Introduction: HPV and cervical cancer
Cervical cancer is attributed to human
papillomaviruses (HPVs). Depending on their oncogenic potential,
HPVs are divided into low-risk types that predominantly cause
benign warts and high-risk types associated with malignant disease
(1). There are ~15 high-risk
mucosal HPV types of which the most important ones are HPV16 and
HPV18, which together account for more than 70% of cervical
carcinomas in women (2). The genome
of HPV16 and HPV18 contains a long control region (LCR) and eight
genes implicated in the virus life cycle. Binding sites for
transcription factors and the viral E1 and E2 proteins are located
in the LCR. The expression of viral mRNAs is controlled by two
promoters, early promoter (PE) and late promoter (PL) in HPV16 and
HPV18 (3).
Viral entry occurs through a minor abrasion of the
cervical epithelium, which allows HPV virions to infect the basal
strata. Upon entry, during the establishment phase, the virus is
maintained as an episome at ~50–100 copies in the infected cells,
inducing early intraepithelial lesions such as CIN1. The first
viral genes expressed are the oncogenes E6 and E7 in the lower
strata cells; this expression results in cell cycle deregulation
and subsequent proliferation, while in the upper layers, genome
amplification occurs through the expression of other proteins such
as E1, E2, E4 and E5, with these cells residing in the S or
G2 phase of the cell cycle. Exit from the cell cycle in
these cells occurs through a viral copy number increase up to many
thousands of copies/cell. Furthermore, the ensuing expression of
the viral L1 and L2 proteins in E4-positive cells, allows the
packaging and generation of novel infectious viral particles. In
the second or the maintenance phase of the viral life cycle, the
virus is integrated into the host genome, leading to a gradual
progression to cervical cancer via the CIN2 and CIN3 stages.
Finally, in the vegetative or productive phase, amplification of
the viral DNA occurs in high copy numbers in the differentiated
cells, resulting in its packaging in viral capsids. In established
cervical cancer, the integration of the viral genome results in the
abnormal overexpression of E6 and E7, and the loss of expression of
full-length E1, E2, E4 and E5 as well as L1 and L2 capsid proteins
(3–5). The identification and assessment of
all the proteins expressed in the HPV genome, has opened the way
for detailed studies of their molecular function, in view of the
limited therapeutic modalities for cervical cancer during the last
20 years (6). Currently, major
efforts focusing on immunotherapy have not yet yielded a
significant breakthrough (7).
Thus, the aim of the present review is to present
the available information on the structural approaches concerning
the various key HPV proteins and discuss how this novel insight
could lead to targeted therapeutic approaches for cervical
cancer.
Structural information on the key HPV
proteins
L1 and L2 proteins
The capsid of HPV is composed of two proteins, L1
and L2. L1 exhibits significant differences among HPV types,
whereas the L2 sequence is more conserved. L1 protein is the major
protein of a virus-like particle (VLP), while the L2 protein
constitutes the minor protein. The identification of a
heparin-binding C-terminus domain of the L1 of HPV11 type, suggests
that the initial attachment of the virion to host cells is mediated
by the exposure of the basal membrane of keratinocytes and binding
of L1 to heparin sulfate proteoglycans (8). The mechanism of viral entry includes
the binding of VLPs to the α6 integrin subunit and in turn, these
complexes utilize β1 or β4 integrin as co-receptors that facilitate
HPV binding and entry into epithelial cells (9). The capsid of the HPV16 VLP has been
studied at different stages of maturation by cryo-electron
microscopy and image analysis. The size of VLPs varies during
maturation. The VLP structures are stabilized by inter-L1 disulfide
bonds and their cross-linking increases towards more mature forms
(10). It has been suggested that
C161, C229 and C379, which are highly conserved in different HPV
types, are actually involved in the early phases of virion
assembly, by creating transient disulfide bonds, as was shown
mainly by the mutational analysis of the above cysteines (11). The VLP surface displays loops
projecting to the exterior surface, containing epitopes that are
recognized by the immune system. Five loops have been identified
(BC, DE, EF, FG and HI) which contain epitopes that generate
specific antibody responses. Three different structural states of
L1 HPV16, an L1 monomer, an L1 pentamer and a VLP consisting of 12
pentamers arranged in an icosahedral structure, exhibit different
immunogenicity. The icosahedral VLP is the most effective in
eliciting an immune response (12).
In another study, crystal structures of L1 pentamers of high-risk
HPV16, HPV18, HPV35 and low-risk HPV11 were resolved. The β-sheets
of all HPV types are identical but important conformational changes
occur on their pentamer surfaces, including a few angstrom (Å)
shift of the surface loop or some substitutions of the surface loop
residues. The structural differences of the pentamers are possibly
responsible for the binding specificity of monoclonal antibodies
that are raised against the VLP of the corresponding HPV type
(13). Furthermore, the properties
of the L1 monomer of cutaneous and mucosal HPVs, i.e. HPV5 and
HPV16, respectively, have been compared and their 3D-structures
were analyzed in order to visualize the number of exposed charged
amino acids of the L1 protein. The number of negatively charged
amino acids was higher in HPV5 than in HPV16. L1 HPV5 is negatively
charged at physiological pH 7.40, compared to positively charged
HPV16 L1 at the same pH. In the same study, the uptake of HPV5 and
HPV16 was determined using pseudoviruses in two cancer cell lines,
the cervical C33A and the cutaneous HaCaT. The negatively charged
heparin did not inhibit the uptake of HPV5 in C33A cells and
slightly inhibited its uptake in HaCaT cells. On the contrary, it
inhibited the HPV16 uptake in HaCaT cells and completely blocked
its uptake in C33A cells. These differences between HPV5 and HPV16
could reflect differences of the HPV tropism between cutaneous and
mucosal HPV types (14).
