Introduction
Pancreatic cancer is a highly malignant disease with
a 5-year survival rate of less than 4% (1). Gemcitabine
(2′-deoxy-2′,2′-difluorocytidine monohydrochloride) is the standard
first-line drug used to treat patients with advanced pancreatic
cancer (2). Its active metabolites,
diphosphorylated and triphosphorylated nucleosides (dFdCDP and
dFdCTP), inhibit both DNA polymerase and ribonucleotide reductase
(RR), leading to impaired DNA synthesis and repair, and then cause
DNA damage and apoptosis (3).
However, its efficacy remains low with a median survival rate of
5.7 months and a 1-year survival rate of 18% (4,5). This
has been attributed, in part, to the presence of a highly effective
DNA damage response in pancreatic cancer.
Checkpoint kinase 1 (CHK1) acts as a master
regulator of DNA damage signaling to regulate cell cycle
progression, DNA repair, and DNA replication (6). CHK1 is activated by diverse stimuli
including DNA-damaging agents via both ATM and Rad3-related (ATR)
and ataxia telangiectasia-mutated (ATM). Activated CHK1 destablizes
CDC25s (e.g., CDC25C) to prevent the activation of CDKs and cause
cell cycle arrest (7). Inhibition
of CHK1 abrogates DNA damage-induced cell cycle arrest allowing
cells to enter mitosis despite the presence of DNA damage, which
can lead to cell death, especially in p53-defective cancer cells.
p53 gene is inactivated in 50 to 75% of pancreatic cancers
(8). Thus inhibition of CHK1 is a
promising cancer therapeutic strategy for increasing the
chemosensitization in pancreatic cancer.
Numerous inhibitors of Chk1 are in pre-clinical and
clinical development with the focus predominantly on their ability
to potentiate the cytotoxicity of chemotherapy drugs. However,
owing to the multiple roles of CHK1 in the DNA damage response
(DDR) pathway, molecular mechanism of chemosensitization by CHK1
inhibitors is not definitive. Both the abrogation of S or G2/M
checkpoint and inhibition of homologous recombination repair (HRR)
have been reported to contribute to chemosensitization by CHK1
inhibitors (9). Noteworthy, a
recent study demonstrated that the ATR-Chk1 pathway promoted RRM2
accumulation by CDK2, limiting DNA replication stress and
generation of single-stranded DNA (ssDNA) (10). Ribonucleotide reductase is composed
of the homodimeric RRM1 and RRM2 subunits that catalyze the
conversion of ribonucleotides to deoxyribonucleotides (dNTPs),
which are used in the synthesis of DNA during replication and
repair (11). We propose that
inhibition of Chk1 may enhance sensitization of DNA-damaging agents
via suppressing the RR level, exhausting dNTP and enhancing DNA
damage.
To explore the contribution of ribonucleotide
reductase and DNA damage on chemosensitization by CHK1 inhibitors,
we selected a potent inhibitor of CHK1, LY2603618 which has been
demonstrated activity both as a monotherapy and in combination with
a range of cytotoxic chemotherapeutic agents (12,13).
We observed the molecular mechanism of cytotoxic effects of
LY2603618 alone and in combination with gemcitabine.
Materials and methods
Drugs
LY2603618 and roscovitine were purchased from
Selleck Chemicals (Houston, TX, USA). Gemcitabine was purchased
from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
ASPC-1, CFPAC-1, HPAC, BxPC-3 and MiaPaCa-2 cell
lines were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA). The cell lines were cultured in RPMI
1640 medium (Invitrogen, for ASPC-1 and BxPC-3), Dulbecco's
modified Eagle's medium (DMEM, Invitrogen, for HPAC and MiaPaCa-2),
or Iscove's Modified Dulbecco's medium (IMDM, Invitrogen, for
CFPAC-1) containing 10% fetal bovine serum, 100 u/ml pencillin, and
100 µg/ml streptomycin in a 37°C humidified atmosphere containing
5% CO2/95% air. All cell lines were authenticated by the
University of Arizona Genetics Core Facility (Tucson, AZ, USA).
Cell viability assay
In vitro cytotoxicities of LY2603618, gemcitabine
and roscovitine, alone or in combination, against the pancreatic
cancer cell lines were measured using MTT.
[3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium-bromide,
Sigma-Aldrich] assays, as previously described (14,15).
IC50 values were calculated as the drug concentrations necessary to
inhibit 50% proliferation as compared to untreated control cells.
The extent and direction of LY2603618 and gemcitabine or
roscovitine cytotoxic interactions were determined by standard
isobologram analyses, as previously described (15–17).
Cell cycle analysis
Cell cycle distribution was determined by using
propidium iodide (PI) staining and flow cytometry analysis with a
FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA), as
previously described (17). Cell
cycle analysis was performed using Multicycle software (Phoenix
Flow Systems, Inc., San Diego, CA, USA).
Western blot analysis
Western blotting was performed using polyvinylidene
difluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL, USA)
and iimmunoblotted with mouse anti-Chk1 (sc8408, 1:500; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), and -β-actin antibodies
(A2228/A5441, mouse, 1:2,500; Sigma-Aldrich), or rabbit anti-PARP
(9542, 1:1,000), -pCDK1(Y15) (9111, 1:2,000), -CDK2 (2546,
1:2,000), -γH2AX (2577, 1:1,000; Cell Signaling Technology,
Danvers, MA, USA), -RRM1 (ab137114, 1:10,000), -RRM2 (ab172476,
1:2,000), -pCHK1 (S345) (ab47318, 1:500), -pCDC25C (S216) (ab32051,
1:1,000), -pCDK2 (Y15) (ab76146, 1:2,000), -CDK1 (ab32094,
1:1,000), and -cleaved-caspase-3 antibodies (ab2302, 1:1,000;
Abcam, Cambridge, MA, USA), as previously described (18). Immunoreactive proteins were
visualized using the Odyssey Infrared Imaging System (Li-Cor), as
described by the manufacturer.
Alkaline comet assay
Pancreatic cancer cells were treated with LY2603618
and gemcitabine, alone or in combination for 8 h and then subjected
to alkaline comet assay, as previously described (19). Slides were stained with SYBR Gold
(Life Technologies), and then visualized using Olympus IX-70
fluorescence microscope (Olympus, Tokyo, Japan). At least 100
comets per gel were scored using CometScore (TriTekCorp, Sumerduck,
VA). The comets were analyzed based on the percentage (%) of DNA in
the tail as the measure of primary DNA damage.
