Introduction
Chronic pancreatitis (CP) is a serious disease that
is characterized by progressive inflammation of the pancreas and
fibrosis, which result in exocrine and endocrine dysfunction
(1). Pancreatic fibrosis is closely
associated with CP and pancreatic cancer (PC) and it induces severe
damage in the pancreas. CP and PC are characterized by a
desmoplastic reaction that involves activated pancreatic stellate
cells (PSCs) (1–3). This reaction promotes the growth and
invasion of tumor cells (3–5). The activation of PSCs has been
previously proposed as the key initiating step in pancreatic
fibrosis (6) and a major source of
extracellular matrix (ECM) deposition during pancreatic injury
(7). Activated PSCs are believed to
significantly contribute to the progression of pancreatic diseases
and may therefore, present beneficial therapeutic targets (3,8–11).
Furthermore, the activation of PSCs is associated with the
secretion of various inflammatory cytokines/chemokines, as well as
collagen (12,13). Understanding the mechanism
underlying the activation of PSCs and the effects of activated PSCs
on pancreatic diseases would help identify treatment targets for
pancreatic fibrosis associated with diseases such as CP/PC.
Transforming growth factor (TGF)-β, a potent
pro-fibrotic factor that plays a functional role in the
pathogenesis of pancreatic fibrosis (14), is responsible for the activation of
PSCs (15). Following stimulation
with TGF-β, PSCs exhibit enhanced expression of significant ECM
proteins, including collagen and fibronectin. Concurrently, TGFβ
inhibits the degradation of ECM by blocking the secretion of
proteases, such as matrix metalloproteinases (MMPs) and stimulating
the production of naturally occurring protease inhibitors, such as
tissue inhibitors of metalloproteinases (TIMPs) (16). To date, previous studies have
revealed that PSCs are the main source of ECM proteins in
pancreatic fibrosis (16) and that
the activation and collagen synthesis of PSCs are highly controlled
by TGF-β1 (17). TGF-β1 is a
subtype of the TGF-β family, which is multifunctional and increases
significantly in CP (in both human and animal models) (18).
Galectins are a growing family of
β-galactoside-binding animal lectins that have been implicated in a
variety of biological processes, including fibrosis, angiogenesis
and immune activation (19).
Galectin-1, a member of the galectin family, is strongly expressed
in fibroblasts, which have been recognized as activated PSCs, in
CP/PC (20). Galectin-1 has high
affinity for β-galactosides and induces collagen synthesis,
chemokine production and proliferation of PSCs in CP/PC (7,20,21).
In addition, previous studies have revealed that galectin-1 plays a
role in the desmoplastic reaction associated with PC (22). Furthermore, it has been reported
that TGF-β1 alone or TGF-β1 together with galectin-1 induces the
transition of human dermal fibroblasts to myofibroblasts (23) and that galectin-1 may promote the
TGF-β1-induced differentiation of fibroblasts by sustaining nuclear
localization of Smad2 in pulmonary fibrotic diseases (17). However, it is not known whether
galectin-1 plays a fibrogenic role in CP/PC and which is the
related underlying mechanism. Therefore, the present study was
designed to investigate the potential fibrogenic role of galectin-1
in activated PSCs in CP/PC using immunohistochemical methods under
in vitro conditions.
Materials and methods
Patients and pancreatic tissues
The clinicopathological characteristics of patients,
the PDAC, CP and normal pancreatic control tissues, as well as the
histological evaluation of these specimens have been previously
described (21,24,25).
From January 2006 to December 2010, PC tissue samples were obtained
from 66 patients undergoing pancreaticoduodenectomy for PC and from
18 patients with CP at the First Affiliated Hospital of Nanjing
Medical University, (Jiangsu, China). The PC patients comprised 45
men and 21 women with a median age of 55 years (range, 37–83 years)
and the CP patients comprised 13 men and five women with a median
age of 54.5 years (range, 27–71 years). Ten normal pancreatic
control tissue samples were obtained from patients undergoing
partial pancreatic resections for bile duct or duodenal ampullary
cancer. All procedures performed involving human participants were
in accordance with the ethical standards of the ethics committee of
The First Affiliated Hospital of Nanjing Medical University and
with the 1964 Helsinki declaration and its later amendments or
comparable ethical standards.
Compliance with ethical standards
Informed consents were obtained from all the
patients for their participation in the study, which was approved
by the ethics committee of The First Affiliated Hospital of Nanjing
Medical University (Jiangsu Provincial People's Hospital). Every
participant provided a written informed consent to participate in
this study and the copies of the written consents of participants
were reserved in our laboratory and can be obtained at any time.
The participants signed the Letter of Information and Consent and
each one held and saved a copy of the informed consent. The ethics
committee approved this consent procedure, which has been recorded
in the Consent Form of the Ethics Committee.
Cell and culture conditions
Primary human PSCs were isolated, identified,
maintained and passaged as previously described (21,24,25).
The cells from passage numbers 0–5 were used for all assays.
Preparation and transduction of
recombinant lentiviruses
The plasmids used for preparing the recombinant
lentiviruses have been previously described (21). Briefly, the galectin-1 gene fragment
was excised from a human cDNA library and cloned into
pHAGE-CMV-MCS-IZsGreen between the BamHI and XhoI
restriction sites. Galectin-1-specific oligonucleotides were
ligated into the pLKO.1-puro vector (21). The study groups were as follows:
overexpression Galectin-1-PSC (Over), normal PSC control group
(Control), knockdown shRNA-Galectin-1-PSC#1 (Sh-1) and
shRNA-Galectin-1-PSC #2 (Sh-2).
Wound healing assay
PSCs were seeded in 24-well culture plates and grown
to reach confluency. After starvation for 12 h, the monolayers were
wounded by scrapping off a strip of cells with a 200-µl pipette
tip. The cells were incubated for 24 h. Subsequently the cells were
fixed, images of three different segments of the ‘wound’ area on
each well were captured at an ×10 magnification, using the Olympus
DP71 camera (Olympus Optical Co. Ltd, Tokyo, Japan) and the cell
numbers inside the wound boundaries were counted.
In vitro migration assay
PSC migration through Matrigel was determined using
6-well Corning Transwell chambers (8.0-µm pore size with a
polycarbonate membrane) as previously described (26). Briefly, the upper chambers were
coated with diluted Matrigel (1 mg/ml, 356243; BD Biosciences,
Bedford, MA, USA) and incubated at 37°C in 5% CO2 for 3
h. After trypsinization, PSCs were suspended in DMEM with 10% FBS
at 1×105 cells/well and immediately placed onto the
upper compartment. After 24-h incubation, non-migrated cells were
removed from the upper surface of the membrane by wiping with
cotton-tipped swabs. The cells on the lower surface of the membrane
were stained with 0.1% crystal violet for 10 min and photographed
at an ×10 magnification using the Olympus DP71 camera (Olympus
Optical). The crystal violet was then bleached using 500 µl 33%
acetic acid. Absorbance at 570 nm was determined using a microtiter
plate reader.
In vitro proliferation assay
PSC proliferation was determined by methyl thiazolyl
tetrazolium (MTT) assay (Sigma-Aldrich, St. Louis, MO, USA) as
previously described (27). PSCs
(5×105) were seeded in 6-well plates, cultured with 10%
fetal culf serum (FCS) for 12 h until the cells adhered to the
plate and then, exchanged the medium and the proliferation was
detected at 24, 48, 72 h. The results were expressed as absorbance
at 570 nm in the microtiter plate reader.
