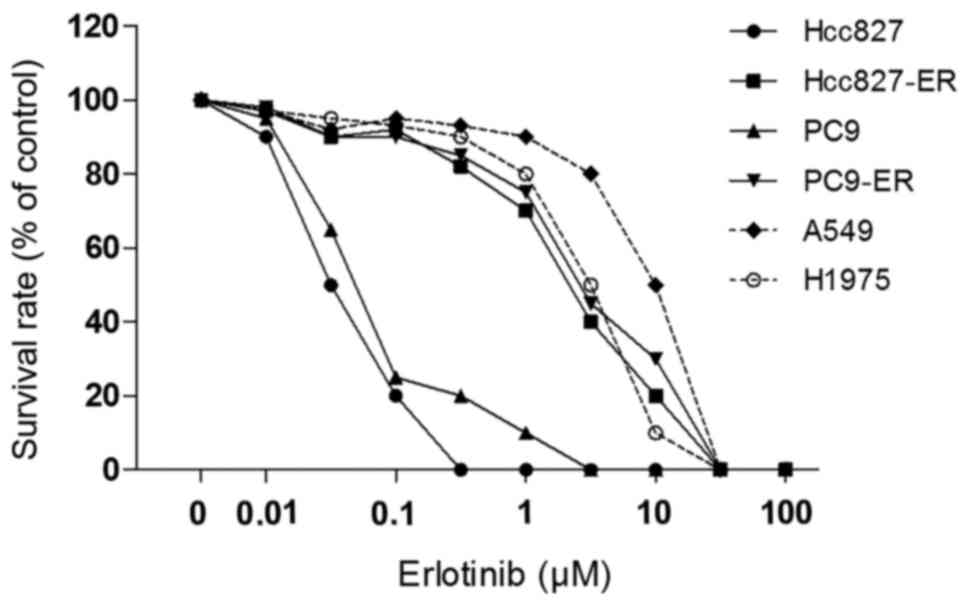
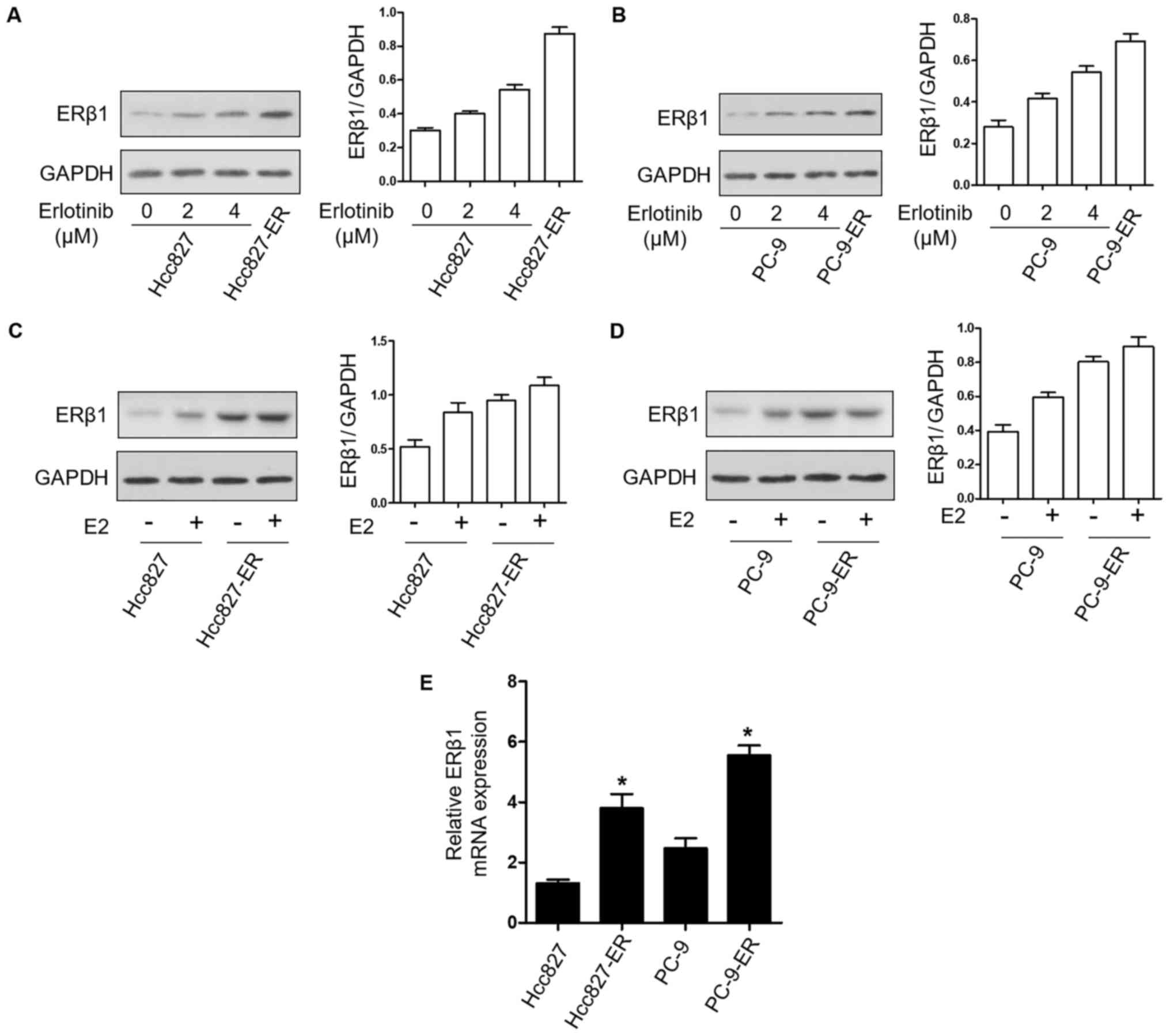
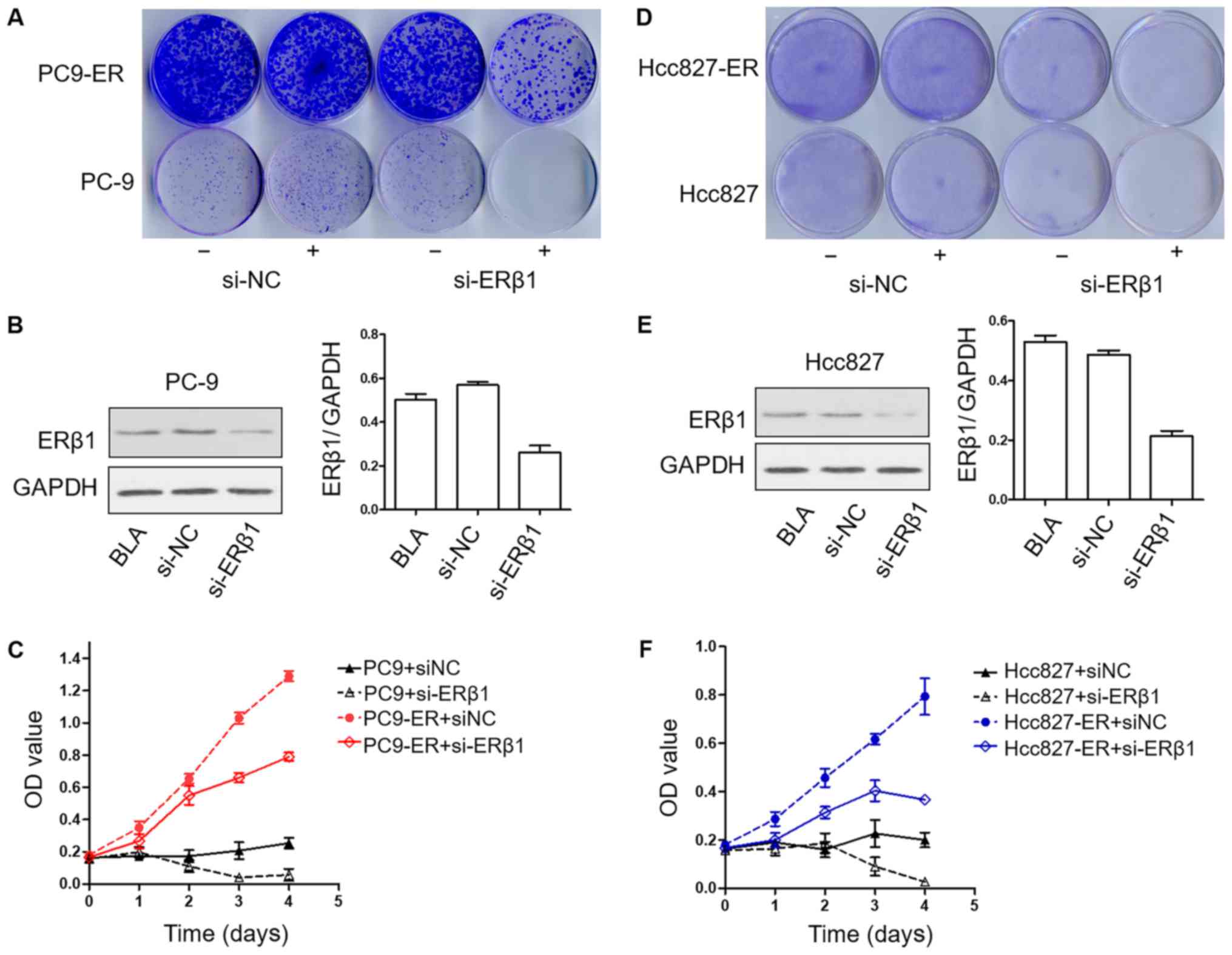
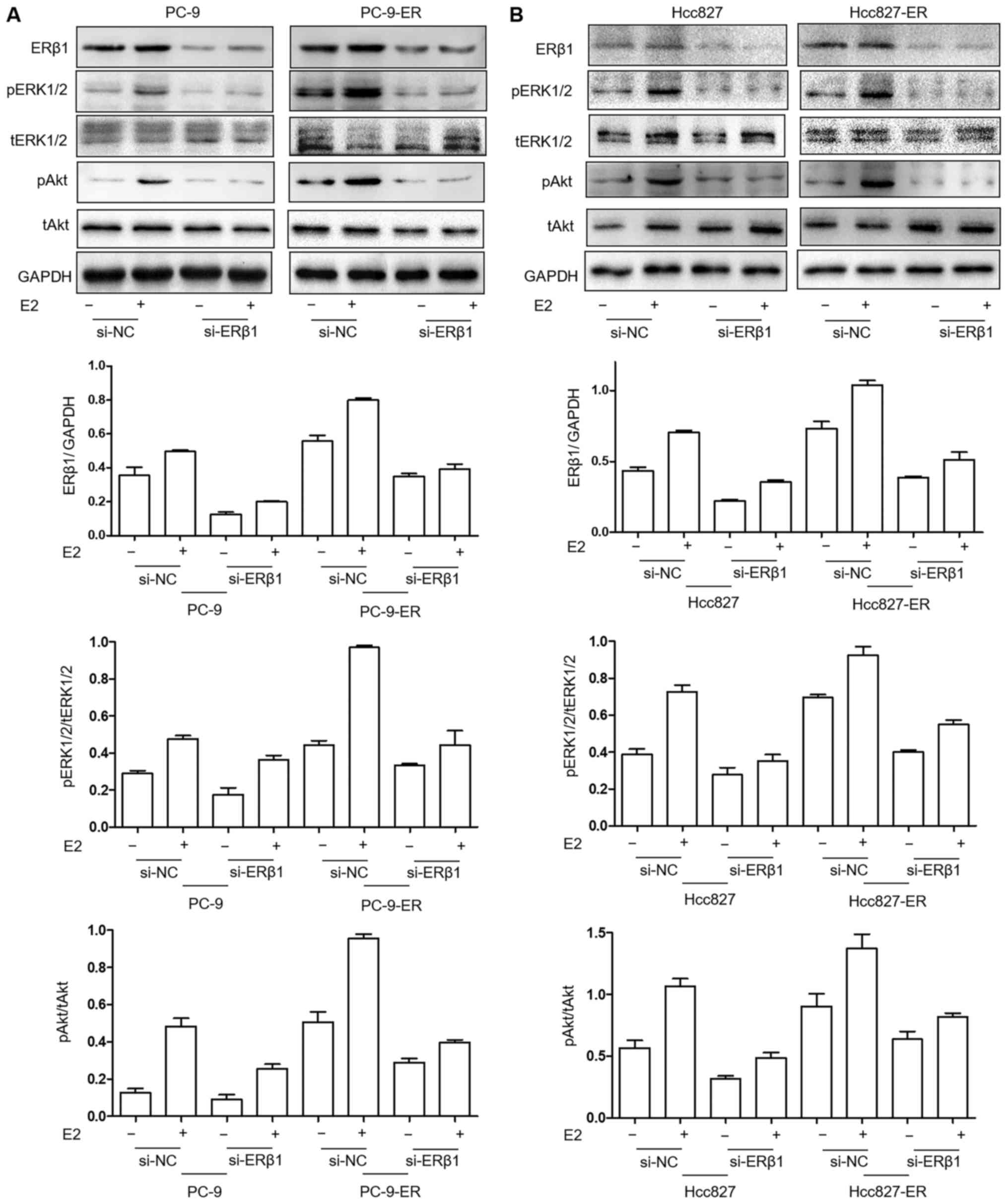
|
1
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al North-East Japan Study Group, : Gefitinib or chemotherapy for
non-small-cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosell R, Carcereny E, Gervais R,
Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R,
Pallares C, Sanchez JM, et al Spanish Lung Cancer Group in
collaboration with Groupe Français de Pneumo-Cancérologie and
Associazione Italiana Oncologia Toracica, : Erlotinib versus
standard chemotherapy as first-line treatment for European patients
with advanced EGFR mutation-positive non-small-cell lung cancer
(EURTAC): A multicentre, open-label, randomised phase 3 trial.