Statistical analysis
Data are expressed as the mean ± standard deviation
of three experiments. Differences in the sample means between test
groups and control groups were analyzed using the pair-wise
two-sample t-test. Statistical analyses were performed with
GraphPad Prism 5.0 (GraphPad Software, Inc.). A P-value of <0.05
was considered as significant and labeled as *P<0.05;
**P<0.01; ***P<0.001.
Results
CHK1 inhibition induces growth
inhibition and cell death in pancreatic cancer cells
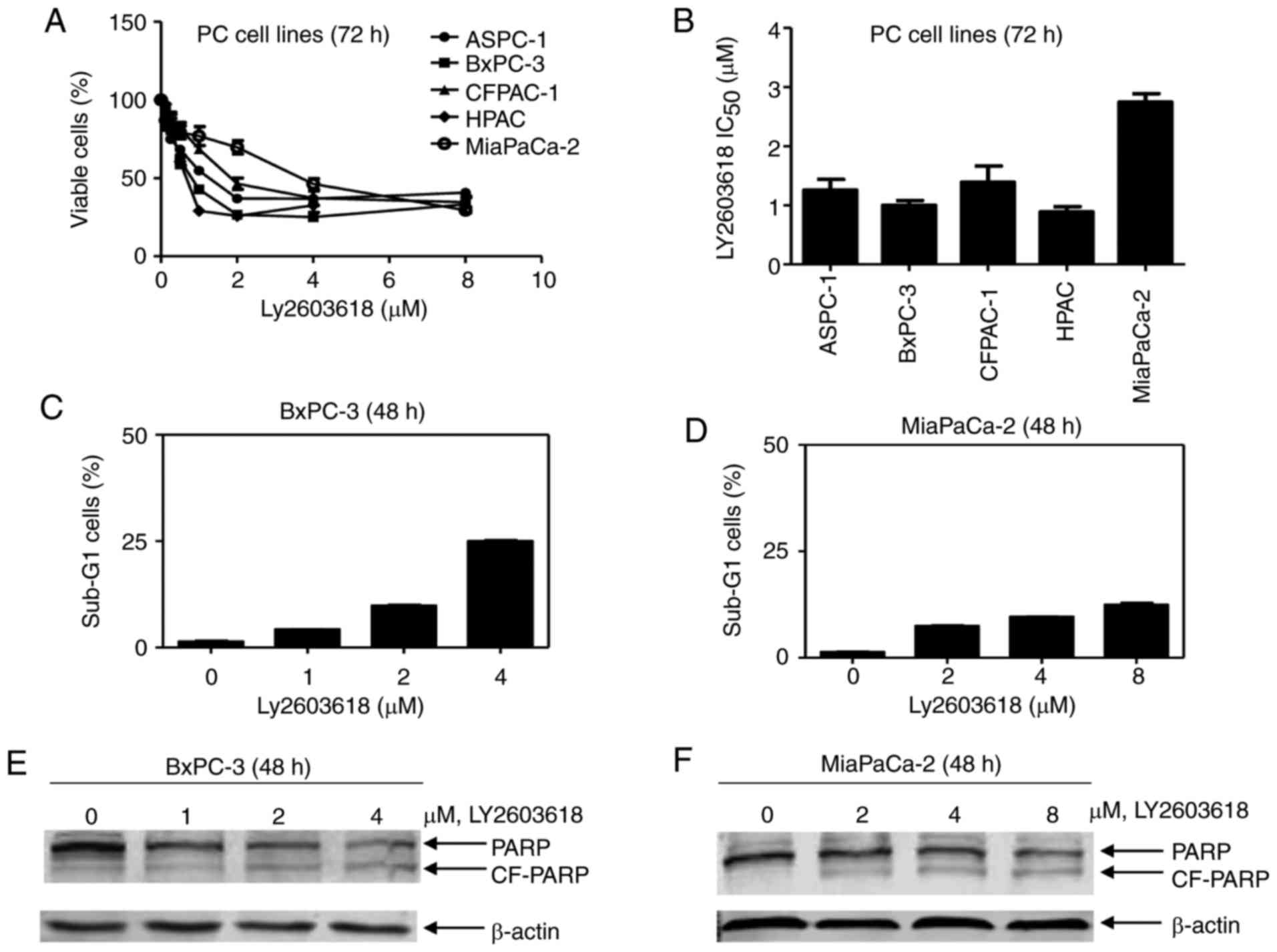
To evaluate the anti-tumor efficacy of LY2603618 in
human pancreatic cancer cells, we selected 5 pancreatic cancer cell
lines with different p53 phenotype, BxPC-3 (p53 mutation),
MiaPaCa-2 (p53 mutation), HPAC (p53 wild-type), CFPAC (p53
mutation) and ASPC-1 (p53 null). The results showed that LY2603618
inhibited cell proliferation in all studied pancreatic cancer cell
lines in a dose-dependent manner after 72 h of treatment (Fig. 1A). The IC50 values of LY2603618
modestly varied from 0.89 µM for HPAC cells to 2.75 µM for
MiaPaCa-2 cells (Fig. 1B), which
are less than the maximum clinically achievable concentration of
LY2603618 (9 µM) (13).
To explore whether LY2603618 causes pancreatic
cancer cell death, we treated BxPC-3 (sensitive to LY2603618 with
IC50 of 1.00 µM) and MiaPaCa-2 cells (low sensitive to LY2603618
with IC50 of 2.75 µM) with varying concentrations of LY2603618 for
48 h. No more than 25% cells with DNA fragments (Sub-G1) were
observed after LY2603618 treatment by PI staining followed by flow
cytometry (Fig. 1C and D),
accompanied by an increased PARP cleavage (Fig. 1E and F). It indicates that LY2603618
causes a small amount of pancreatic cancer cell death.
Inhibition of CHK1 causes
CDK-dependent RRM1/2 downregulation and DNA damage in pancreatic
cancer cells
To confirm CHK1 inhibition by LY2603618, we analyzed
CHK1 signaling in LY2603618-treated pancreatic cancer cells by
western blot analysis. First, we determined the phosphorylated and
total protein levels of CHK1 after 48 h of treatment with LY2603618
in clinically achievable concentrations. LY2603618 decreased the
total CHK1 level but increased the pCHK1S345 level in BxPC-3 or
MiaPaCa-2 cells (Fig. 2A). Since
Ser345 phosphoylation is predominantly catalyzed by ATR in response
to DNA damage (20), our results
suggest that LY2603618 treatment may cause DNA damage-mediated
phosphorylation of CHK1 at Ser345. Generally, CDC25C
phosphorylation by CHK1 may predit CHK1 activity. Next, we observed
CHK1 downstream signaling effectors, including pCDC25C, CDK1,
pCDK1, CDK2, and pCDK2. The results showed that LY2603618 treatment
reduced the phosphorylated protein level of CDC25C, CDK1, and CDK2
without altering the total protein levels of CDK1 and CDK2 in
BxPC-3 or MiaPaCa-2 cells (Fig.