Western blot analysis
Western blotting was performed as previously
described (21,24,25).
The following antibodies were used: mouse anti-Galectin-1
antibodies (1:200, sc-166618; Santa Cruz Biotechnology, Santa Cruz,
CA, USA), anti-fibronectin antibodies (1:200, sc-59824; Santa Cruz
Biotechnology), anti-collagen type I antibodies (1:200, sc-376350;
Santa Cruz Biotechnology), anti-α-SMA antibodies (1:200, MA1-37027;
Thermo Fisher Scientific Inc., Fremont, CA, USA), anti-TIMP-1
antibodies (1:200, sc-21734; Santa Cruz Biotechnology), anti-MMP-2
antibodies (1:200, sc-13595; Santa Cruz Biotechnology) or
anti-Smad2 antibodies (1:200, sc-101153; Santa Cruz
Biotechnology).
Quantitative reverse
transcription-polymerase chain reaction
Total RNA was extracted from all the cultured groups
of PSCs using TRIzol reagent (Invitrogen Life Technologies,
Beijing, China) according to the manufacturer's instructions.
Quantitative reverse transcription-polymerase chain reaction
(qRT-PCR) was performed as previously described (21,24,25).
The sequences of primers used in the present study are shown in
Table I.
 | Table I.Primers used for quantitative
real-time RT-PCR. |
Table I.
Primers used for quantitative
real-time RT-PCR.
| Primers | Forward sequence
5′-3′ | Reverse sequence
5′-3′ |
|---|
| Galectin-1 |
GAGGTGGCTCCTGACGCTAA |
CCTTGCTGTTGCACACGATG |
| TGF-β1 |
GAAACCCACAACGAAATCTATGAC |
GCTGAGGTATCGCCAGGAAT |
| Smad2 |
TCTTGATGGTCGTCTCCAGGTA |
AGAGGCGGAAGTTCTGTTAGG |
| MMP-2 |
CCTTTGCTCGTGCCTTCCA |
TCGGCGTTCCCATACTTCA |
| TIMP-1 |
GGCTTCTGGCATCCTGTTGT |
GTGGTCTGGTTGACTTCTGGTG |
| α-SMA |
GGTGACGAAGCACAGAGCAA |
ACCGCCTGGATAGCCACATAC |
| Collagen type
I |
GCATTCGTGGCGATAAGGG |
ACCAGCGATACCAGGCAGA |
| Fibronectin |
CGACTGTGGACCAAGTTGATGAC |
AAGGTTGAGTTCTGTGCTGCTAC |
| β-actin |
AGAAAATCTGGCACCACACC |
TAGCACAGCCTGGATAGCAA |
Immunohistochemical staining and
evaluation
Pancreatic tissue samples were fixed by immersion in
4% paraformaldehyde overnight at 4°C and then embedded in regular
paraffin wax and cut into 4-µm sections. Immunohistochemical
detection and analyses were performed as previously described
(21,24,25).
The primary antibodies used were as follows: mouse monoclonal
anti-Galectin-1, anti-fibronectin antibodies, anti-collagen type I
antibodies, anti-α-SMA antibodies, or anti-Desmin antibodies. The
results of the immunohistochemical staining were evaluated by two
experienced pathologists.
Statistical analysis
Values are expressed as the mean ± standard
deviation (SD). All experiments were performed in triplicate. One
way ANOVA and t-tests were performed using SPSS 13.0 software (SPSS
Inc., Chicago, IL, USA) to compare differences between groups. All
P-values were two-sided and P-values <0.05 were considered to
indicate a statistically significant difference.
Results
Role of activated PSCs in fibrosis
associated with CP/PC
Desmin and α-SMA are important markers of the
quiescent and activation statuses of PSCs, respectively (21,28).
Activation of PSCs is regulated by a complex network of growth
factors and cytokines and is associated with increase in the
expression and release of collagen I and II, fibronectin and other