The L2 protein in HPVs represents the minor protein
of the capsid (15). It promotes
the transportation of the virion into the nucleus of the host cell
by interacting with host dynein following endosomal entry (16). HPV16 L2 contains 462 amino acids and
can enter the nucleus via Kapα2β1,
Kapβ2 and Kapβ3 receptors (17). The structures of different L2
proteins and their interaction with L1 have been studied in
silico. Different Alpha-HPV sequences were evaluated and it was
shown that five loops of L1 (BC, DE, EF, FG and HI), are the most
variable regions among the Alpha-HPVs, whereas specific regions of
L2 interacting with L1, are evolutionarily conserved. Furthermore,
specific regions of L1 that interact with L2 regions are also
conserved. A predicted 3D model indicated the interaction between
L1 and L2, via the direct involvement of DE and FG L1 loops and L2
proline rich areas (18). In order
to study in vitro the interaction of L1 and L2, HPV16
capsids or pseudoviruses were produced in 293T cells. Capsids were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and the evaluation of L1 and L2 band
intensities yielded a L1:L2 ratio of 5:1. Computerized
reconstructions of L1 and L2 capsids revealed pentavalent
capsomers, whereas cryo-electron microscopy showed an icosahedrally
ordered L2-specific density beneath the axial lumen of each L1
capsomer. Using bimolecular fluorescence and ‘split GFP’
technology, L2 proteins were shown to be located in close proximity
in the virion structure (15). A
proposed model for the entry of HPV16 in keratinocytes, involves
binding via the L1 major capsid protein to heparin sulfate
proteoglycans (HSPGs) on the epithelial cell surface or the
basement membrane. Growth factor receptors form complexes with
HSPGs and HPV16, thus inducing conformational changes of the
virion. Subsequently, isomerization of the virion by cyclophilin B
and proprotein convertase occurs, leading to L2 minor capsid
protein cleavage and the increase of L2 N-terminus exposure. The
binding of HPV16 to α6 integrins promotes further signaling
cascades inside the cell, by binding to L2-specific receptors, such
as Annexin A2 heterotetramers, leading to clathrin-, caveolin-,
lipid raft-, flotillin-, cholesterol- and dynamin-independent
endocytosis of HPV16 (19). The
role of Annexin A2 heterotetramer has been studied in terms of
viral infection, since the S100A10 subunit of the heterotetramer
based on immunoprecipitation studies, interacts with HPV16 L2,
facilitating viral entry in knockdown experiments (20). In addition, the entry of HPV51 was
found to be mediated by the transport protein particle complex
subunit 8 (TRAPPC8) following HPV binding to HSPGs. The transport
protein particle (TRAPP) complex regulates multiple membrane
trafficking pathways and TRAPPC8 is one of its subunits expressed
in human tissues. As proposed, TRAPPC8-dependent endocytosis
transports the capsid into the cell. After conformational
rearrangements of the capsid, the L2 N-terminus is exposed and
interacts with TRAPPC8, blocking the function of TRAPPC8. As a
result, the endosome is degraded and the viral genome is released
from the trans-Golgi network (21). In conclusion, L1 and L2 proteins are
responsible for the virion assembly and are involved in crucial
interactions with cellular macromolecules that facilitate viral
entry to keratinocytes.
E1 protein
The E1 protein acts as a DNA-dependent adenosine
triphosphatase (ATPase) and DNA helicase; these enzymatic
activities are required for the initiation of viral DNA
replication. E1 protein forms a complex with E2 protein that binds
to the origin of replication of the viral DNA, which contains
binding sites for both proteins. In particular, binding of an E1
dimer to an E2 dimer occurs, forming a complex that binds to the
origin of replication with high specificity. The E2 dimer is
displaced by a second E1 dimer, in an ATP-dependent manner,
resulting in E1 tetramer formation. The next step is the binding of
two E1 monomers to each half of the origin of replication, which
results in the formation of two E1 trimers. The final result is the
formation of two hexamers to the viral origin, each hexamer located
to each half of the origin of replication, that act as a helicase,
unwinding the viral DNA and eventually recruiting the host DNA
polymerase to initiate replication (22,23). A
highly conserved N-terminal region of E1 among different HPV types
was found by deletion analysis, to be necessary for efficient
replication of viral DNA. The above region forms an amphipathic
α-helix (AH) and a specific peptide within binds to the p62 protein
subunit of the human homolog of transcription factor TFHII.
Mutational analysis of the three conserved hydrophobic residues in
the E1 AH, revealed a 50% reduction of the ability of E1 to support
transient replication of DNA in C33A cells (24). Human p80 is an important co-partner
of all mucosal HPV E1 proteins. P80 binds to the N-terminal 40
amino acids of HPV31 E1. Amino acid substitutions in this region,
cause 70% reduction of viral DNA replication. The structure of the
N-terminal 40-amino acid-long peptide originating from HPV31 E1,
was solved by NMR. Overexpression of this 40-amino acid-long
p80-binding peptide, derived from HPV31 E1, led to the inhibition
of viral DNA replication preventing the recruitment of endogenous
p80 to the origin of replication. Thus, the E1-p80 interaction
represents a promising new target for antiviral therapy (22). In another study, the structure of
the DNA-binding domain of E1 HPV18 was resolved by crystallography
and was compared to the respective structure of DNA-binding domain
from E1 of the bovine papillomavirus type 1 (BPV1). The DNA-binding
loop in HPV18 E1 displays a disordered pattern of organization in
contrast to the respective loop in E1 BPV1. The α3 helix of the
HPV18 E1 DNA-binding domain (DBD) is a residue shorter than the
analogous BPV1 E1-DBD α3 helix. The α3 helix is the dimerization
helix and the process of dimerization occurs through its
hydrophobic residues of each monomer, as confirmed by mutational
analysis and ChiP assays (25). In
conclusion, the structure of E1 HPV protein allows binding to
specific origins of replication of viral DNA and its distinct
domains display an ATP-dependent DNA helicase activity that
facilitates viral DNA replication.
E2 protein
Regulatory protein E2 is involved in the initiation
of viral DNA replication through the interaction with E1. E2 acts
as a transcription factor by binding to the E2 response element
(E2RE), which is the major E2-dependent enhancer in the LCR, as
shown in the BPV1 genome (23). The
structure of E2 protein reveals its discrete functions, as far as
cell transformation is concerned. There are three basic domains in
E2 which include the DBD at the C-terminus, the flexible hinge in
the middle and the transactivation domain (TAD) at the N-terminus
of the protein (26). The
structures of the DBDs of different high- and low-risk HPVs were
resolved by crystallography. Moreover, the interaction of the DBD
of E2 proteins with the LCR of the viral DNA was evaluated. It is
interesting to note that the structure of the DBD from the HPV6 E2
protein, shows higher structural homology to the equivalent DBD
from the HPV18 E2 and BPV1 E2 proteins than the DBD from HPV16 E2.
The most common DNA binding site for HPV16 and HPV6 E2 DBDs is the
sequence 5′-AACCGN4CGGTT-3′, where N4
represents a 4-bp central spacer sequence. This central spacer
contains sequences rich in AT that promote DNA bending and
increases protein binding (27).