2A), indicating that LY2603618 inhibites CHK1 activity and
activated CDK1/2.
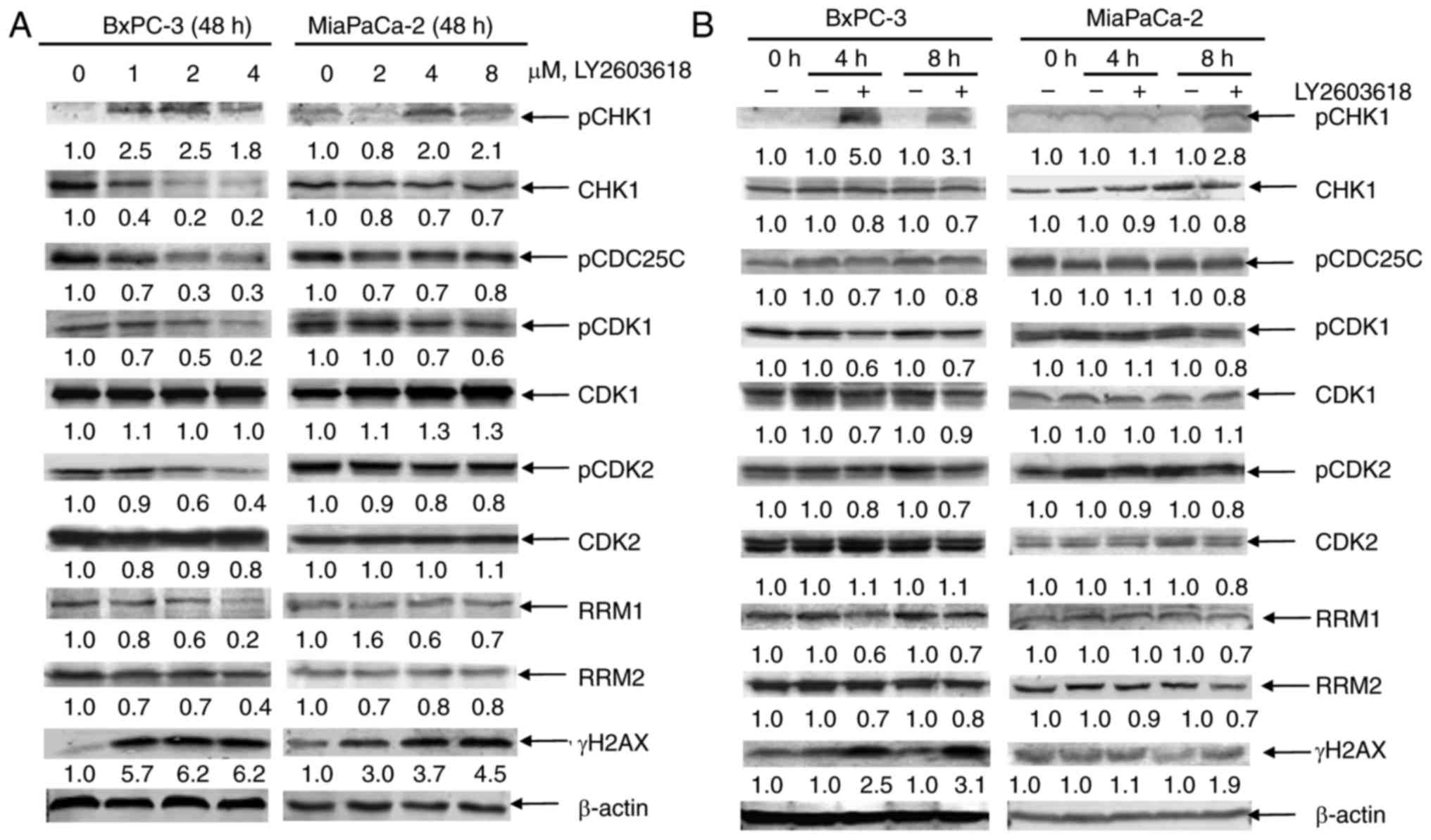 | Figure 2.LY2603618 causes CDK-dependent RRM1/2
downregulation and DNA damage enhancement in pancreatic cancer
cells. (A) BxPC-3 and MiaPaCa-2 cells were treated with varying
concentrations of LY2603618 for 48 h. Whole cell lysates were
subjected to western blotting and probed with anti-pCHK1, -CHK1,
-pCDC25C, -pCDK1, -CDK1, -pCDK2, -CDK2, -RRM1, -RRM2, -γH2AX or
-β-actin antibodies. (B) After 4 and 8 h of LY2603618 treatment of
BxPC-3 (1 µM) and MiaPaCa-2 (4 µM), cells were harvested and lysed.
Protein extracts were subjected to western blotting and probed with
anti-pCHK1, -CHK1, -pCDC25C, -pCDK1, -CDK1, -pCDK2, -CDK2, -RRM1,
-RRM2, -γH2AX or -β-actin antibodies. All experiments were
performed at least 3 independent times, and representative western
blots are shown. (C-F) BxPC-3 or MiaPaCa-2 cells were treated with
1 or 4 µM LY2603618 in the absence or presence of 20 µM roscovitine
for 48 h, respectively. RRM1, RRM2, γH2AX or -β-actin protein
levels in BxPC-3 (C) and MiaPaCa-2 (D) cells were shown by western
blot analysis. All experiments were performed at least 3
independent times, and representative western blots are shown. The
percentage of PI+ cells with sub-G1 DNA content in BxPC-3 (E) and
MiaPaCa-2 (F) cells was measured by PI staining and flow cytometry
analyses. ***P<0.001. BxPC-3 (G) and MiaPaCa-2 (H) cells were
treated with LY2603618 and roscovitine, alone or in combination,
for 72 h and then viable cells were determined using MTT reagent.