components of the ECM in PSCs (29–31).
With the development in the deposition of ECM components,
pancreatic fibrosis gradually increases. To understand the role of
PSCs in pancreatic fibrosis, we performed immunohistochemical
staining for desmin, α-SMA, fibronectin and collagen type I in
normal pancreatic, CP and PC tissues. Immunohistochemical staining
revealed that desmin was positively expressed and that α-SMA,
fibronectin and collagen type I were negatively or weakly expressed
in the normal pancreas, in which the expression profile was
consistent with that of PSCs in the quiescent stage (Fig. 1). However, desmin was weakly
expressed and α-SMA, fibronectin and collagen type I were
positively expressed in CP/PC tissues, in which the expression
profile was consistent with that of PSCs in the activation stage
(Fig. 1). In addition, galectin-1
was negatively expressed in normal pancreatic tissue and quiescent
PSCs and was positively expressed in CP tissue and activated PSCs.
Thus, galectin-1 expression was also associated with the activation
stage of PSCs and the degree of fibrosis of pancreatic tissue
(Fig. 1).
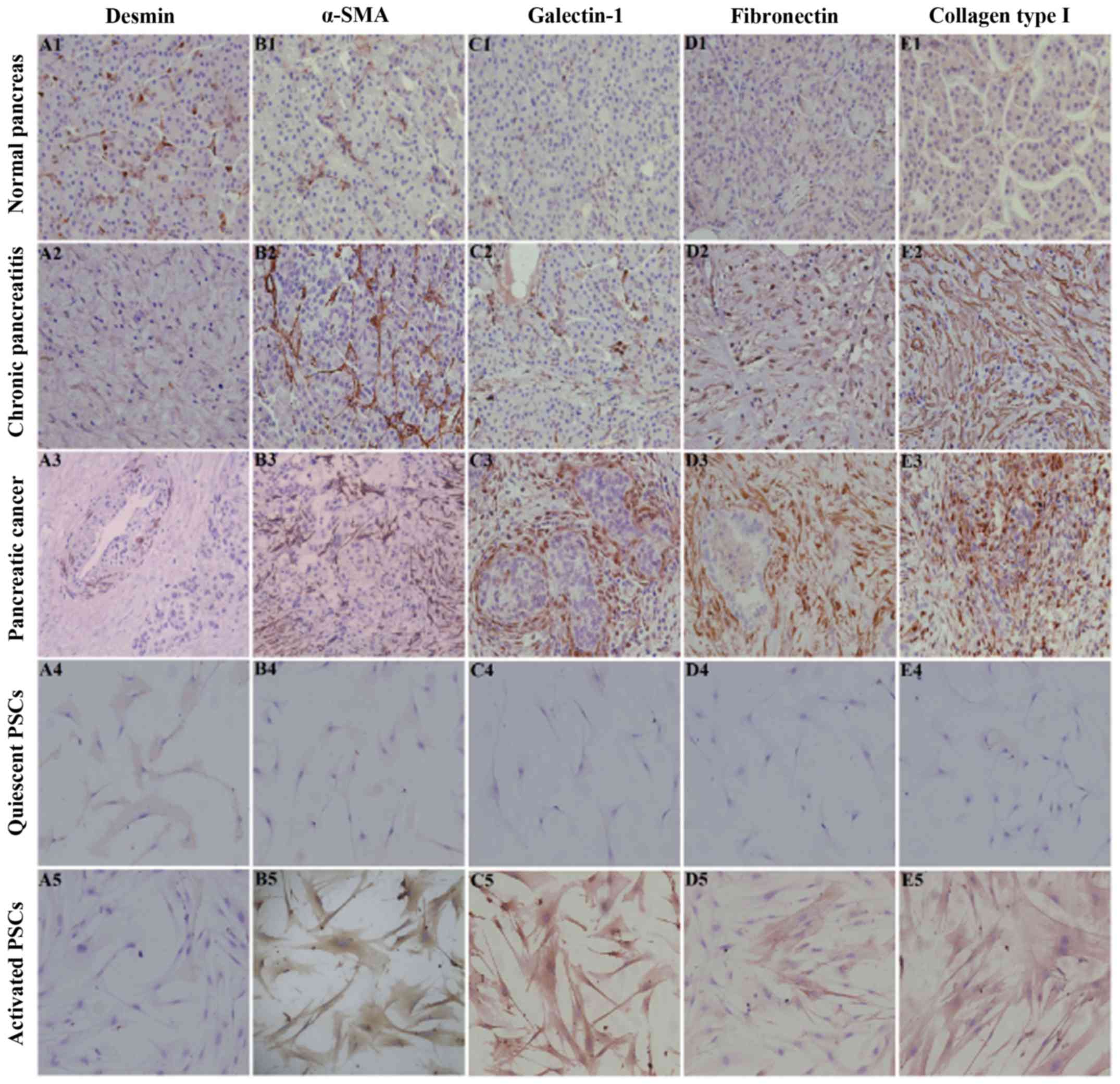 | Figure 1.Immunohistochemical staining of
desmin (A), α-SMA (B), galectin-1 (C), fibronectin (D) and collagen
type I (E) in normal pancreatic tissue (A1-E1), chronic pancreatic
tissue (A2-E2), PC tissue (A3-E3), quiescent PSCs (A4-E4) and
activated PSCs (A5-E5). Immunohistochemical staining revealed that
desmin (A1) was positively expressed while α-SMA (B1), fibronectin
(C1) and collagen type I (D1) were negatively or weakly expressed
in the normal pancreas, in which the expression profile was
consistent with that of quiescent PSCs (A4-E4). In contrast, desmin
(A2) was weakly expressed and α-SMA (B2), fibronectin (D2) and
collagen type I (E2) were positively expressed in CP and PC tissues
(A3-E3), in which the expression profile was consistent with that
of activated PSCs (A5-E5). Quiescent PSCs were obtained on the
second day and activated PSCs were obtained on the seventh day of
the primary culture. In all the images, immunohistochemical
staining is indicated by the brown diaminobenzidine color reaction
and the sections are counterstained with hematoxylin. Original
magnification, ×200. CP, chronic pancreatitis; PC, pancreatic
cancer. |
Effect of galectin-1 on the
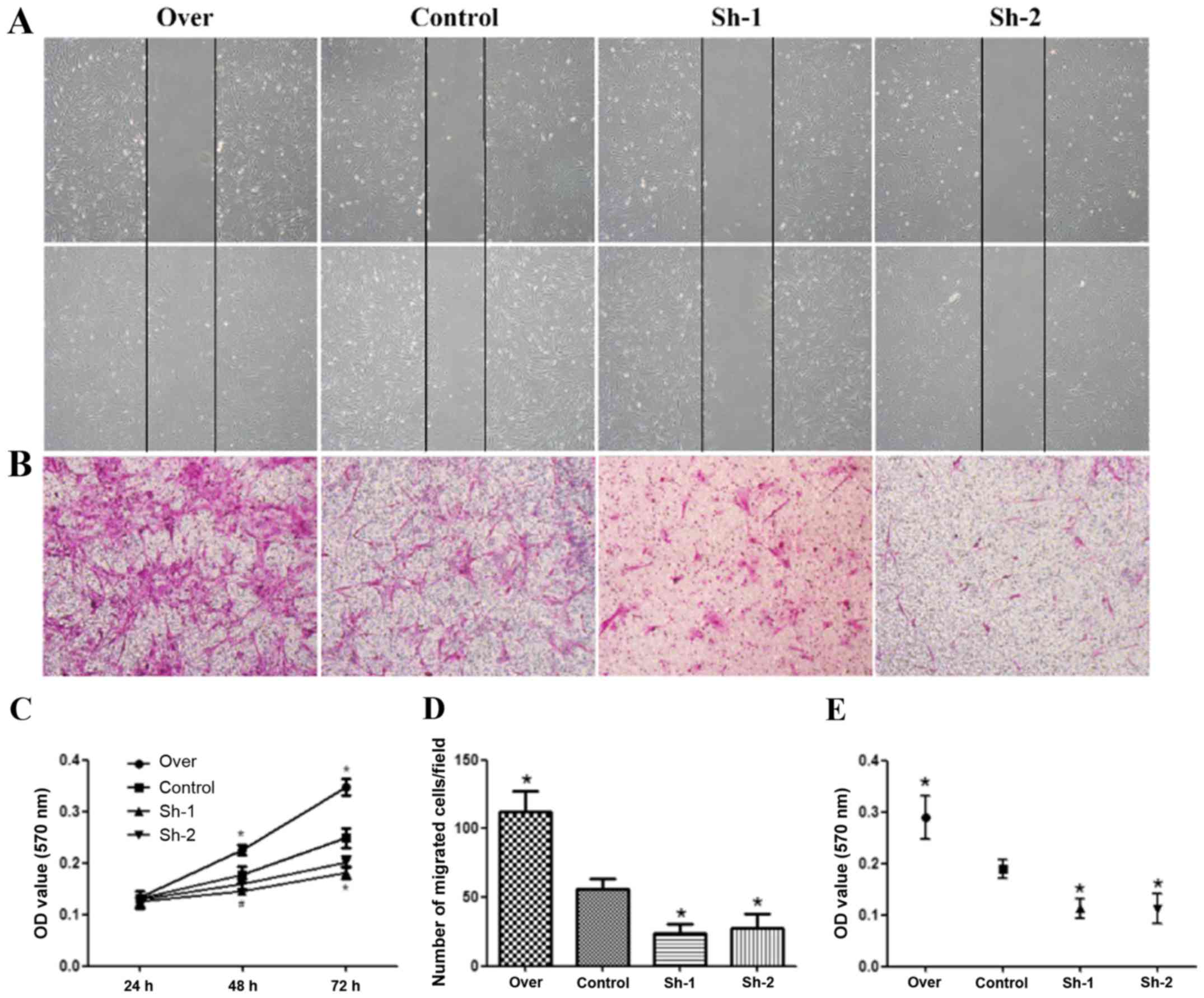
proliferation of PSCs
In order to obtain PSCs with different expression
levels of galectin-1 that reflect different pancreatic conditions,
lentiviral sh1RNA-galectin-1 and sh2RNA-galectin-1 transduction in
PSCs was performed. PSCs that overexpressed galectin-1 were
purified by screening for green fluorescent protein (GFP) using
flow cytometry and PSCs that contained the galectin-1-silenced
plasmids (sh1RNA- and sh2RNA-transduced galectin-1-knockdown PSCs)
were selected with puromycin. The MTT assay results revealed that
galectin-1-overexpressing PSCs had a significantly higher
proliferation rate than the control PSCs (P<0.05). Furthermore,
the galectin-1-silenced PSCs had a significantly lower
proliferation rate than the control PSCs (P<0.05) (Fig. 2C).