The interaction of E2 with E1 was studied by the use of small
molecules that bind to the N-terminal TAD of HPV11 E2 and inhibit
its interaction with E1. These inhibitors contain an indandione
system spirofused onto a substituted tetrahydrofuran ring, and
abolish DNA replication. The study of the tertiary structure of the
low-risk HPV11 E2 TAD is interesting, as it resembles the
corresponding domain of HPV16. The crystal structures of the HPV11
E2 TAD with the above inhibitors, were resolved by crystallography
and the specific amino acids that form the inhibitor binding pocket
have been identified. These amino acids are located in a
three-helix formation and amino acid substitutions in this area
alter the binding pocket, by changing the packing of these three
helices (28). DNA binding
properties of HPV6 E2 DBD and a corresponding mutated DBD lacking
two C-terminal leucine residues that form part of the hydrophobic
core of the protein, were compared by employing NMR. It was proven
that the hydrophobic core and loop regions that guide the DNA
binding helices are highly flexible in the mutant. This increased
flexibility resulted in non-specific DNA binding and loss of E2
protein function (29). In
conclusion, the current data indicate that E2 protein facilitates
initiation of viral replication via its interactions with the E1
protein.
E4 protein
E4 protein of HPV16 is expressed as an E1^E4
transcript of 92 amino acids (30).
The process of alternative splicing generates the E1^E4 protein, in
which the initiation codon and the first few amino acids originate
from the E1 open reading frame (31). E4 is detectable in the upper layers
of the epithelium, expressed at the late phases of the HPV life
cycle (23). The N-terminus of
HPV16 E2 protein was proven to interact with the E1^E4 protein
through direct binding, leading to the stabilization of E2 and
translocation from the nucleus to the cytoplasm (32). A mammalian expression system was
used to demonstrate that HPV16 E1^E4 arrests HeLa
(HPV18+) and SiHa (HPV16+) cervical
epithelial cells in the G2 phase. Mutagenesis analysis
revealed an important region rich in prolines in HPV16 E1^E4
responsible for the G2 arrest, which contains a putative
nuclear localization signal, a cyclin-binding motif, and a single
cyclin-dependent kinase phosphorylation site. The arrest domain of
HPV16 E1^E4 is similar to the corresponding E1^E4 of HPV11, a
low-risk mucosal type, which also causes G2 arrest.
However, E1^E4 from HPV1, a low-risk cutaneous type which does not
mediate G2 arrest, exhibits significant differences at
the sequence level when its arrest domain was aligned with E1^E4
from HPV16 and HPV11. This structural difference is responsible for
the inability of HPV1 E1^E4 to induce arrest at the G2
phase (33). Mutational analysis of
HPV1 E4 and HPV16 E4, revealed that the C-terminal domain of HPV16
E1^E4 is crucially involved in the disruption of the keratin
filament network (34). HPV16 E1^E4
protein associates with the keratin intermediate filament network
as it was shown by immunofluorescence microscopy. This conserved
function amongst the HPV Alpha-group E1^E4 proteins, has been
demonstrated by amino acid sequence alignment. Hyperphosphorylated
and ubiquitinated E1^E4-keratin structures have been documented,
resulting in the impairment of the proteasome and causing the
disruption of the keratin network (35). In conclusion, E4 protein is
expressed in the upper layers of the cervical epithelium and its
biological functions involve cell cycle arrest and disruption of
keratin filaments.
E5 protein
HPV16 E5 is a transmembrane protein of 83 amino
acids, localized in the Golgi apparatus (GA) and the endoplasmic
reticulum (ER). The protein folds in three hydrophobic domains
which contain a high percentage (37%) of α-helical structure
(36). HPV16 E5 regulates growth
signaling pathways as it activates the epidermal growth factor
receptor (EGFR) and the downstream Ras-Raf-MAP kinase pathway or
the PI3K-Akt pathway that lead to altered cell proliferation,
angiogenesis and inhibition of apoptosis (37). The HPV E5 protein has been shown to
contribute to immune evasion as it interacts with MHC/HLA class I,
at the GA, where it accumulates. As a result, HPV16 E5 prevents
transport of MHC-I to the cell surface and retains the complex in
the GA, attenuating the recognition of a viral epitope by natural
killer (NK) cells (37,38). The biological effect of E5 is mainly
mediated by its interaction with the HLA-I heavy chain.
Furthermore, the first hydrophobic domain is absolutely required
for the downregulation of surface HLA-I and interaction with the
heavy chain (38). In another
study, the pH status of different intracellular compartments was
tested upon the presence of the E5 protein. The transduction of
human keratinocytes with an E5 vector increased endosomal pH from
5.9 to 6.9, but it did not affect the normal trans-Golgi pH
of 6.3. Endosomal location of E5 increases pH and presumably
enhances EGFR signaling (39). The
expression pattern of HPV16 and HPV18 E5 was evaluated by creating
specific 3H-labeled riboprobes targeting the
corresponding mRNAs. The E5 mRNA and protein of these HPV types
were found in anogenital LSIL but not in cervical tumors,
suggesting a role of E5 in the early stages of HPV infection
(40). A proposed mechanism for the
interference of HPV16 E5 in apoptotic pathways has been described.
HPV16 E5 is responsible for an antiapoptotic effect by impairing
tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and
Fas ligand (FasL)-mediated apoptosis in two different ways.
Firstly, HPV16 E5 downregulates the levels of Fas receptor in the
cell surface, thus preventing its interaction to its corresponding
ligand. Secondly, HPV16 E5 prevents the formation of the
death-inducing signalling complex (DISC) which is induced by TRAIL
(41). One important target of
HPV16 E5 was proven to be interferon β-1 (IFN-β1), its expression
known to be facilitated by the increased interferon regulatory
factor 1 (IRF-1) protein accumulation in human keratinocytes. A
proposed model of action during the episomal infection of HPV,
assumes that HPV16 E5 stimulates IFN-β1 and IRF-1 expression that
lead to episomal clearance. Following this, only transcripts of
integrated viral genes remain, E5 expression is abolished and the
IFN-IRF-1 mediated response is shut down. Meanwhile, high levels of
E6 and E7 oncogenes suppress the signaling pathway of IFN (42). In a comparative study of BPV1 E5 and
HPV16 E5, the transforming properties of the two different E5
proteins were evaluated. Epitope-tagged BPV1 E5 and HPV16 E5
proteins were expressed at the same level in fibroblast cell lines.