Standard isobologram was used to analyze the antagonistic cytotoxic
effect of LY2603618 and roscovitine. The IC50 values of each drug
are plotted on the axes; the solid line represents the additive
effect, while the points represent the concentrations of each drug
resulting in 50% inhibition of proliferation. Points falling below
the line indicate synergism whereas those above the line indicate
antagonism. |
Since inhibition of CHK1 may suppress RRM2
expression, leading to DNA replication stress and DNA damage, we
next observed effect of LY2603618 on RRM1 and RRM2 levels in BxPC-3
and MiaPaCa-2 cells. As shown in Fig.
2A, LY2603618 treatment decreased the protein levels of RRM1
and RRM2, accompanied by a dose-dependent increase of
phosphorylated H2AX (γH2AX, an established biomarker for DNA
double-strand breaks) in both cell lines. Time course experiments
demonstrated that decreases of CHK1, pCDC25C, pCDK1, pCDK2 and
RRM1/2 protein levels and increases of pCHK1 and γH2AX were
simultaneously detected as early as 4 h in BxPC-3 cells and as
early as 8 h in MiaPaCa-2 cells (Fig.
2B). This finding indicates that LY2603618-induced CHK1
inhibition, CDK activation, RRM1/2 downregulation and DNA damage
simultaneously occur at an earlier time in pancreatic cancer
cells.
To determine whether RRM1/2 downregulation and DNA
damage are dependent on CDK activation in response to LY2603618, we
selected a CDK1/2/5 inhibitor, roscovitine. Noteworthy, roscovitine
almost completely restored the levels of RRM1/2 and γH2AX in
LY2603618-treated cells (Fig. 2C and
D), suggesting that CHK1 inhibition causes CDK-dependent RRM1/2
downregulation and DNA damage in pancreatic cancer cells.
Furthermore, we observed the effect of roscovitine on
LY2603618-induced cytotoxicity in BxPC-3 and MiaPaCa-2 cells. As
expected, roscovitine significantly decreased the amount of Sub-G1
cells in the presence of LY2603618 (Fig. 2E and F). Moreover, when administered
simultaneously, roscovitine significantly attenuated LY2603618
sensitivities reflected by increased IC50 values in BxPC-3 and
MiaPaCa-2 cells (Fig. 2G and H).
The combined effects of LY2603618 with roscovitine on cell
proliferation were clearly antagonistic, reflected by all points
falling above the line using standard isobologram analysis. Taken
together, our data indicate that CHK1 inhibition by LY2603618
causes CDK-dependent RRM1/2 downregulation, DNA damage, and
cytotoxicity in pancreatic cancer cells.
CHK1 inhibition synergizes with
gemcitabine treatment to induce growth inhibition and apoptosis in
pancreatic cancer cells
Since interference with DNA damage checkpoints has
been demonstrated preclinically to be a highly effective means of
increasing the cytotoxicity of DNA-damaging drugs, we then
investigated the combination of LY2603618 with gemcitabine. When
simultaneously administered with LY2603618, gemcitabine
significantly enhanced LY2603618 sensitivity, reflected by the
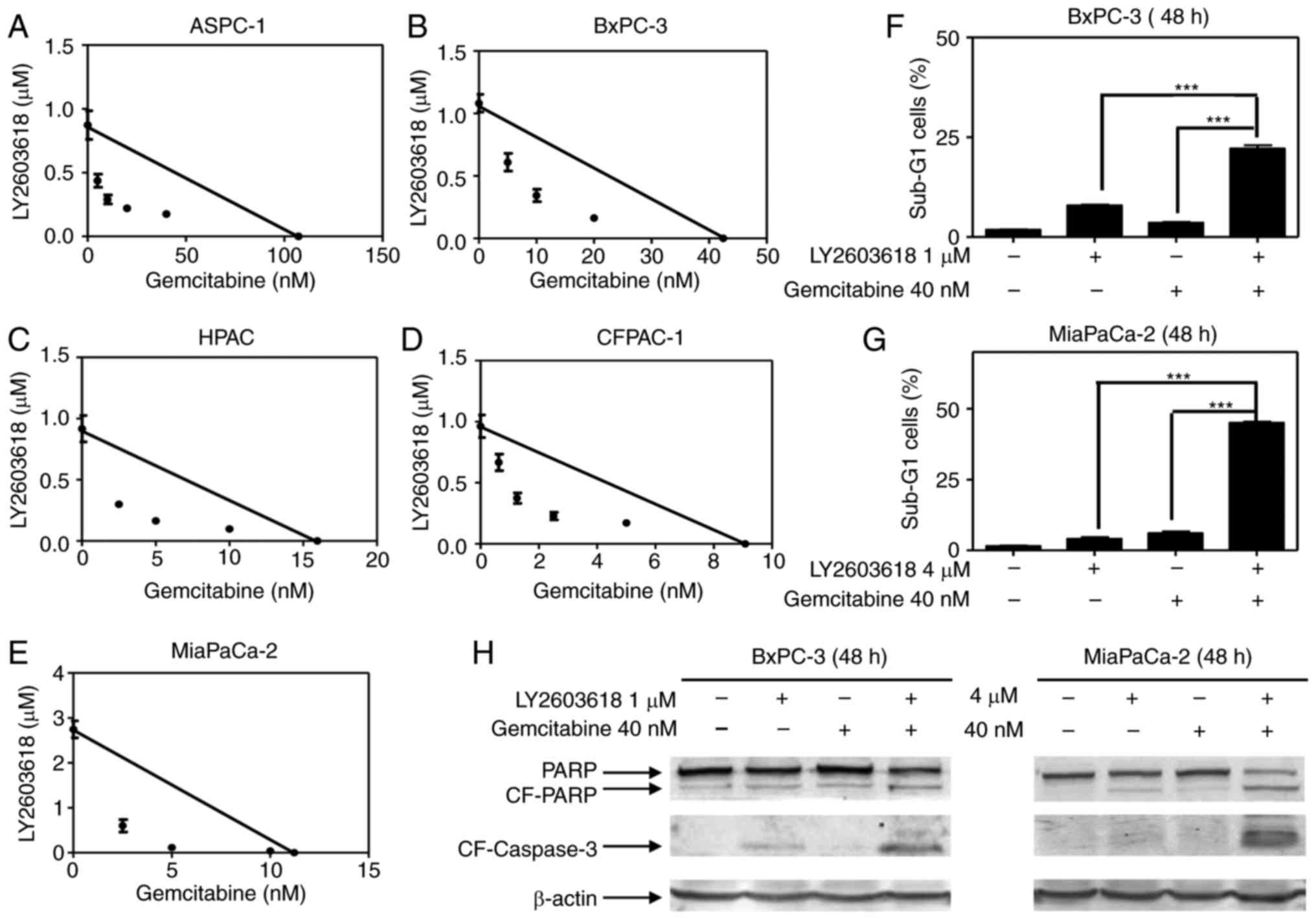
decreased IC50 values in 5 pancreatic cancer cell lines (Fig. 3A-E). The combined effects of
LY2603618 with gemcitabine on cell proliferation were clearly
synergistic, reflected by all the points falling below the line by
standard isobologram analysis (Fig.