Effect of galectin-1 on the migration
of PSCs
PSC migration was assessed by the wound healing
assay, a well-established in vitro system for assessing cell
motility. A confluent monolayer growing in 24-well plates was
wounded by scraping off the cells with a pipette tip, thus creating
a space free of cells. The cells were allowed to migrate into the
cell-free area. In the presence of 5% FCS, the migration of
galectin-1-overexpressing PSCs (P<0.05) was significantly
greater than that of the control PSCs. The migration of
galectin-1-silenced PSCs (transduced with sh1RNA and sh2RNA) was
significantly lower than that of the control PSCs (P<0.05)
(Fig. 2A and D). The migration
ability of the different groups of PSCs was further confirmed by
the Transwell assay. The results of this assay also revealed that
the migration ability of galectin-1-overexpressing PSCs was
significantly greater than that of the control PSCs, and that the
migration ability of the galectin-1-silenced PSCs was significantly
lower than that of the control PSCs (P<0.05; Fig. 2B and E).
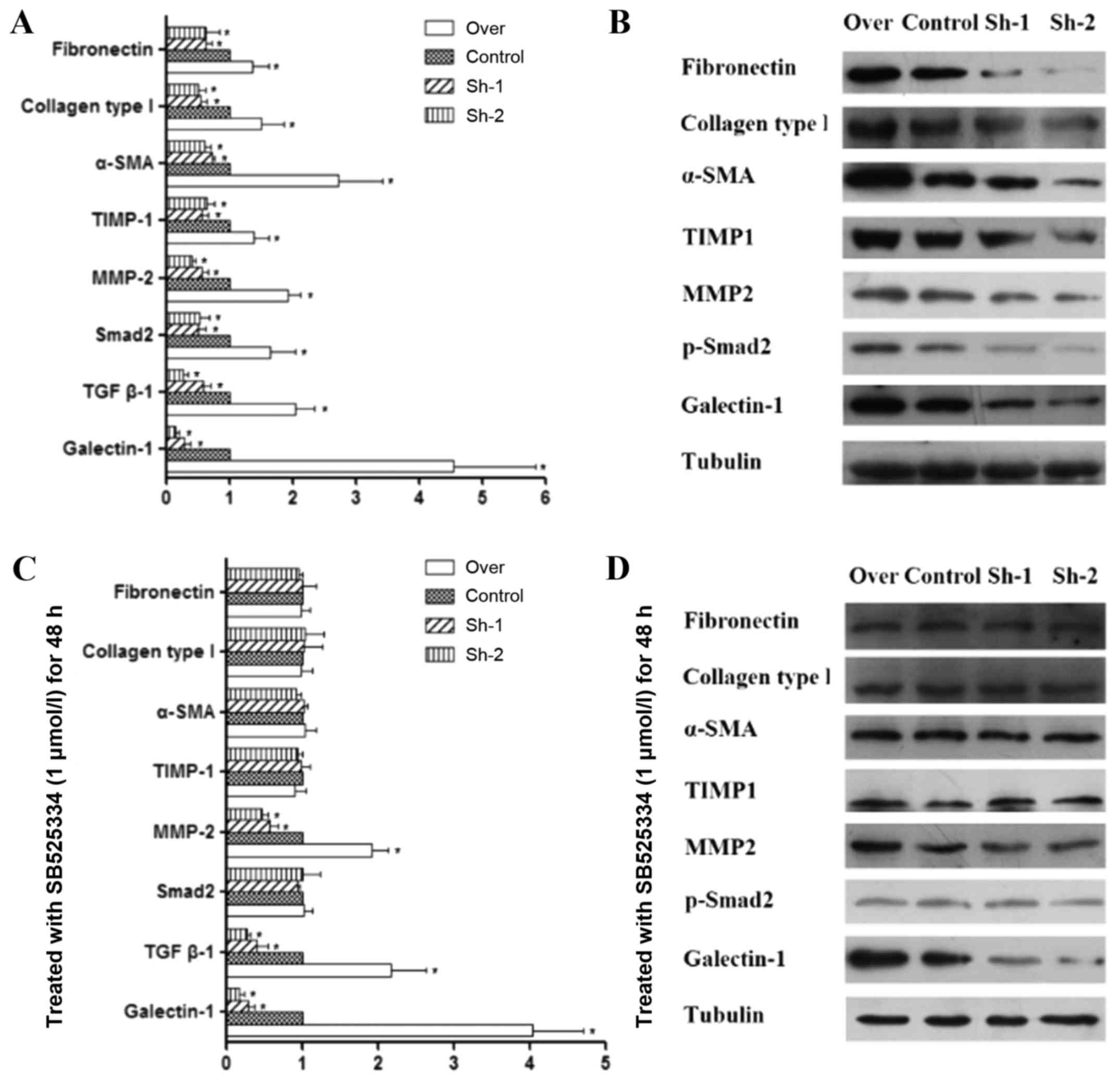
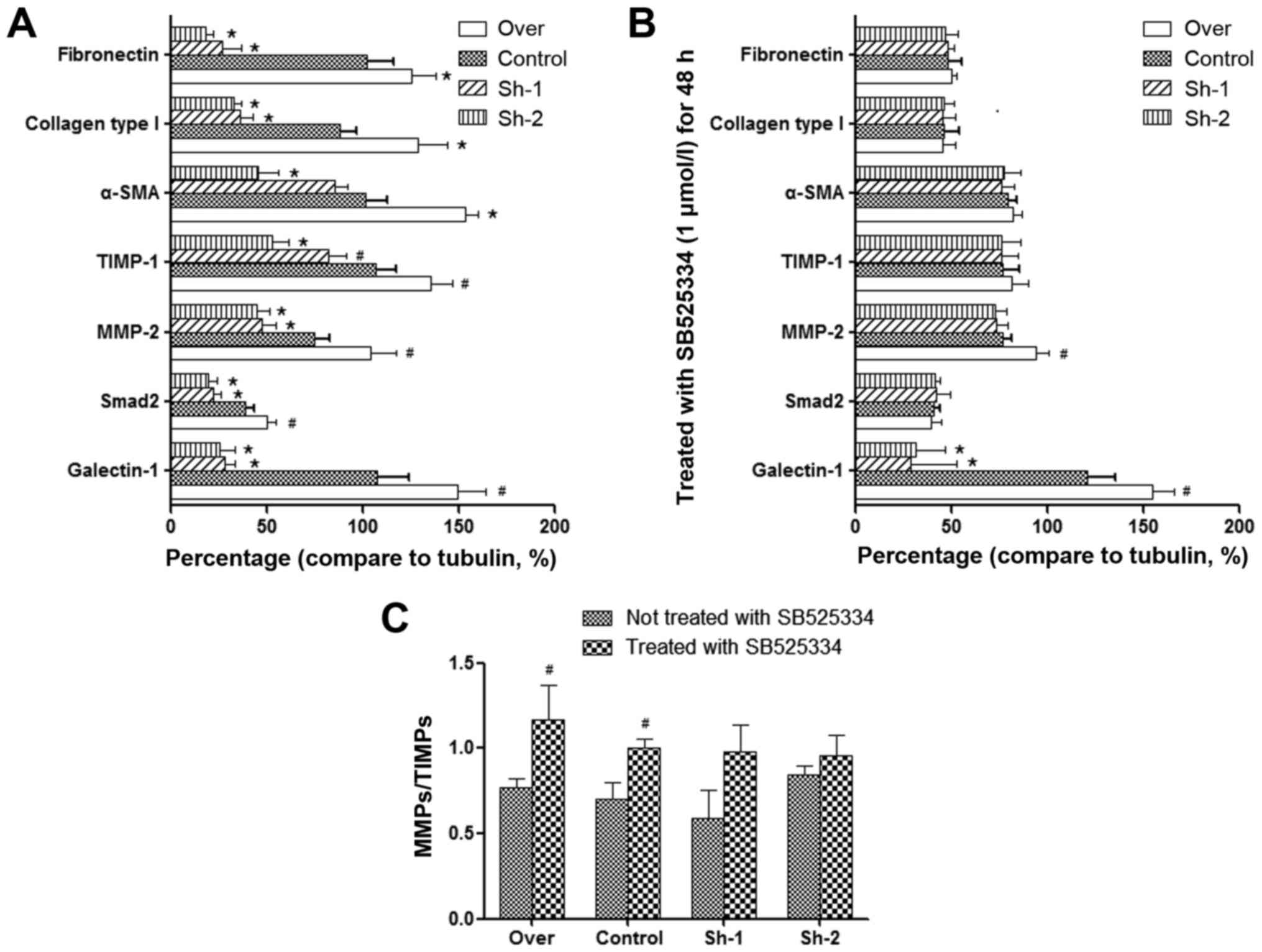
Effect of galectin-1 expression on the
level of MMP-2, TIMP-1 and other fibrosis-associated factors in
activated PSCs
In pancreatic fibrosis, there is an imbalance in the
synthesis and degradation of the ECM. MMPs and TIMPs are mainly
responsible for the degradation of the pancreatic ECM. MMPs play a
role in the degradation of collagen, while TIMPs have an inhibitory
effect on MMPs (32–34). Our quantitative PCR and western blot
analysis results indicated that in galectin-1-overexpressing PSCs,
the expression of both MMP-2 and TIMP-1 was increased, but the
expression of TIMP-1 increased to a higher degree than the
expression of MMP-2 (P<0.01; Fig. 3A
and B). In addition, galectin-1-overexpressing PSCs also
revealed an increase in the expression of fibronectin, collagen
type I and α-SMA (Fig. 3A and B),
which are fibroblast markers that are strongly expressed in
activated PSCs, as well as TGF-β1 expression and Smad2
phosphorylation. In contrast, in galectin-1-silenced PSCs, the
opposite effects were observed (Fig. 3A
and B); lower galectin-1 expression resulted in lower MMP-2 and
slightly lower TIMP-1 expression in galectin-1-silenced PSCs than
in the control PSCs (P<0.05; Fig. 3A
and B), but a significant decrease was observed in the
expression of fibronectin, collagen type I and α-SMA. Collectively
these results indicated that overexpression of galectin-1 promoted
the fibrosis of activated PSCs by tilting the balance of MMP/TIMP
expression in favor of TIMP and that galectin-1 silencing reversed
this effect on the progression of fibrosis.
Endogenous galectin-1-induced fibrosis
of activated PSCs via the TGF-β1/Smad signaling pathway
As aforementioned, galectin-1 overexpression in PSCs
resulted in an increase in both TGF-β1 and Smad2 expression and was
also associated with MMP/TIMP imbalance. It is not clear whether
the TGF-β1/Smad2 signaling pathway is directly associated with
MMP/TIMP imbalance and fibrosis of PSCs. Therefore, in order to
shed light on the role of this pathway in pancreatic fibrosis,
control, galectin-1-overexpressing and galectin-1-silenced PSCs
were treated for 48 h with SB525334, which is a selective inhibitor
of TGF-β receptor I (ALK5, TGF-βRI). This agent inhibits
TGF-β-induced ALK5 serine/threonine kinase activity, thus
preventing the phosphorylation of Smad transcription factors and
their subsequent gene activation (35,36).
The following results were observed: although SB525334 had no