However, only BPV1 E5 but not HPV16 E5 could activate growth factor
receptors, phosphoinositide 3-kinase or c-Src. In fibroblast cell
line NIH 3T3 co-cultured with vectors expressing either BPV1 E5 or
HPV16 E5, only cells that expressed BPV1 E5 formed visible foci,
underscoring the limited proliferating capacity of cells expressing
HPV16 E5 (43). E5 has also been
shown to be responsible for cell transformation when acting with
other oncoproteins. For example, with the contribution of E6 it
results in the formation of koilocytes, as was demonstrated in
human cervical cells in vitro. Koilocytes, which are
squamous epithelial cells that contain an acentric, hyperchromatic
nucleus that is displaced by a large perinuclear halo, constitute a
morphological marker of HPV infection (44). A proposed model for the formation of
koilocytes includes E5-induced translocation of calpactin I to the
perinuclear region which promotes perinuclear membrane fusion
(45). A novel partner for HPV31 E5
seems to be the proteolipid protein A4 which is localized to the ER
with E5. This interaction facilitates the differentiation of
keratinocytes, as was confirmed in organotypic raft cultures, with
these cells expressing high levels of A4 (46).
The role of HPV16 E5 was also demonstrated in the
HaCaT human keratinocyte model. E5-expressing cells formed a highly
abnormal epithelium, where many cells exhibited characteristics of
cells derived from the basal membrane, such as production of matrix
metalloproteases (MMPs). It was documented by mutational analysis,
that the first hydrophobic domain of E5 is required for invasion.
The four C-terminal amino acids induce the production of MMP9,
presumably by increasing the levels of transcription factor AP-1
that binds to the MMP9 promoter (47). In conclusion, E5 protein is
responsible for immune system evasion of the HPV, induction of
apoptosis, cell transformation and episomal clearance.
E6 protein
HPV E6 proteins are quite small in size of ~150
amino acids, and include two zinc-binding domains E6N and E6C
(48). The structure of the E6
homodimer was characterized by NMR where it was shown that E6 dimer
disruption through mutations at the E6N domain, enhances E6
solubility and subsequent reduction of p53 degradation potential.
The E6N and E6C domain structures were resolved by NMR studies
demonstrating that E6 specifically dimerizes through its N-terminal
domain. It has been proposed that each E6 molecule of the dimer
binds to one molecule of ubiquitin ligase E6-associated protein
(E6AP) and to one molecule of p53 in order to promote the
polyubiquitination of p53 by E6AP, resulting in p53 degradation
(Fig. 1). Dimers of E6 are formed
in high-risk mucosal HPV16 and HPV18 types, as well as in low-risk
mucosal type HPV11, whereas E6 from cutaneous strains HPV5 and BPV1
is monomeric. However, only the dimeric E6 protein from oncogenic
types HPV16 and HPV18 has the ability to polyubiquitinate p53. The
structural basis for the inability of the HPV11 E6 dimer to target
p53 for degradation is currently under investigation (49). E6 protein from high-risk HPV types
also binds to proteins containing PDZ domains and targets them for
degradation. The interaction of the complex of the human homologue
of the Drosophila discs large tumor suppressor protein
(hDlg) bound to HPV18 E6 was analyzed by NMR (50). The structure of the second
zinc-binding domain (51Z2) of the high-risk HPV51 E6 protein was
also resolved by NMR spectroscopy. Specifically, circular dichroism
spectroscopy revealed an increased flexibility of the above area of
E6, indicating that this may be the structural feature of E6
oncoproteins. Another study investigated the E6AP binding site of
E6 protein in HPV16. The conclusion was that an inhibitory ligand
can bind to E6 protein at its E6AP binding pocket, restore p53
expression and induce apoptosis in HPV16 positive cells. This
important finding opens new perspectives for drug development
against HPV (51). In a subsequent
study, more details were provided concerning the specific
interactions of E6, E6AP and p53. In particular, the crystal
structure of the ternary complex containing the proteins of
interest HPV16 E6, the LxxLL motif of E6AP and the core domain of
p53, was determined. The LxxLL motif is a short leucine (L)-rich
LxxLL consensus sequence within the ubiquitin ligase domain of E6AP
that binds to E6. The formation of the ternary complex results in
ubiquitin-mediated p53 degradation. Mutations of E6 residues that
establish interactions with p53 abolish complex formation and p53
degradation (52). An equally
important function of the E6 protein is the upregulation of hTERT
telomerase expression; only high-risk HPV E6 proteins bind to the
promoter of hTERT telomerase and induce its expression. Another
interesting function of E6 proteins both in high- and in low-risk
HPV types, is their ability to interact with caspase 8, through the
DED region of caspase. The result is the localization of caspase 8
to the nucleus, as shown by binding assays and immunofluorescence
experiments. The nuclear localization of caspase 8 is required for
the facilitation of the viral cycle and the proliferation of cells
that express E6, presumably through regulation of apoptotic signals
(53). In conclusion, E6 interacts
with p53 and proteins with PDZ domains leading to their
degradation, while increases hTERT expression, and thus promoting
cell cycle deregulation and subsequent malignant
transformation.