3A-E).
To further address the synergism of LY2603618 and
gemcitabine, we treated the BxPC-3 or MiaPaCa-2 cells with both
drugs alone or in combination and looked at their effects on cell
apoptosis. The results showed that LY2603618 and gemcitabine
cooperatively induced apoptosis, as indicated by the high
percentage of cells with sub-G1 DNA content and the increased
cleavage of PARP and caspase-3 (Fig.
3F-H).
Gemcitabine sensitization by CHK1
inhibition is associated with CDK-dependent RRM1/2 downregulation
and DNA damage enhancement
To explore the molecular mechanism of gemcitabine
sensitization by LY2603618, cell cycle progression was analyzed by
flow cytometry. Gemcitabine treatment led to S and G2/M arrest,
which was abrogated to some extent by the addition of LY2603618 in
both pancreatic cancer cells (Fig. 4A
and B). It is in accordance with the western blotting results
that LY2603618 inhibited gemcitabine-induced pCDC25C, CDK1/2 and
pCDK1/2 protein levels (Fig. 4C).
Collectively, it suggests that inhibition of Chk1 abrogates
gemcitabine-activated S and G2/M checkpoints.
 | Figure 4.LY2603618 abrogates
gemcitabine-activated S and G2/M checkpoint and inhibits
gemcitabine-induced RRM1/2 level in pancreatic cancer cells. BxPC-3
(A) and MiaPaCa-2 (B) cells were treated with vehicle control,
gemcitabine (40 nM), LY2603618 (1 µM for BxPC-3 or 4 µM for
MiaPaCa-2) or gemcitabine plus LY2603618 for 48 h. Cell cycle
distribution was detected by PI staining and flow cytometry
analyses. (C and D) Cells were treated as shown in (A and B).
pCHK1, CHK1, pCDC25C, pCDK1, CDK1, pCDK2, CDK2or β-actin protein
levels in BxPC-3 and MiaPaCa-2 cells were detected by western blot
analysis (C). RRM1, RRM2, γH2AX or β-actin protein levels in BxPC-3
and MiaPaCa-2 cells were detected by western blot analysis (D).
Experiments were performed at least 3 independent times, and
representative western blots are shown. |
We next looked at DNA damage in the combined
treatment of pancreatic cancer cells. As expected, gemcitabine
treatment increased γH2AX level in both BxPC-3 and MiaPaCa-2 cell
lines, which was further increased by the addition of LY2603618,
indicating that DNA damage was enhanced by the combined treatment