obvious effect on the expression of TGF-β1, it inhibited the
phosphorylation of Smad2 that was induced by TGF-β1 and reversed
the imbalance of MMPs/TIMPs. Furthermore, it decreased the
expression of fibronectin, collagen type I and α-SMA. These
findings were observed in all corresponding PSC groups (over,
control, Sh-1, Sh-2), which have significant differences compared
with prior to using the TGF-β inhibitor (Fig. 3C and D; Fig. 4). This could prove that TGF-β1/Smad
pathway is essential for the fibrosis. As aforementioned we have
clarified the relationship between galectin-1 and TGF-β1/Smad2
expression. In addition, in the present study, upon applying the
TGF-β1 inhibitor, we observed that the subsequent effects were not
dependent on whether galectin-1 expression was up- or
downregulated. Thus, these results indicated that it was endogenous
galectin-1 that induced TIMP expression by stimulating the
TGF-β1/Smad signaling pathway, which decreased the degradation of
ECM and increased the expression of fibronectin, collagen type I
and α-SMA, thus promoting the progression of PSC fibrosis.
Discussion
This study aimed to investigate the effect of
PSC-derived galectin-1 on fibrogenesis in CP/PC tissues and the
underlying mechanisms. Fibrosis plays a vital role in the formation
of tumor microenvironment and initiation of tumor angiogenesis
(37), therefore the reversal of
pancreatic fibrosis could be efficient for CP/PC treatment
(8). However, to date there are few
therapeutic targets for the treatment of pancreatic fibrosis. We
found that galectin-1 was expressed in CP/PC tissue and activated
PSCs in vitro and that negative galectin-1 expression was
observed in normal pancreatic tissue and PSCs in the quiescent
state. Furthermore, we demonstrated that PSC-derived galectin-1
promoted the progression of fibrosis in PSCs via stimulation of the
TGF-β1/Smad signaling pathway. Collectively, these results
indicated that PSC-derived galectin-1 may serve as a potential
biomarker in therapeutic interventions for CP/PC.
Previous studies have revealed that endogenous
galectin-1 expression is strongly induced upon activation of PSCs
(21,24,25).
In accordance with these results, the immunohistochemical results
of the present study also revealed strong galectin-1 staining in
CP/PC tissues and activated PSCs. These findings indicated that
PSCs play a role in pancreatic fibrosis in CP/PC and that
galectin-1 derived from PSCs may play a critical role in the
progression of fibrosis in CP/PC. Furthermore, endogenous
galectin-1 was found to significantly increase the mRNA expression
of collagen type I and fibronectin in PSCs. These results indicated
that endogenous galectin-1 induced the expression of soluble
collagen and fibronectin by increasing their secretion or
increasing the rate of their degradation. Thus, by altering the
balance between ECM protein secretion and synthesis, endogenous
galectin-1 may induce collagen and fibronectin synthesis in the
ECM. Previous studies have revealed that collagen degradation
promoted the regenerative response of hepatocytes during resolution
of liver fibrosis (38). Thus,
endogenous galectin-1 in PSCs may induce pancreatic degradation in
patients with pancreatic injury by accelerating collagen
synthesis.
Previous studies have revealed a 4.5-fold increase
in galectin-1 mRNA expression (p<0.01) in CP/PC samples compared
with normal controls, as well as upregulation of galectin-1 in
fibroblasts. These findings indicated that galectin-1 plays a role
in tissue remodeling in CP (39).