E7 protein
The structure of the HPV18 E7 protein contains three
discrete domains, CD1, CD2 and CD3. These domains are also called
CR1, CR2 and CR3, respectively. E7 protein has one Zn2+
ion which coordinates to four cysteines at the C-terminal domain
through the two Cys-X–X-Cys motifs located in the CD3 domain, thus
forming a hydrophobic core. Zn2+ plays a crucial role in
the maintenance of structural integrity of HPV18 E7 and is
absolutely required for proper folding and thermal stability of the
protein. The N-terminal domain is flexible and binds to the pRb
protein via its LXCXE motif, which is located in the CD2 region
(54). In a crystallography study
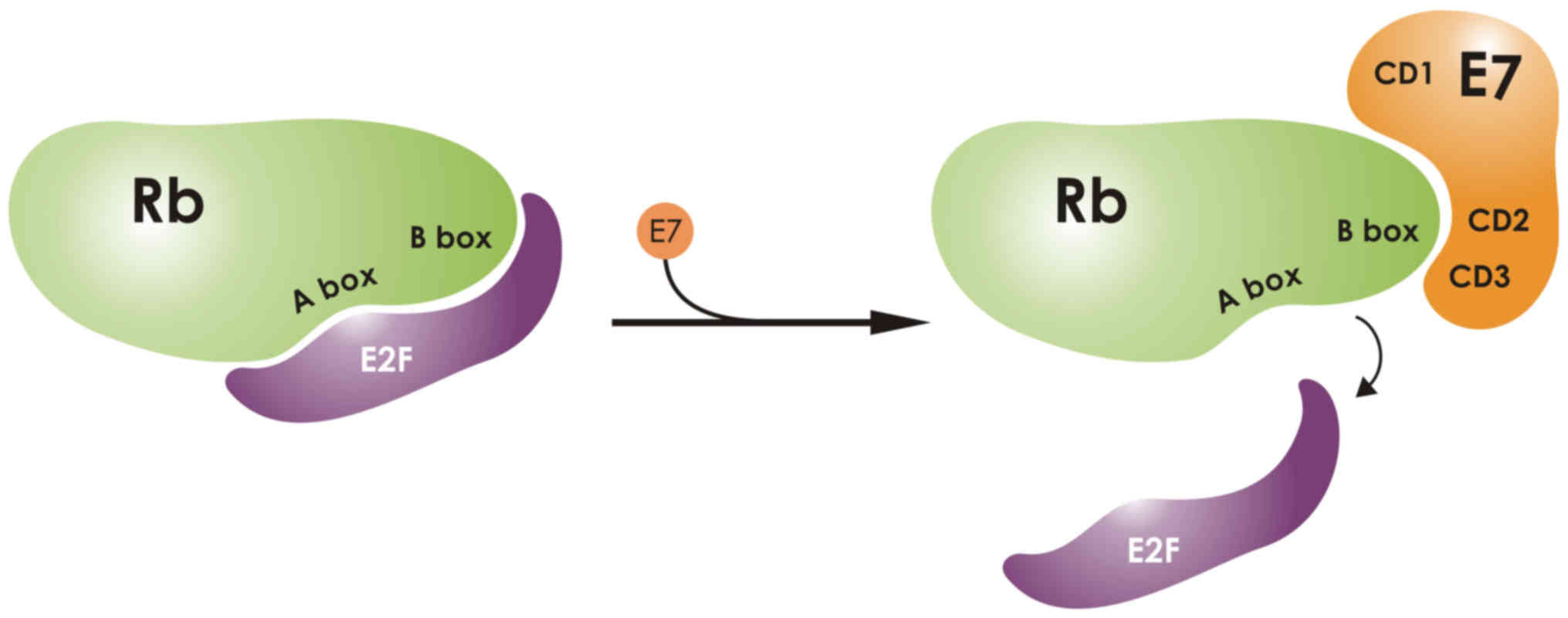
of the CD3 domain of E7, a zinc binding domain was characterized
and was implicated in the dissociation of pRb from E2F
transcription factors and the early cell progression into the S
phase of the cell cycle. By performing mutational analysis, two
specific binding sites were discovered in the CD3 region. The first
is necessary for pRb binding, whereas the other is necessary for
E2F binding (Fig. 2). Interaction
of pRb with E7 is necessary to disrupt the pRb-E2F complex
(55). A crystallography analysis
of the pocket domain of the retinoblastoma (Rb) protein, revealed
its interaction with a nine-residue E7 peptide containing the LxCxE
motif. The crystallography study revealed the presence of two boxes
in the Rb pocket, the A- and the B-box. The E7 LxCxE was shown to
bind to an extended peptide onto a conserved groove of the B Box
(Fig. 2). A recent study links the
conformational status of HPV16 E7 to functional redox roles, as the
protein contains seven highly conserved cysteines. These are
modified under oxidative stress that characterizes HPV-transformed
cells. There are two distinct redox centers, cysteine 24 located in
the E7N domain, which contains the Rb binding site, and also
cysteines 59/68 located in the E7C domain, which contains the
Zn2+ binding site. Under oxidative stress,
glutathionylation of cysteine 24 abolishes Rb-binding. Binding of
Zn2+ to cysteine residues 59 and 68, mediates structural
rearrangements leading to disulfide bridging of the above cysteines
(56). Moreover, it was
demonstrated that HPV8 E7 is imported in the nucleus of HeLa cells,
following transfection with enhanced green fluorescent protein 8E7
(EGFP-8E7), and that the actual role of cysteine residues seems to
be significant. Mutations in cysteine residues involved in zinc
coordination alter the nuclear localization of EGFP-8E7 (57). The entire structure of HPV16 E7
protein was recently resolved by NMR studies, which revealed that
the E7 heterogeneity and dynamics, containing a flexible N-terminus
and a more structured C-terminus, may be responsible for the
oncogenic potential of E7 and its interactions with other important
biomolecules (58). These
interactions of E7 with its protein targets result in uncontrolled
cell division and subsequent malignant transformation.
Potential for targeted therapy
Delineation of the interactions of HPV oncoproteins
E6 and E7 with their multiple cellular targets, can provide the
impetus for an eventual targeted drug therapy for cervical cancer.
Specifically, an in silico study suggested that plant
compounds could be used to treat cervical cancer since they can
block the binding of HPV18 E6 to p53, and thus prevent its
degradation (59). Moreover,
competitive antagonist ligands, i.e. benzopyranone derivatives,
that disrupt the E6-E6AP interaction, were developed in order to
inhibit the formation of the complex containing E6, E6AP and p53
(Fig. 1). These promising
anticancer agents were used in binding and functional assays
(60). Another promising agent for
cervical cancer therapy is a peptide that targets the E6AP binding
pocket of E6 from HPV16 and prevents p53 degradation (61). As far as therapy via targeting of E7
is concerned, an in vitro characterization and screening of
compounds that inhibited the ability of HPV-E7 to disrupt pRB/E2F
complexes, has been recently conducted. Thiadiazolidinedione-based
molecules exhibited the highest affinity for E7 and effectively
prevented its binding to pRB. In addition, some of these compounds
were proven to induce apoptosis in HeLa and SiHa cervical cell
lines. It is important to note that one molecule was tested in mice
injected with the TC-1 lung cancer-derived cell line and was found
to significantly reduce the tumor volume without any adverse
effects (62).