(Fig. 4D). Meanwhile, gemcitabine
treatment caused modest increase of both RRM1 and RRM2, which were
further decreased by LY2603618 (Fig.
4D). Noteworthy, roscovitine treatment almost completely
restored the levels of RRM1/2 and γH2AX in the combined treatment
of pancreatic cancer cells (Fig.
5A). More importantly, after the combined treatment of
gemcitabine and LY2603618, the amount of Sub-G1 cells was
significantly decreased by roscovitine, accompanied with the
downregulation of cleaved PARP (Fig.
5A-C). Taken together, it suggests that gemcitabine
sensitization by CHK1 inhibition is associated with CDK-dependent
RRM1/2 downregulation and DNA damage enhancement.
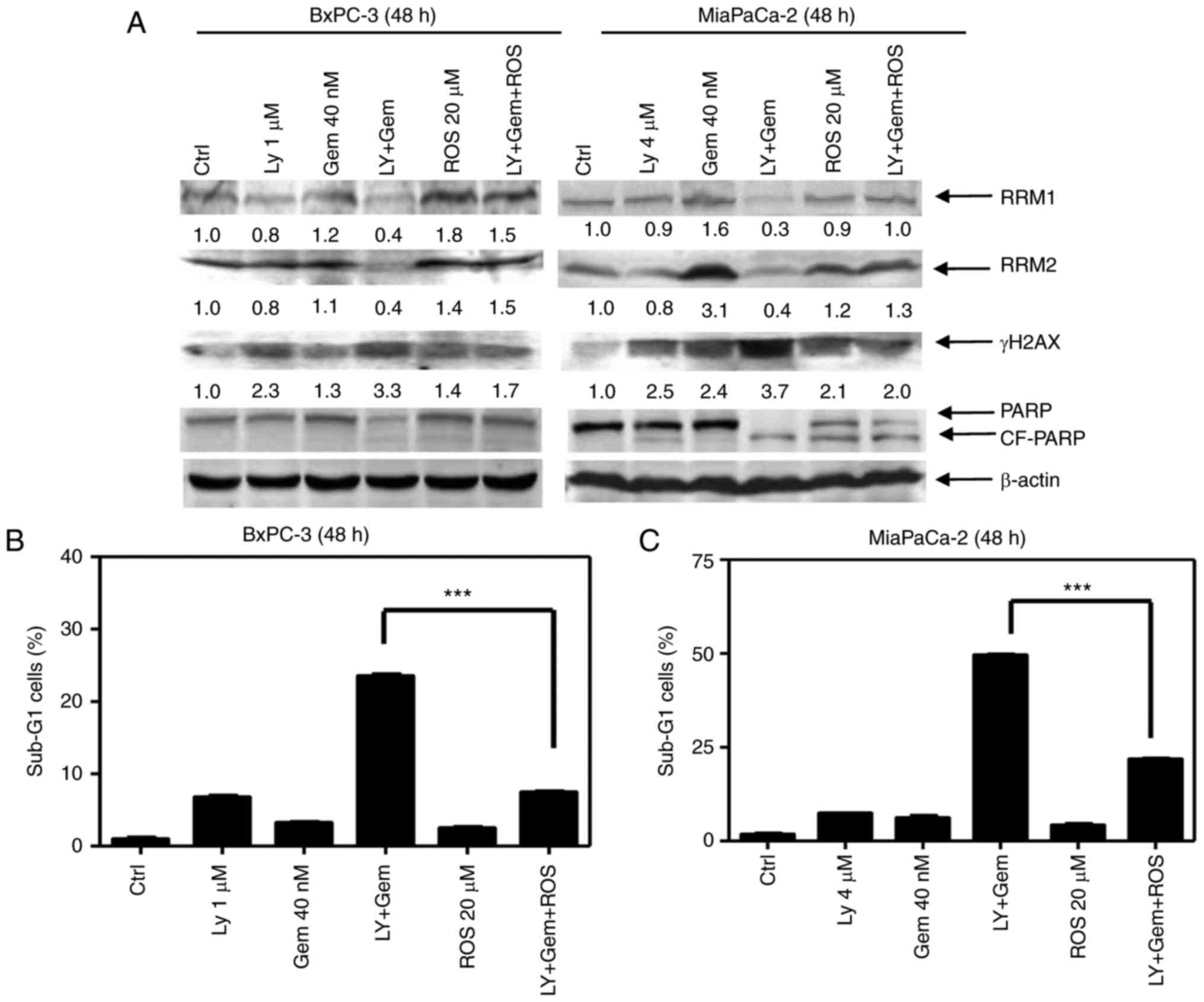 | Figure 5.Roscovitine treatment reverses the
cytotoxic effects of gemcitabine combined with LY2603618. (A-C)
BxPC-3 and MiaPaCa-2 cells were treated with vehicle control
(Ctrl), gemcitabine (Gem, 40 nM), LY2603618 (LY, 1 µM for BxPC-3 or
4 µM for MiaPaCa-2), gemcitabine plus LY2603618 (Gem+LY),
roscovitine (ROS, 20 µM), or gemcitabine plus LY2603618 plus
roscovitine (Gem+LY+ROS) for 48 h. RRM1, RRM2, γH2AX, PARP or
β-actin protein levels in BxPC-3 and MiaPaCa-2 cells were detected
by western blot analysis (A). Experiments were performed at least 3
independent times, and representative western blots are shown. The
percentage of Sub-G1 cells in BxPC-3 (B) and MiaPaCa-2 cells (C)
were calculated by PI staining and flow cytometry analyses. Data
are presented as the mean of triplicate experiments ± SEM.
***P<0.001. |
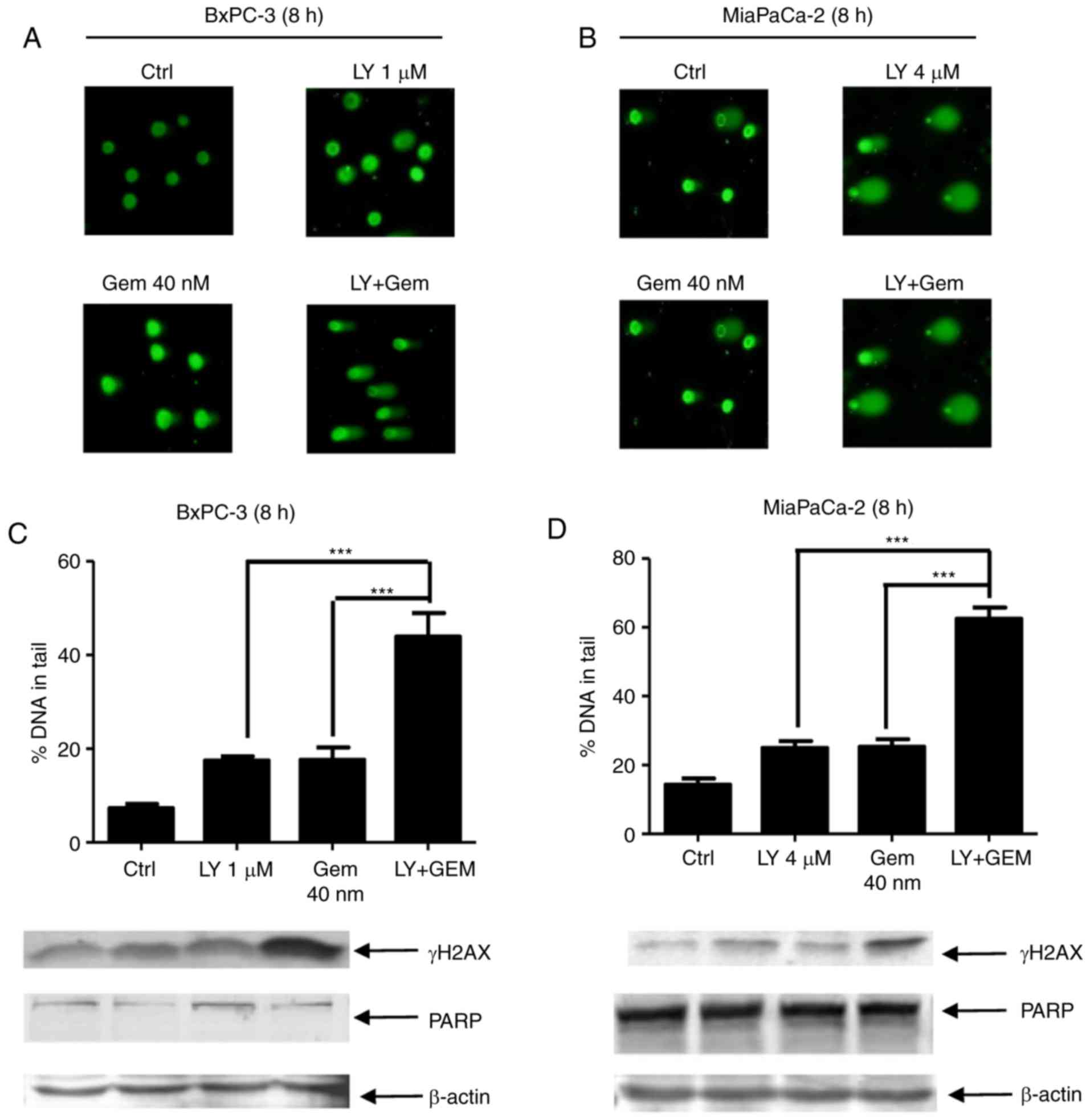
To determine whether DNA damage occurs prior to
induction of apoptosis in response to LY2603618 and gemcitabine,
the cells were treated for a shorter time, 8 h, and then DNA damage
was determined by the alkaline comet assay. As demonstrated in
Fig. 6A-D, LY2603618 significantly
increased the percentage of DNA in the tail and the γH2AX level in
gemcitabine-treated pancreatic cancer cells. However, cleaved PARP
was not detected after the combination treatment, providing
evidence that gemcitabine combined with LY2603618 caused increased
DNA damage, prior to induction of apoptosis.