In addition, PSCs exposed to exogenous galectin-1 proliferated at a
higher rate and synthesized more collagen than the control cells
(7). The present study also
revealed that endogenous overexpression of galectin-1 in PSCs
resulted in a significant increase in the proliferation and
migration of PSCs. Furthermore, it resulted in an increase in the
expression of TGF-β1 [a known key pro-fibrogenic factor (2)] and the phosphorylation of Smad2 and a
consequent increase in the expression of fibronectin, collagen type
I and α-SMA. Contrasting results were observed in PSCs in which the
expression of endogenous galectin-1 was silenced. Furthermore, the
treatment of PSCs with SB525334, a selective inhibitor of TGF-β
receptor I, resulted in the inhibition of the phosphorylation of
Smad2 (induced by TGF-β1) and as a result, the expression of
fibronectin, collagen type I and α-SMA was also decreased. In
accordance with these findings, it has been reported that
galectin-1 may promote the TGF-β1-induced differentiation of
fibroblasts by sustaining nuclear localization of Smad2 and that
knockdown of galectin-1 could decrease the phosphorylation and
nuclear retention of Smad2, which may prevent the differentiation
of fibroblasts (40). The
TGF-β/Smad signaling pathway has also been implicated in fibrosis
development in a previous study (41). Collectively, all these results
indicated that endogenous galectin-1 in PSCs significantly
interacted with the TGF-β1/Smad2 pathway in a positive feedback
loop, which may accelerate fibrosis of activated PSCs. Furthermore,
TGF-β1 may be a key promoter of ECM production and deposition of
collagen type I and could trigger a Smad-dependent pathway to
control galectin-1-induced pancreatic fibrosis in PSCs. Thus,
TGF-β1 and galectin-1 may work in synergy to tilt the balance
towards fibrosis in CP/PC (42).
Therefore, galectin-1 expression in PSCs may present a potential
therapeutic target for the anti-fibrosis treatment of CP/PC.
In the present study, we also examined MMP-2 and
TIMP-1 synthesis by transformed cultured PSCs and their regulation
by TGF-β1. A significant role in the pathogenesis of PC and CP may
be attributed to metalloproteinases (MMPs). The cellular basement
membrane (BM) and ECM consist of collagen, laminin, elastin,
fibronectin and proteoglycans which are subject to MMPs degradation
(43). Degradation of the ECM is an
essential step in tumor invasion and metastasis. Each MMP has
different substrate specificities within the ECM and is important
in the degradation of ECM. MMP activity is dependent on the levels
of activated MMP and TIMPs (44).
In the present study, our data revealed that galectin-1 induced an
increase in MMP-2 secretion and an even greater increase in TIMP-1
expression in PSCs. Thus, galectin-1 facilitates the synthesis of
ECM proteins by enhancing MMP activity, which has a pro-fibrogenic
effect. There are several explanations for this effect. Firstly,
MMP-2 is considered as an autocrine growth factor for stellate
cells. Thus, increasing MMP-2 secretion may result in an increase
in the number of PSCs and consequently induce collagen synthesis.
Secondly, MMP-2 degrades normal basement membrane collagen (type
IV); an increase in MMP-2 secretion may therefore, induce the
deposition of pathological fibrillar collagen in the gland.
However, TIMP-1 counteracts these effects and contributes to the
development of pancreatic fibrosis. Therefore, when TIMP-1
expression is higher than MMP-2 expression, as observed in the
activated PSCs in the present study, MMP activity is inhibited to
some degree and the balance between ECM synthesis and degradation
tilts towards fibrogenesis. These findings were similar to the
above-mentioned studies and demonstrated that galectin-1 may play
an important and hitherto unappreciated role in inducing pancreatic
fibrosis (40).
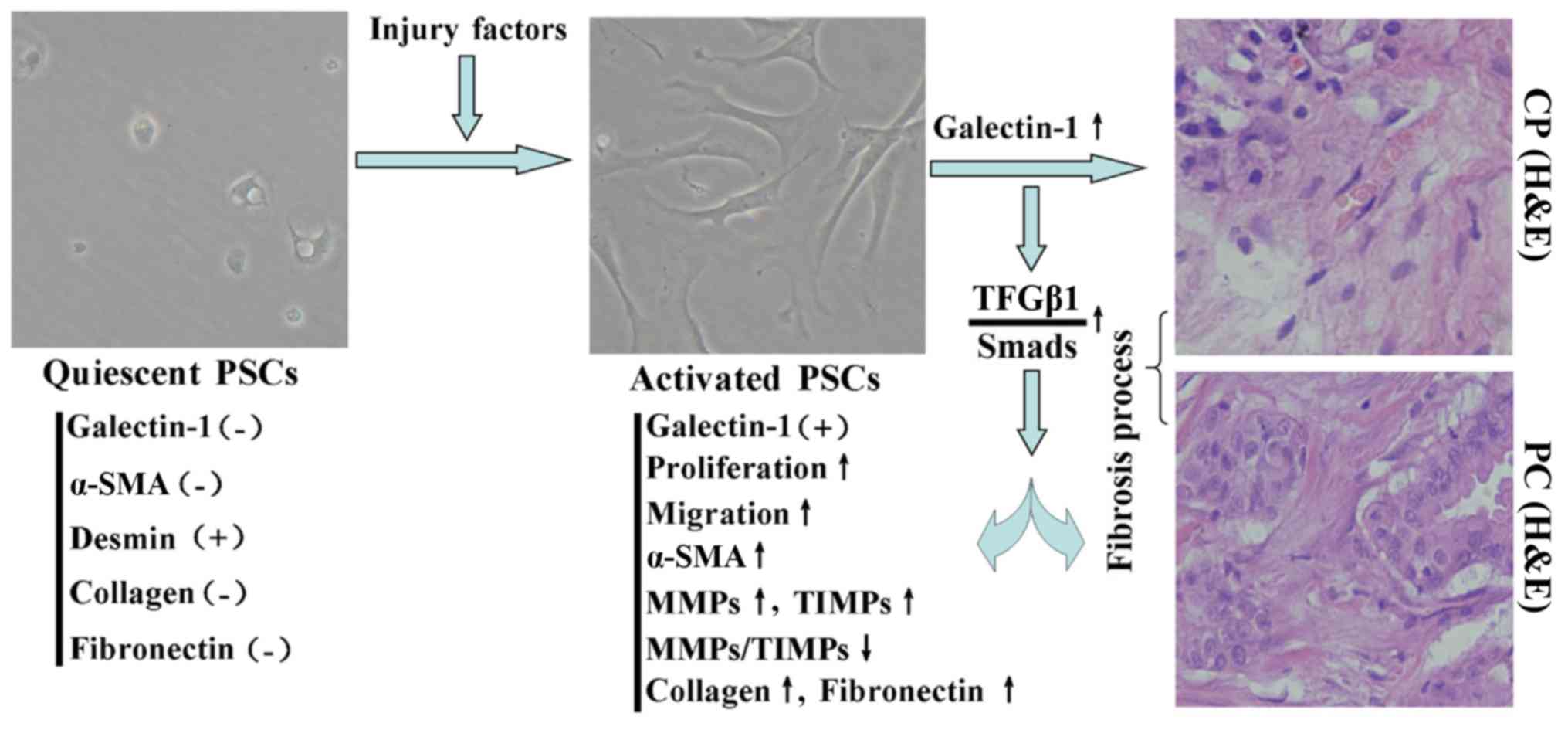
This is the first study to investigate the effect of
endogenous galectin-1 expression in PSCs in the fibrotic pancreatic
tissue. By increasing the number of PSCs in the injured area and by
promoting ECM synthesis, galectin-1 may act as a pro-fibrogenic
protein in the process of injury of pancreatitis (Fig. 5). Further investigation into the
molecular mechanisms and pathogenesis of pancreatic fibrosis using
animal models would provide a better understanding of the process
of fibrosis and clarify the adverse role of galectin-1 in
pancreatic injury. This would be important in the development of
novel approaches to antifibrotic therapy. Furthermore, the present
study highlighted the role of galectin-1 in regulating the level of
the fibrogenic cytokine marker TGF-β1 and MMPs in the pathogenesis
of CP/PC. This could mean that genotypes corresponding to high
TGF-β1 production may be associated with fibrogenesis in CP/PC
(45,46).