The elucidation of the structural features of the E6
oncoprotein responsible for establishing interactions with protein
targets, has provided the basis for potential pharmacological
interventions. In particular, the PDZ-binding motif (PBM) of E6, is
involved in the formation of complexes with proteins containing PDZ
domains and leads to their degradation (50). An in silico analysis revealed
four compounds that could disrupt the interaction of the E6 PBM
with the PDZ domain. These compounds target specifically the PDZ
domain via the interaction with Gly463 and Phe464 (63). However, the in silico
prediction has to be validated experimentally and thus, the
molecular therapy of targeting the PBM, is currently at an early
stage of development. As previously demonstrated by Zanier et
al (51), eventual molecular
therapy for cervical cancer can also be developed by disrupting the
interactions of the E6AP binding site and the E6 protein of HPV16,
via an inhibitory ligand that binds to E6 protein. Furthermore,
in vitro experiments by the same group, indicate that p53
expression can be restored by treating HPV16-positive cells with
selected compounds, leading to apoptosis (49). In a related study, the interaction
of the HPV16 E6 oncoptotein with regulators of apoptosis caspase 8
and E6AP was targeted, in order to reveal putative drugs. Screening
of compound libraries for such putative inhibitors was performed
and two compounds, myricetin and spinacine, were found to inhibit
the binding of E6 to caspase 8 and E6AP. These two molecules had a
significant cytotoxic effect specifically for the HPV16-positive
cervical cancer cell line SiHa (64). These compounds exhibit
IC50 value in the µΜ range and offer a proof of
principle for therapeutic targeting of HPV E6. Finally, an
additional study was based on screening a library of 88,000
compounds for inhibitors of the E6-E6AP interaction. Seven
inhibitors were identified by cell-free ELISA assays and two
compounds exhibited significant cytotoxic activity towards SiHa and
HeLa (HPV18-positive) cells. It is important to note that treatment
of these cervical cancer cell lines with the two compounds restored
p53 expression and the IC50 values reached the nM range
(65). This seminal study opens the
way for testing the efficacy of such promising compounds for the
treatment of cervical cancer.
Therefore, besides the functional studies on
informative cervical cell lines in vitro (66,67),
further efforts are also needed employing appropriate in
vivo models of cervical cancer in order to fully evaluate the
effectiveness of these novel targeted therapies.
Perspectives
Structural studies have revealed important
differences between HPV proteins from high- and low-risk HPV types.
The structural differences are responsible for different binding
partners and offer insight into the mechanisms of HPV-mediated
carcinogenesis. In particular, E6 and E7 proteins from high-risk
HPV types are responsible for inducing uncontrolled cell
proliferation. Only high-risk E6 binds p53 and targets it for
ubiquitination, whereas high-risk E7 binds the non-phosphorylated
form of the Rb protein and facilitates the cell to bypass the
G1/S checkpoint.
The structural information on HPV proteins can be
used to facilitate research for the development of novel
therapeutic targets and new types of vaccines for the prevention
and eventual therapy of cervical cancer. In conclusion, insight
from the structural studies of HPV proteins has contributed to the
elucidation of molecular mechanisms associated with cervical
carcinogenesis and may open the way for the development of novel
therapeutic approaches.
Acknowledgements
The present review was co-funded by the European
Union [European Social Fund and Greek National Fund (ESF)], through
the Program THALIS, under the Operational Program Education and
Lifelong Learning of the National Strategic Reference Framework
(NSRF), project no. 383418, grant no. 70-3-11830 to K.I.P.
References
|
1
|
Hoory T, Monie A, Gravitt P and Wu TC:
Molecular epidemiology of human papillomavirus. J Formos Med Assoc.
107:198–217. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Muñoz N, Bosch FX, Castellsagué X, Díaz M,
de Sanjose S, Hammouda D, Shah KV and Meijer CJ: Against which
human papillomavirus types shall we vaccinate and screen? The
international perspective. Int J Cancer. 111:278–285. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Doorbar J, Quint W, Banks L, Bravo IG,
Stoler M, Broker TR and Stanley MA: The biology and life-cycle of
human papillomaviruses. Vaccine. 30 Suppl 5:F55–F70. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stanley MA: Epithelial cell responses to
infection with human papillomavirus. Clin Microbiol Rev.
25:215–222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McBride AA: Replication and partitioning
of papillomavirus genomes. Adv Virus Res. 72:155–205. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dueñas-González A and Campbell S: Global
strategies for the treatment of early-stage and advanced cervical
cancer. Curr Opin Obstet Gynecol. 28:11–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schiffman M, Doorbar J, Wentzensen N, de
Sanjosé S, Fakhry C, Monk BJ, Stanley MA and Franceschi S:
Carcinogenic human papillomavirus infection. Nat Rev Dis Primers.
2:160862016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Joyce JG, Tung JS, Przysiecki CT, Cook JC,
Lehman ED, Sands JA, Jansen KU and Keller PM: The L1 major capsid
protein of human papillomavirus type 11 recombinant virus-like
particles interacts with heparin and cell-surface
glycosaminoglycans on human keratinocytes. J Biol Chem.
274:5810–5822. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Evander M, Frazer IH, Payne E, Qi YM,
Hengst K and McMillan NA: Identification of the alpha6 integrin as
a candidate receptor for papillomaviruses. J Virol. 71:2449–2456.
1997.PubMed/NCBI
|
|
10
|
Cardone G, Moyer AL, Cheng N, Thompson CD,
Dvoretzky I, Lowy DR, Schiller JT, Steven AC, Buck CB and Trus BL:
Maturation of the human papillomavirus 16 capsid. MBio.
5:e01104–e01114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ryndock EJ, Conway MJ, Alam S, Gul S,
Murad S, Christensen ND and Meyers C: Roles for human
papillomavirus type 16 l1 cysteine residues 161, 229, and 379 in
genome encapsidation and capsid stability. PLoS One. 9:e994882014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Joshi H, Cheluvaraja S, Somogyi E, Brown
DR and Ortoleva P: A molecular dynamics study of loop fluctuation
in human papillomavirus type 16 virus-like particles: A possible
indicator of immunogenicity. Vaccine. 29:9423–9430. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bishop B, Dasgupta J, Klein M, Garcea RL,
Christensen ND, Zhao R and Chen XS: Crystal structures of four
types of human papillomavirus L1 capsid proteins: Understanding the
specificity of neutralizing monoclonal antibodies. J Biol Chem.