Discussion
In the past several years, various specific small
molecule CHK1 inhibitors have been developed. Understanding the
mechanisms of action of these inhibitors may help to guide their
application in clinical settings. In the study, we demonstrate that
inhibition of CHK1 by LY2603618 potentiates anti-pancreatic cancer
activity of gemcitabine through promoting CDK-dependent
ribonucleotide reductase downregulation and DNA damage.
First, our results showed that LY2603618 treatment
of pancreatic cancer cells caused growth inhibition and a small
amount of cell death, which were alleviated by a CDK1/2/5
inhibitor, roscovitine. This indicates that the cytotoxic effect of
LY2603618 is dependent on CDK activation. Consistently, CDK1/2
activation was observed in LY2603618-treated pancreatic cancer
cells, as evidenced by the reduced pCDC25C, pCDK1, and pCDK2
levels. Further, we found that LY2603618 treatment reduced the
levels of RRM1 and RRM2, resulting in DNA damage, which was also
completely reversed by roscovitine. It suggests that activation of
CDK by CHK1 inhibition may reduce RRM1/2 accumulation, leading to
DNA damage, consistent with a previous report (10). Collectively, we infer that Chk1
inhibition causes cytotoxicity in pancreatic cancer cells by
promoting CDK-dependent RRM1/2 downregulation and DNA damage.
Of note, we found that LY2603618 significantly
increased the phosphorylated CHK1S345 level, consistent with a
previous study (20). It is
reported that phosphorylation of Chk1 at S345 is predominantly
catalyzed by ATR in response to DNA damage (21,22),
indicating that enhanced pCHK1S345 level may be attributed to DNA
damage caused by LY2603618. As expected, we observed the enhanced
γH2AX levels in dose-dependant manner after 48 h of LY2603618
treatment. Furthermore, a time course experiment showed that the
protein levels of γH2AX and pCHK1S345 were simultaneously increased
by LY2603618 treatment (4 h for BxPC-3 cells and 8 h for MiaPaCa-2
cells). Noteworthy, in contrast to the increased pCHK1S345 protein
level, the decreased total CHK1 levels were also detected in
LY2603618-treated cells. This may be explained by a previous report
demonstrating that ATR-mediated phosphorylation of CHK1 at S345
induced the polyubiquitination and degradation of CHK1 in human
cells (23). Taken together, these
data support our hypothesis that LY2603618 causes CDK-dependent DNA
damage, which further activates the ATR-CHK1 pathway, resulting in
CHK1 phosphorylation at S345 and subsequently CHK1 degradation in
pancreatic cancer cells.
As a key activator of the S- or G2/M-phase DNA
damage response, CHK1 may be involved in gemcitabine resistance in
cancer therapy. Thus interference with DNA damage checkpoints by
CHK1 inhibition has been demonstrated to be an effective means of
increasing the cytotoxicity of gemcitabine. As expected, we
observed synergistic anti-pancreatic cancer activities of LY2603618
and gemcitabine in 5 pancreatic cancer cell lines with different
p53 phenotype, consistent with several preclinical studies
(24–26), suggesting that p53 status does not
play a major role in sensitization of gemcitabine by LY2603618.
Moreover, the combination induced pronounced levels of apoptosis as
indicated by an increase in the fraction of sub-G1 cells or in the
levels of cleaved PARP and caspase-3. Mechanistic investigations
showed that CHK1 inhibition by LY2603618 partially abrogated S and
G2/M phase checkpoints and significantly enhanced DNA damage in
gemcitabine-treated pancreatic cancer cells, which is consistent
with an in vivo study (26). This
suggests that the abrogation of S or G2/M checkpoint contributes to
sensitization of gemcitabine by LY2603618. In addition, it is
reported that one of molecular mechanisms of gemcitabine resistance
includes upregulation of gemcitabine targets, RRM1 and RRM2, which
play an essential role in the maintenance of the dNTPs level
(27,28). RRM1 has been identified as the major
determinant of gemcitabine efficacy in patients treated with
gemcitabine. Interestingly, our results showed that RRM1 and RRM2
protein levels were much lower after the combined treatment than
after LY2603618 or gemcitabine treatment alone. It suggests that
downregulation of RRM1/2 by LY2603618 potentiates the effect of
gemcitabine by decreasing the competition between gemcitabine and
deoxycytidine, and then increasing DNA damage. To test whether CDK
activation contributes to RRM1/2 downregulation and DNA damage by
the combined treatment, we added CDK inhibitor, roscovitine.
Surprisingly, RRM1/2 and γH2AX levels were almost completely
restored after 48 h of roscovitine treatment. Furthermore,
roscovitine significantly decreased the amount of Sub-G1 cells in
combined treatment of pancreatic cancer cells. These data reveal
that activation of CDK at least partly contributes to synergistic
cytotoxicity of gemcitabine and LY2603618 by decreasing RRM1/2
protein level and enhancing DNA damage.
In conclusion, CHK1 inhibition by LY2603618
treatment caused a CDK-dependent cell death via downregulating
RRM1/2 levels and enhancing DNA damage. Combined use of gemcitabine
and LY2603618 synergistically reduced pancreatic cancer cell
viability relative to either single treatment, which was also
associated with CDK-dependent RRM1/2 downregulation and DNA damage
enhancement. These findings suggest that CDK activation plays an
important role in cytotoxicities of CHK1 inhibitor alone or in
combination with gemcitabine in pancreatic cancer cells.
Acknowledgements
This project was supported by the Science and
Technology Development Program of Jilin Province (no.
20150101185JC).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang P, Chubb S, Hertel LW, Grindey GB
and Plunkett W: Action of 2′,2′-difluorodeoxycytidine on DNA
synthesis. Cancer Res. 51:6110–6117. 1991.PubMed/NCBI
|
|
4
|
Kullmann F, Hollerbach S, Dollinger MM,
Harder J, Fuchs M, Messmann H, Trojan J, Gäbele E, Hinke A,
Hollerbach C and Endlicher E: Cetuximab plus
gemcitabine/oxaliplatin (GEMOXCET) in first-line metastatic
pancreatic cancer: A multicentre phase II study. Br J Cancer.