Based on the data presented in this study, we
propose a mechanism for the induction of fibrosis by activated
PSC-derived galectin-1 in CP/PC (Fig.
5): upon stimulation with a variety of pancreatic injury
factors, quiescent PSCs were activated and their proliferation and
migration were induced. Furthermore, the TGF-β1/Smad signaling
pathway was induced, which increased the expression of fibronectin,
collagen type I and α-SMA and altered the MMP/TIMP ratio. These
events resulted in the reduction of ECM degradation and promoted
fibrosis in CP/PC.
In conclusion, the findings of the present study
indicated that PSCs and galectin-1 may have a potential for
selective therapeutic treatments targeting fibrosis in CP/PC.
Acknowledgements
We thank Professor Lu Chun (Department of
Microbiology and Immunology, Nanjing Medical University, Nanjing,
China) for kindly providing the lentiviral packaging system
consisting of pHAGE-CMVMCS-IZs Green, psPAX2 and pMD2.G. We would
like to thank the native English speaking scientists of Elixigen
Company (Huntington Beach, CA, USA) for editing the study. The
present study was supported by grants from the National Natural
Science Funding of China (no. 81572344), the Postdoctoral Science
Foundation of China (no. 2013M530243), the Science and Technology
Development Funding of Yangzhou City (no. 2012123), the Jiangsu
Province Natural Science Foundation of China (no. BK20140495), the
Six Big Talent Peak Projects of Jiangsu Province (no.
2014-WSW-078), the Postdoctoral Science Foundation of Jiangsu
Province (2013), the training project of key talents of youth
medicine in Jiangsu Province, (no. QNRC2016330), the ‘Promote
Health Development by Science and Technology’ program of Jiangsu
Province (no. KF201225) and the academic science and technology
innovation fund for college students (×20160750, ×20160753,
×20160774 and ×20160783).
References
|
1
|
Xue J, Sharma V, Hsieh MH, Chawla A,
Murali R, Pandol SJ and Habtezion A: Alternatively activated
macrophages promote pancreatic fibrosis in chronic pancreatitis.
Nat Commun. 6:71582015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Staloch D, Gao X, Liu K, Xu M, Feng X,
Aronson JF, Falzon M, Greeley GH, Rastellini C, Chao C, et al:
Gremlin is a key pro-fibrogenic factor in chronic pancreatitis. J
Mol Med (Berl). 93:1085–1093. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tian L, Lu ZP, Cai BB, Zhao LT, Qian D, Xu
QC, Wu PF, Zhu Y, Zhang JJ, Du Q, et al: Activation of pancreatic
stellate cells involves an EMT-like process. Int J Oncol.
48:783–792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mahadevan D and Von Hoff DD: Tumor-stroma
interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther.
6:1186–1197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hamada S, Masamune A, Yoshida N, Takikawa
T and Shimosegawa T: IL-6/STAT3 plays a regulatory role in the
interaction between pancreatic stellate cells and cancer cells. Dig
Dis Sci. 61:1561–1571. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin Z, Zheng LC, Zhang HJ, Tsang SW and
Bian ZX: Anti-fibrotic effects of phenolic compounds on pancreatic
stellate cells. BMC Complement Altern Med. 15:2592015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fitzner B, Walzel H, Sparmann G, Emmrich
J, Liebe S and Jaster R: Galectin-1 is an inductor of pancreatic
stellate cell activation. Cell Signal. 17:1240–1247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Apte M, Pirola RC and Wilson JS:
Pancreatic stellate cell: Physiologic role, role in fibrosis and
cancer. Curr Opin Gastroenterol. 31:416–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiao W, Jiang W, Shen J, Yin G, Fan Y, Wu
D, Qiu L, Yu G, Xing M, Hu G, et al: Retinoic acid ameliorates
pancreatic fibrosis and inhibits the activation of pancreatic
stellate cells in mice with experimental chronic pancreatitis via
suppressing the Wnt/β-catenin signaling pathway. PLoS One.
10:e01414622015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Incio J, Suboj P, Chin SM, Vardam-Kaur T,
Liu H, Hato T, Babykutty S, Chen I, Deshpande V, Jain RK, et al:
Metformin reduces desmoplasia in pancreatic cancer by reprogramming
stellate cells and tumor-associated macrophages. PLoS One.
10:e01413922015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takikawa T, Masamune A, Hamada S, Nakano
E, Yoshida N and Shimosegawa T: miR-210 regulates the interaction
between pancreatic cancer cells and stellate cells. Biochem Biophys
Res Commun. 437:433–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsang SW, Zhang HJ, Chen YG, Auyeung KK
and Bian ZX: Eruberin A, a natural flavanol glycoside, exerts
anti-fibrotic action on pancreatic stellate cells. Cell Physiol
Biochem. 36:2433–2446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakamura T, Ito T, Uchida M, Hijioka M,
Igarashi H, Oono T, Kato M, Nakamura K, Suzuki K, Jensen RT, et al:
PSCs and GLP-1R: Occurrence in normal pancreas, acute/chronic
pancreatitis and effect of their activation by a GLP-1R agonist.
Lab Invest. 94:63–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He J, Sun X, Qian KQ, Liu X, Wang Z and
Chen Y: Protection of cerulein-induced pancreatic fibrosis by
pancreas-specific expression of Smad7. Biochim Biophys Acta.
1792:56–60. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao X, Cao Y, Yang W, Duan C, Aronson JF,
Rastellini C, Chao C, Hellmich MR and Ko TC: BMP2 inhibits
TGF-β-induced pancreatic stellate cell activation and extracellular
matrix formation. Am J Physiol Gastrointest Liver Physiol.
304:G804–G813. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fitzner B, Brock P, Nechutova H, Glass A,
Karopka T, Koczan D, Thiesen HJ, Sparmann G, Emmrich J, Liebe S, et
al: Inhibitory effects of interferon-gamma on activation of rat
pancreatic stellate cells are mediated by STAT1 and involve
down-regulation of CTGF expression. Cell Signal. 19:782–790. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Böhm K, Teich N, Hoffmeister A, Mössner J,
Keim V, Bödeker H and Gress TM: Transforming growth factor-beta-1
variants are not associated with chronic nonalcoholic pancreatitis.