282:31803–31811. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mistry N, Wibom C and Evander M: Cutaneous
and mucosal human papillomaviruses differ in net surface charge,
potential impact on tropism. Virol J. 5:1182008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Buck CB, Cheng N, Thompson CD, Lowy DR,
Steven AC, Schiller JT and Trus BL: Arrangement of L2 within the
papillomavirus capsid. J Virol. 82:5190–5197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schneider MA, Spoden GA, Florin L and
Lambert C: Identification of the dynein light chains required for
human papillomavirus infection. Cell Microbiol. 13:32–46. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Darshan MS, Lucchi J, Harding E and
Moroianu J: The l2 minor capsid protein of human papillomavirus
type 16 interacts with a network of nuclear import receptors. J
Virol. 78:12179–12188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lowe J, Panda D, Rose S, Jensen T, Hughes
WA, Tso FY and Angeletti PC: Evolutionary and structural analyses
of alpha-papillomavirus capsid proteins yields novel insights into
L2 structure and interaction with L1. Virol J. 5:1502008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raff AB, Woodham AW, Raff LM, Skeate JG,
Yan L, Da Silva DM, Schelhaas M and Kast WM: The evolving field of
human papillomavirus receptor research: A review of binding and
entry. J Virol. 87:6062–6072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Woodham AW, Da Silva DM, Skeate JG, Raff
AB, Ambroso MR, Brand HE, Isas JM, Langen R and Kast WM: The
S100A10 subunit of the annexin A2 heterotetramer facilitates
L2-mediated human papillomavirus infection. PLoS One. 7:e435192012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ishii Y, Nakahara T, Kataoka M,
Kusumoto-Matsuo R, Mori S, Takeuchi T and Kukimoto I:
Identification of TRAPPC8 as a host factor required for human
papillomavirus cell entry. PLoS One. 8:e802972013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lehoux M, Fradet-Turcotte A, Lussier-Price
M, Omichinski JG and Archambault J: Inhibition of human
papillomavirus DNA replication by an E1-derived p80/UAF1-binding
peptide. J Virol. 86:3486–3500. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peter M: Howley DRL: Papillomaviruses.
Journal II. 1–2354. 2007.
|
|
24
|
Morin G, Fradet-Turcotte A, Di Lello P,
Bergeron-Labrecque F, Omichinski JG and Archambault J: A conserved
amphipathic helix in the N-terminal regulatory region of the
papillomavirus E1 helicase is required for efficient viral DNA
replication. J Virol. 85:5287–5300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Auster AS and Joshua-Tor L: The
DNA-binding domain of human papillomavirus type 18 E1. Crystal
structure, dimerization, and DNA binding. J Biol Chem.
279:3733–3742. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bellanger S, Tan CL, Xue YZ, Teissier S
and Thierry F: Tumor suppressor or oncogene? A critical role of the
human papillomavirus (HPV) E2 protein in cervical cancer
progression. Am J Cancer Res. 1:373–389. 2011.PubMed/NCBI
|
|
27
|
Dell G, Wilkinson KW, Tranter R, Parish J,
Leo Brady R and Gaston K: Comparison of the structure and
DNA-binding properties of the E2 proteins from an oncogenic and a
non-oncogenic human papillomavirus. J Mol Biol. 334:979–991. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Coulombe R, Cameron DR, Thauvette
L, Massariol MJ, Amon LM, Fink D, Titolo S, Welchner E, Yoakim C,
et al: Crystal structure of the E2 transactivation domain of human
papillomavirus type 11 bound to a protein interaction inhibitor. J
Biol Chem. 279:6976–6985. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brown C, Campos-León K, Strickland M,
Williams C, Fairweather V, Brady RL, Crump MP and Gaston K: Protein
flexibility directs DNA recognition by the papillomavirus E2
proteins. Nucleic Acids Res. 39:2969–2980. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakahara T, Peh WL, Doorbar J, Lee D and
Lambert PF: Human papillomavirus type 16 E1circumflexE4 contributes
to multiple facets of the papillomavirus life cycle. J Virol.
79:13150–13165. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Doorbar J: The E4 protein; structure,
function and patterns of expression. Virology. 445:80–98. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Davy C, McIntosh P, Jackson DJ, Sorathia
R, Miell M, Wang Q, Khan J, Soneji Y and Doorbar J: A novel
interaction between the human papillomavirus type 16 E2 and E1^E4
proteins leads to stabilization of E2. Virology. 394:266–275. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Davy CE, Jackson DJ, Wang Q, Raj K,
Masterson PJ, Fenner NF, Southern S, Cuthill S, Millar JB and
Doorbar J: Identification of a G2 arrest domain in the
E1 wedge E4 protein of human papillomavirus type 16. J Virol.
76:9806–9818. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Roberts S, Ashmole I, Rookes SM and
Gallimore PH: Mutational analysis of the human papillomavirus type
16 E1^E4 protein shows that the C terminus is dispensable for
keratin cytoskeleton association but is involved in inducing
disruption of the keratin filaments. J Virol. 71:3554–3562.
1997.PubMed/NCBI
|
|
35
|
McIntosh PB, Laskey P, Sullivan K, Davy C,
Wang Q, Jackson DJ, Griffin HM and Doorbar J: E1^E4-mediated
keratin phosphorylation and ubiquitylation: A mechanism for keratin
depletion in HPV16-infected epithelium. J Cell Sci. 123:2810–2822.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang DH, Wildeman AG and Sharom FJ:
Overexpression, purification, and structural analysis of the
hydrophobic E5 protein from human papillomavirus type 16. Protein
Expr Purif. 30:1–10. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Venuti A, Paolini F, Nasir L, Corteggio A,
Roperto S, Campo MS and Borzacchiello G: Papillomavirus E5: The
smallest oncoprotein with many functions. Mol Cancer. 10:1402011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ashrafi GH, Haghshenas M, Marchetti B and
Campo MS: E5 protein of human papillomavirus 16 downregulates HLA
class I and interacts with the heavy chain via its first
hydrophobic domain. Int J Cancer. 119:2105–2112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Disbrow GL, Hanover JA and Schlegel R:
Endoplasmic reticulum-localized human papillomavirus type 16 E5
protein alters endosomal pH but not trans-Golgi pH. J Virol.
79:5839–5846. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stoler MH, Rhodes CR, Whitbeck A, Wolinsky
SM, Chow LT and Broker TR: Human papillomavirus type 16 and 18 gene
expression in cervical neoplasias. Hum Pathol. 23:117–128. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kabsch K and Alonso A: The human
papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated
apoptosis in HaCaT cells by different mechanisms. J Virol.
76:12162–12172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Muto V, Stellacci E, Lamberti AG, Perrotti
E, Carrabba A, Matera G, Sgarbanti M, Battistini A, Liberto MC and
Focà A: Human papillomavirus type 16 E5 protein induces expression
of beta interferon through interferon regulatory factor 1 in human
keratinocytes. J Virol. 85:5070–5080. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Suprynowicz FA, Disbrow GL, Simic V and
Schlegel R: Are transforming properties of the bovine
papillomavirus E5 protein shared by E5 from high-risk human
papillomavirus type 16? Virology. 332:102–113. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Krawczyk E, Suprynowicz FA, Liu X, Dai Y,
Hartmann DP, Hanover J and Schlegel R: Koilocytosis: A cooperative
interaction between the human papillomavirus E5 and E6
oncoproteins. Am J Pathol. 173:682–688. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Krawczyk E, Suprynowicz FA, Hebert JD,
Kamonjoh CM and Schlegel R: The human papillomavirus type 16 E5
oncoprotein translocates calpactin I to the perinuclear region. J
Virol. 85:10968–10975. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kotnik Halavaty K, Regan J, Mehta K and
Laimins L: Human papillomavirus E5 oncoproteins bind the A4
endoplasmic reticulum protein to regulate proliferative ability
upon differentiation. Virology 452–453. 1–230. 2014.
|
|
47
|
Barbaresi S, Cortese MS, Quinn J, Ashrafi
GH, Graham SV and Campo MS: Effects of human papillomavirus type 16
E5 deletion mutants on epithelial morphology: Functional
characterization of each transmembrane domain. J Gen Virol.