100:1032–1036. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li J and Saif MW: Advancements in the
management of pancreatic cancer. JOP. 10:109–117. 2009.PubMed/NCBI
|
|
6
|
Dai Y and Grant S: New insights into
checkpoint kinase 1 in the DNA damage response signaling network.
Clin Cancer Res. 16:376–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Löffler H, Rebacz B, Ho AD, Lukas J,
Bartek J and Kramer A: Chk1-dependent regulation of Cdc25B
functions to coordinate mitotic events. Cell Cycle. 5:2543–2547.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scarpa A, Capelli P, Mukai K, Zamboni G,
Oda T, Iacono C and Hirohashi S: Pancreatic adenocarcinomas
frequently show p53 gene mutations. Am J Pathol. 142:1534–1543.
1993.PubMed/NCBI
|
|
9
|
Morgan MA, Parsels LA, Zhao L, Parsels JD,
Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D,
Simeone DM, et al: Mechanism of radiosensitization by the Chk1/2
inhibitor AZD7762 involves abrogation of the G2 checkpoint and
inhibition of homologous recombinational DNA repair. Cancer Res.
70:4972–4981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buisson R, Boisvert JL, Benes CH and Zou
L: Distinct but concerted roles of ATR, DNA-PK, and Chk1 in
countering replication stress durings phase. Mol Cell.
59:1011–1024. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nordlund P and Reichard P: Ribonucleotide
reductases. Annu Rev Biochem. 75:681–706. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
King C, Diaz H, Barnard D, Barda D,
Clawson D, Blosser W, Cox K, Guo S and Marshall M: Characterization
and preclinical development of LY2603618: A selective and potent
Chk1 inhibitor. Invest New Drugs. 32:213–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calvo E, Chen VJ, Marshall M, Ohnmacht U,
Hynes SM, Kumm E, Diaz HB, Barnard D, Merzoug FF, Huber L, et al:
Preclinical analyses and phase I evaluation of LY2603618
administered in combination with pemetrexed and cisplatin in
patients with advanced cancer. Invest New Drugs. 32:955–968. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Taub JW, Huang X, Matherly LH, Stout ML,
Buck SA, Massey GV, Becton DL, Chang MN, Weinstein HJ and
Ravindranath Y: Expression of chromosome 21-localized genes in
acute myeloid leukemia: Differences between Down syndrome and
non-Down syndrome blast cells and relationship to in vitro
sensitivity to cytosine arabinoside and daunorubicin. Blood.
94:1393–1400. 1999.PubMed/NCBI
|
|
15
|
Xie C, Edwards H, Xu X, Zhou H, Buck SA,
Stout ML, Yu Q, Rubnitz JE, Matherly LH, Taub JW and Ge Y:
Mechanisms of synergistic antileukemic interactions between
valproic acid and cytarabine in pediatric acute myeloid leukemia.
Clin Cancer Res. 16:5499–5510. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang G, He J, Zhao J, Yun W, Xie C, Taub
JW, Azmi A, Mohammad RM, Dong Y, Kong W, et al: Class I and class
II histone deacetylases are potential therapeutic targets for
treating pancreatic cancer. PLoS One. 7:e520952012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Edwards H, Xie C, La Fiura KM, Dombkowski
AA, Buck SA, Boerner JL, Taub JW, Matherly LH and Ge Y: RUNX1
regulates phosphoinositide 3-kinase/AKT pathway: Role in
chemotherapy sensitivity in acute megakaryocytic leukemia. Blood.
114:2744–2752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie C, Drenberg C, Edwards H, Caldwell JT,
Chen W, Inaba H, Xu X, Buck SA, Taub JW, Baker SD and Ge Y:
Panobinostat enhances cytarabine and daunorubicin sensitivities in
AML cells through suppressing the expression of BRCA1, CHK1, and
Rad51. PLoS One. 8:e791062013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang FZ, Fei HR, Cui YJ, Sun YK, Li ZM,
Wang XY, Yang XY, Zhang JG and Sun BL: The checkpoint 1 kinase
inhibitor LY2603618 induces cell cycle arrest, DNA damage response
and autophagy in cancer cells. Apoptosis. 19:1389–1398. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kasahara K, Goto H, Enomoto M, Tomono Y,
Kiyono T and Inagaki M: 14-3-3gamma mediates Cdc25A proteolysis to
block premature mitotic entry after DNA damage. EMBO J.
29:2802–2812. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Clarke CA and Clarke PR: DNA-dependent
phosphorylation of Chk1 and Claspin in a human cell-free system.
Biochem J. 388:705–712. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang YW, Otterness DM, Chiang GG, Xie W,
Liu YC, Mercurio F and Abraham RT: Genotoxic stress targets human
Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell.
19:607–618. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parsels LA, Morgan MA, Tanska DM, Parsels
JD, Palmer BD, Booth RJ, Denny WA, Canman CE, Kraker AJ, Lawrence
TS and Maybaum J: Gemcitabine sensitization by checkpoint kinase 1
inhibition correlates with inhibition of a Rad51 DNA damage
response in pancreatic cancer cells. Mol Cancer Ther. 8:45–54.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matthews DJ, Yakes FM, Chen J, Tadano M,
Bornheim L, Clary DO, Tai A, Wagner JM, Miller N, Kim YD, et al:
Pharmacological abrogation of S-phase checkpoint enhances the
anti-tumor activity of gemcitabine in vivo. Cell Cycle. 6:104–110.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barnard D, Diaz HB, Burke T, Donoho G,
Beckmann R, Jones B, Barda D, King C and Marshall M: LY2603618, a
selective CHK1 inhibitor, enhances the anti-tumor effect of
gemcitabine in xenograft tumor models. Invest New Drugs. 34:49–60.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Davidson JD, Ma L, Flagella M, Geeganage
S, Gelbert LM and Slapak CA: An increase in the expression of
ribonucleotide reductase large subunit 1 is associated with
gemcitabine resistance in non-small cell lung cancer cell lines.
Cancer Res. 64:3761–3766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bergman AM, Eijk PP, Ruiz van Haperen VW,
Smid K, Veerman G, Hubeek I, Van den Ijssel P, Ylstra B and Peters
GJ: In vivo induction of resistance to gemcitabine results in
increased expression of ribonucleotide reductase subunit M1 as the
major determinant. Cancer Res. 65:9510–9516. 2005. View Article : Google Scholar : PubMed/NCBI
|