Pancreatology. 5:75–80. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu J, Akanuma N, Liu C, Naji A, Halff GA,
Washburn WK, Sun L and Wang P: TGF-β1 promotes acinar to ductal
metaplasia of human pancreatic acinar cells. Sci Rep. 6:309042016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan R, Liu X, Wang J, Lu P, Han Z, Tao J,
Yin C and Gu M: Alternations of galectin levels after renal
transplantation. Clin Biochem. 47:83–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masamune A, Satoh M, Hirabayashi J, Kasai
K, Satoh K and Shimosegawa T: Galectin-1 induces chemokine
production and proliferation in pancreatic stellate cells. Am J
Physiol Gastrointest Liver Physiol. 290:G729–G736. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang
H, Chen M, An Y, Wei J, Zhu Y, et al: High expression of Galectin-1
in pancreatic stellate cells plays a role in the development and
maintenance of an immunosuppressive microenvironment in pancreatic
cancer. Int J Cancer. 130:2337–2348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Berberat PO, Friess H, Wang L, Zhu Z, Bley
T, Frigeri L, Zimmermann A and Büchler MW: Comparative analysis of
galectins in primary tumors and tumor metastasis in human
pancreatic cancer. J Histochem Cytochem. 49:539–549. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mifková A, Kodet O, Szabo P, Kučera J,
Dvořánková B, André S, Koripelly G, Gabius HJ, Lehn JM and Smetana
K Jr: Synthetic polyamine BPA-C8 inhibits TGF-β1-mediated
conversion of human dermal fibroblast to myofibroblasts and
establishment of galectin-1-rich extracellular matrix in vitro.
ChemBioChem. 15:1465–1470. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang D, Zhang J, Yuan Z, Gao J, Wang S, Ye
N, Li P, Gao S, Miao Y, Wang D, et al: Pancreatic satellite cells
derived galectin-1 increase the progression and less survival of
pancreatic ductal adenocarcinoma. PLoS One. 9:e904762014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang D, Gao J, Wang S, Yuan Z, Ye N, Chong
Y, Xu C, Jiang X, Li B, Yin W, et al: Apoptosis and anergy of T
cell induced by pancreatic stellate cells-derived galectin-1 in
pancreatic cancer. Tumour Biol. 36:5617–5626. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang D, Gao J, Wang S, Ye N, Chong Y,
Huang Y, Wang J, Li B, Yin W and Wang D: Cancer-associated
fibroblasts promote angiogenesis in gastric cancer through
galectin-1 expression. Tumour Biol. 37:1889–1899. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xue X, Lu Z, Tang D, Yao J, An Y, Wu J, Li
Q, Gao W, Xu Z, Qian Z, et al: Galectin-1 secreted by activated
stellate cells in pancreatic ductal adenocarcinoma stroma promotes
proliferation and invasion of pancreatic cancer cells: An in vitro
study on the microenvironment of pancreatic ductal adenocarcinoma.
Pancreas. 40:832–839. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Apte MV, Haber PS, Applegate TL, Norton
ID, McCaughan GW, Korsten MA, Pirola RC and Wilson JS: Periacinar
stellate shaped cells in rat pancreas: Identification, isolation,
and culture. Gut. 43:128–133. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ellenrieder V, Schneiderhan W, Bachem M
and Adler G: Fibrogenesis in the pancreas. Rocz Akad Med Bialymst.
49:40–46. 2004.PubMed/NCBI
|
|
30
|
Jaskiewicz K, Nalecz A, Rzepko R and
Sledzinski Z: Immunocytes and activated stellate cells in
pancreatic fibrogenesis. Pancreas. 26:239–242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Menke A and Adler G: TGFbeta-induced
fibrogenesis of the pancreas. Int J Gastrointest Cancer. 31:41–46.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Bimmler D, Graf R, Zhou S, Sun Z,
Chen J, Siech M and Bachem MG: PSP/reg inhibits cultured pancreatic
stellate cell and regulates MMP/ TIMP ratio. Eur J Clin Invest.
41:151–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Phillips PA, McCarroll JA, Park S, Wu MJ,
Pirola R, Korsten M, Wilson JS and Apte MV: Rat pancreatic stellate
cells secrete matrix metalloproteinases: Implications for
extracellular matrix turnover. Gut. 52:275–282. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li L, Bachem MG, Zhou S, Sun Z, Chen J,
Siech M, Bimmler D and Graf R: Pancreatitis-associated protein
inhibits human pancreatic stellate cell MMP-1 and −2, TIMP-1 and −2
secretion and RECK expression. Pancreatology. 9:99–110. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Laping NJ: Therapeutic uses of smad
protein inhibitors: Selective inhibition of specific TGF-beta
activities. IDrugs. 2:907–914. 1999.PubMed/NCBI
|
|
36
|
Laping NJ, Everitt JI, Frazier KS, Burgert
M, Portis MJ, Cadacio C, Gold LI and Walker CL: Tumor-specific
efficacy of transforming growth factor-beta RI inhibition in Eker
rats. Clin Cancer Res. 13:3087–3099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Erkan M, Reiser-Erkan C, Michalski CW,
Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H and Kleeff
J: Cancer-stellate cell interactions perpetuate the
hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma.
Neoplasia. 11:497–508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Suárez-Cuenca JA, Chagoya de Sánchez V,
Aranda-Fraustro A, Sánchez-Sevilla L, Martínez-Pérez L and
Hernández-Muñoz R: Partial hepatectomy-induced regeneration
accelerates reversion of liver fibrosis involving participation of
hepatic stellate cells. Exp Biol Med (Maywood). 233:827–839. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang L, Friess H, Zhu Z, Frigeri L,
Zimmermann A, Korc M, Berberat PO and Büchler MW: Galectin-1 and
galectin-3 in chronic pancreatitis. Lab Invest. 80:1233–1241. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lim MJ, Ahn J, Yi JY, Kim MH, Son AR, Lee
SL, Lim DS, Kim SS, Kang MA, Han Y, et al: Induction of galectin-1
by TGF-β1 accelerates fibrosis through enhancing nuclear retention
of Smad2. Exp Cell Res. 326:125–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu LZ, Sun H and Chen JH: Histone
deacetylases 3 deletion restrains PM2.5-induced mice lung injury by
regulating NF-κB and TGF-β/Smad2/3 signaling pathways. Biomed
Pharmacother. 85:756–762. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Daroqui CM, Ilarregui JM, Rubinstein N,
Salatino M, Toscano MA, Vazquez P, Bakin A, Puricelli L, Bal de
Kier Joffé E and Rabinovich GA: Regulation of galectin-1 expression
by transforming growth factor beta1 in metastatic mammary
adenocarcinoma cells: Implications for tumor-immune escape. Cancer
Immunol Immunother. 56:491–499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lekstan A, Olakowski M, Jabłońska B,
Łabuzek K, Olakowska E, Filip I and Lampe P: Concentration of
gelatinases and their tissue inhibitors in pancreatic inflammatory
and neoplastic tumors and their influence on the early
postoperative course. Pol Przegl Chir. 85:65–72. 2013.PubMed/NCBI
|
|
44
|
Bramhall SR, Neoptolemos JP, Stamp GW and
Lemoine NR: Imbalance of expression of matrix metalloproteinases
(MMPs) and tissue inhibitors of the matrix metalloproteinases
(TIMPs) in human pancreatic carcinoma. J Pathol. 182:347–355. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bendicho MT, Guedes JC, Silva NN, Santana
GO, dos Santos RR, Lyra AC, Lyra LG, Meyer R and Lemaire DC:
Polymorphism of cytokine genes (TGF-beta1, IFN-gamma, IL-6, IL-10,
and TNF-alpha) in patients with chronic pancreatitis. Pancreas.
30:333–336. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shek FW, Benyon RC, Walker FM, McCrudden
PR, Pender SL, Williams EJ, Johnson PA, Johnson CD, Bateman AC,
Fine DR, et al: Expression of transforming growth factor-beta 1 by
pancreatic stellate cells and its implications for matrix secretion
and turnover in chronic pancreatitis. Am J Pathol. 160:1787–1798.
2002. View Article : Google Scholar : PubMed/NCBI
|