91:521–530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nominé Y, Charbonnier S, Ristriani T,
Stier G, Masson M, Cavusoglu N, Van Dorsselaer A, Weiss E, Kieffer
B and Travé G: Domain substructure of HPV E6 oncoprotein:
Biophysical characterization of the E6 C-terminal DNA-binding
domain. Biochemistry. 42:4909–4917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zanier K, Ould M'hamed ould Sidi A,
Boulade-Ladame C, Rybin V, Chappelle A, Atkinson A, Kieffer B and
Travé G: Solution structure analysis of the HPV16 E6 oncoprotein
reveals a self-association mechanism required for E6-mediated
degradation of p53. Structure. 20:604–617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu Y, Henry GD, Hegde RS and Baleja JD:
Solution structure of the hDlg/SAP97 PDZ2 domain and its mechanism
of interaction with HPV-18 papillomavirus E6 protein. Biochemistry.
46:10864–10874. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zanier K, Stutz C, Kintscher S, Reinz E,
Sehr P, Bulkescher J, Hoppe-Seyler K, Travé G and Hoppe-Seyler F:
The E6AP binding pocket of the HPV16 E6 oncoprotein provides a
docking site for a small inhibitory peptide unrelated to E6AP,
indicating druggability of E6. PLoS One. 9:e1125142014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Martinez-Zapien D, Ruiz FX, Poirson J,
Mitschler A, Ramirez J, Forster A, Cousido-Siah A, Masson M, Vande
Pol S, Podjarny A, et al: Structure of the E6/E6AP/p53 complex
required for HPV-mediated degradation of p53. Nature. 529:541–545.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Manzo-Merino J, Massimi P, Lizano M and
Banks L: The human papillomavirus (HPV) E6 oncoproteins promotes
nuclear localization of active caspase 8. Virology 450–451. 1–152.
2014.
|
|
54
|
Liu S, Tian Y, Greenaway FT and Sun MZ: A
C-terminal hydrophobic, solvent-protected core and a flexible
N-terminus are potentially required for human papillomavirus 18 E7
protein functionality. Biochimie. 92:901–908. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu X, Clements A, Zhao K and Marmorstein
R: Structure of the human Papillomavirus E7 oncoprotein and its
mechanism for inactivation of the retinoblastoma tumor suppressor.
J Biol Chem. 281:578–586. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chemes LB, Camporeale G, Sánchez IE, de
Prat-Gay G and Alonso LG: Cysteine-rich positions outside the
structural zinc motif of human papillomavirus E7 provide
conformational modulation and suggest functional redox roles.
Biochemistry. 53:1680–1696. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Onder Z and Moroianu J: Nuclear import of
cutaneous beta genus HPV8 E7 oncoprotein is mediated by hydrophobic
interactions between its zinc-binding domain and FG nucleoporins.
Virology. 449:150–162. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Calçada EO, Felli IC, Hošek T and
Pierattelli R: The heterogeneous structural behavior of E7 from
HPV16 revealed by NMR spectroscopy. ChemBioChem. 14:1876–1882.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kumar S, Jena L, Sahoo M, Kakde M, Daf S
and Varma AK: In silico docking to explicate interface between
plant-originated inhibitors and E6 oncogenic protein of highly
threatening human papillomavirus 18. Genomics Inform. 13:60–67.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rietz A, Petrov DP, Bartolowits M, DeSmet
M, Davisson VJ and Androphy EJ: Molecular probing of the HPV-16 E6
protein alpha helix binding groove with small molecule inhibitors.
PLoS One. 11:e01498452016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stutz C, Reinz E, Honegger A, Bulkescher
J, Schweizer J, Zanier K, Travé G, Lohrey C, Hoppe-Seyler K and
Hoppe-Seyler F: Intracellular analysis of the interaction between
the human papillomavirus type 16 E6 oncoprotein and inhibitory
peptides. PLoS One. 10:e01323392015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Fera D, Schultz DC, Hodawadekar S,
Reichman M, Donover PS, Melvin J, Troutman S, Kissil JL, Huryn DM
and Marmorstein R: Identification and characterization of small
molecule antagonists of pRb inactivation by viral oncoproteins.
Chem Biol. 19:518–528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tian YS, Kawashita N, Arai Y, Okamoto K
and Takagi T: Pharmacophore modeling and molecular docking studies
of potential inhibitors to E6 PBM-PDZ from human papilloma virus
(HPV). Bioinformation. 11:401–406. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yuan CH, Filippova M, Krstenansky JL and
Duerksen-Hughes PJ: Flavonol and imidazole derivatives block HPV16
E6 activities and reactivate apoptotic pathways in HPV+
cells. Cell Death Dis. 7:20602016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Malecka KA, Fera D, Schultz DC,
Hodawadekar S, Reichman M, Donover PS, Murphy ME and Marmorstein R:
Identification and characterization of small molecule human
papillomavirus E6 inhibitors. ACS Chem Biol. 9:1603–1612. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kontostathi G, Zoidakis J, Makridakis M,
Lygirou V, Mermelekas G, Papadopoulos T, Vougas K, Vlamis-Gardikas
A, Drakakis P, Loutradis D, et al: Cervical cancer cell line
secretome analysis highlights the role of transforming growth
factor-beta-induced protein ig-h3, peroxiredoxin-2 and NRF2 on
cervical cancer carcinogenesis. BioMed Res Int. 2017:41807032017.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pappa KI, Lygirou V, Kontostathi G,
Zoidakis J, Makridakis M, Vougas K, Daskalakis G, Polyzos A and
Anagnou NP: Proteomic analysis of normal and cancer cervical cell
lines reveals deregulation of cytoskeleton-associated proteins.
Cancer Genomics Proteomics. 14:253–266. 2017. View Article : Google Scholar : PubMed/NCBI
|