Lancet Oncol. 13:239–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin JJ, Cardarella S, Lydon CA, Dahlberg
SE, Jackman DM, Jänne PA and Johnson BE: Five-year survival in
EGFR-mutant metastatic lung adenocarcinoma treated with EGFR-TKIs.
J Thorac Oncol. 11:556–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bouchardy C, Benhamou S, Schaffar R,
Verkooijen HM, Fioretta G, Schubert H, Vinh-Hung V, Soria JC,
Vlastos G and Rapiti E: Lung cancer mortality risk among breast
cancer patients treated with anti-estrogens. Cancer. 117:1288–1295.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chlebowski RT, Schwartz AG, Wakelee H,
Anderson GL, Stefanick ML, Manson JE, Rodabough RJ, Chien JW,
Wactawski-Wende J, Gass M, et al: Women's Health Initiative
Investigators: Oestrogen plus progestin and lung cancer in
postmenopausal women (Women's Health Initiative trial): A post-hoc
analysis of a randomised controlled trial. Lancet. 374:1243–1251.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tang H, Liao Y, Xu L, Zhang C, Liu Z, Deng
Y, Jiang Z, Fu S, Chen Z and Zhou S: Estrogen and insulin-like
growth factor 1 synergistically promote the development of lung
adenocarcinoma in mice. Int J Cancer. 133:2473–2482. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Słowikowski BK, Lianeri M and Jagodziński
PP: Exploring estrogenic activity in lung cancer. Mol Biol Rep.
44:35–50. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kohno M, Okamoto T, Suda K, Shimokawa M,
Kitahara H, Shimamatsu S, Konishi H, Yoshida T, Takenoyama M, Yano
T, et al: Prognostic and therapeutic implications of aromatase
expression in lung adenocarcinomas with EGFR mutations. Clin Cancer
Res. 20:3613–3622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen L, Li Z, Shen S, Niu X, Yu Y, Li Z,
Liao M, Chen Z and Lu S: The synergistic effect of EGFR tyrosine
kinase inhibitor gefitinib in combination with aromatase inhibitor
anastrozole in non-small cell lung cancer cell lines. Lung Cancer.
78:193–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garon EB, Pietras RJ, Finn RS, Kamranpour
N, Pitts S, Márquez-Garbán DC, Desai AJ, Dering J, Hosmer W, von
Euw EM, et al: Antiestrogen fulvestrant enhances the
antiproliferative effects of epidermal growth factor receptor
inhibitors in human non-small-cell lung cancer. J Thorac Oncol.
8:270–278. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Siegfried JM and Stabile LP: Estrongenic
steroid hormones in lung cancer. Semin Oncol. 41:5–16. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang H, Liao Y, Chen G, Xu L, Zhang C, Ju
S and Zhou S: Estrogen upregulates the IGF-1 signaling pathway in
lung cancer through estrogen receptor-β. Med Oncol. 29:2640–2648.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu C, Liao Y, Fan S, Tang H, Jiang Z,
Zhou B, Xiong J, Zhou S, Zou M and Wang J: G protein-coupled
estrogen receptor (GPER) mediates NSCLC progression induced by
17β-estradiol (E2) and selective agonist G1. Med Oncol.
32:1042015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shien K, Toyooka S, Yamamoto H, Soh J,
Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stabile LP, Dacic S, Land SR, Lenzner DE,
Dhir R, Acquafondata M, Landreneau RJ, Grandis JR and Siegfried JM:
Combined analysis of estrogen receptor beta-1 and progesterone
receptor expression identifies lung cancer patients with poor
outcome. Clin Cancer Res. 17:154–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo Z, Wu R, Jiang Y, Qiu Z, Chen W and Li
W: Overexpression of estrogen receptor beta is a prognostic marker
in non-small cell lung cancer: A meta-analysis. Int J Clin Exp Med.
8:8686–8697. 2015.PubMed/NCBI
|
|
17
|
Mauro LV, Dalurzo M, Carlini MJ, Smith D,
Nuñez M, Simian M, Lastiri J, Vasallo B, Bal de Kier Joffé E,
Pallotta MG, et al: Estrogen receptor β and epidermal growth factor
receptor as early-stage prognostic biomarkers of non-small cell
lung cancer. Oncol Rep. 24:1331–1338. 2010.PubMed/NCBI
|