lines. Oral Oncol.
46:317–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Z, Yu T, Zhou B, Wei J, Fang Y, Lu J,
Guo L, Chen W, Liu ZP and Luo J: Mg(II)-Catechin nanoparticles
delivering siRNA targeting EIF5A2 inhibit bladder cancer cell
growth in vitro and in vivo. Biomaterials. 81:125–134. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tian SB, Yu JC, Liu YQ, Kang WM, Ma ZQ, Ye
X and Yan C: MiR-30b suppresses tumor migration and invasion by
targeting EIF5A2 in gastric cancer. World J Gastroenterol.
21:9337–9347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng QB, Kang WM, Yu JC, Liu YQ, Ma ZQ,
Zhou L, Cui QC and Zhou WX: Overexpression of eukaryotic
translation initiation factor 5A2 (EIF5A2) correlates with cell
aggressiveness and poor survival in gastric cancer. PLoS One.
10:e01192292015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Du F, Chen W, Yao M, Lv K and Fu P:
EIF5A2 is a novel chemoresistance gene in breast cancer. Breast
Cancer. 22:602–607. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee WY, Shin DY, Kim HJ, Ko YH, Kim S and
Jeong HS: Prognostic significance of epithelial-mesenchymal
transition of extracapsular spread tumors in lymph node metastases
of head and neck cancer. Ann Surg Oncol. 21:1904–1911. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harada K, Ferdous T and Ueyama Y:
Establishment of 5-fluorouracil-resistant oral squamous cell
carcinoma cell lines with epithelial to mesenchymal transition
changes. Int J Oncol. 44:1302–1308. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aggarwal BB, Sethi G, Ahn KS, Sandur SK,
Pandey MK, Kunnumakkara AB, Sung B and Ichikawa H: Targeting
signal-transducer-and-activator-of-transcription-3 for prevention
and therapy of cancer: Modern target but ancient solution. Ann NY
Acad Sci. 1091:151–169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gkouveris I, Nikitakis N, Karanikou M,
Rassidakis G and Sklavounou A: Erk1/2 activation and modulation of
STAT3 signaling in oral cancer. Oncol Rep. 32:2175–2182. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Buettner R, Mora LB and Jove R: Activated
STAT signaling in human tumors provides novel molecular targets for
therapeutic intervention. Clin Cancer Res. 8:945–954.
2002.PubMed/NCBI
|
|
40
|
Zhu W, Cai MY, Tong ZT, Dong SS, Mai SJ,
Liao YJ, Bian XW, Lin MC, Kung HF, Zeng YX, et al: Overexpression
of EIF5A2 promotes colorectal carcinoma cell aggressiveness by
upregulating MTA1 through C-myc to induce
epithelial-mesenchymaltransition. Gut. 61:562–575. 2012. View Article : Google Scholar : PubMed/NCBI
|