Introduction
In recent years, breast cancer has become the
leading cause of death among Chinese females, with increasing
morbidity and mortality (1).
Although molecular subtype, grade and lymph node involvement all
influence the prognosis of breast cancer patients, uncontrolled
metastasis coupled with treatment failure, rather than the primary
tumour, is the primary cause of death and the 5-year survival rate
is <25% (2). The treatment of
metastatic breast cancer remains a challenge. Circulating tumour
cells (CTCs) are metastasis-initiating precursors and independent
prognostic factors (3) that closely
correlate with treatment response and tumour recurrence.
Circulating breast cancer cells possess increased
mobility and epithelial-mesenchymal biphenotypic status compared
with anchored tumour cells. These differences may be due to the
different selection pressures placed on the local and disseminated
cancer cells by the different microenvironments. Avoiding
extinction through the selected clonal expansion that occurs in the
tumour bulk, disseminated CTCs not only retain several genetic
characteristics from primary or metastatic tumours but also exhibit
a large number of unique mutations, which may be a reasonable
explanation for the observed extreme genomic heterogeneity
exhibited by the disseminated cancer cells before the manifestation
of metastasis (4–6). Such CTC restricted mutations, which
are often low-frequency ‘driver’ mutations and undetectable by bulk
sequencing, are probably the primary mutations that drive CTCs to
detach from their original location and survive in the entirely
unfamiliar flowing blood. Thus, these driver mutations may serve as
latent drug targets.
Studies on bulk breast cancer sequencing identified
the following pathways altered by somatic mutations in cancer
cells: DNA repair, cell cycle or proliferation and lipid metabolism
defects (7). Recent studies on the
molecular profiles of CTCs have focused on shared mutations via
bulk tissue sequencing, rather than decoding whole genome
alterations de novo (5,8), which
may not be conducive to uncovering the mutation characteristics of
CTCs. Hence, it is necessary to investigate the unique mutations in
CTCs, which probably contribute to CTC-specific properties, such as
intravasation, survival in blood, interactions with immune cells
and re-attachment at the sites of metastasis. However, two major
obstacles hinder the programme progression: isolating rare CTCs
from millions of haematocytes and amplifying trace genomes without
bias and maximum coverage of the whole genome. Therefore, CTCs have
not been precisely defined until recently.
In the present study, we constructed a method for
isolating viable CTCs from breast cancer using the oncolytic herpes
simplex virus (oHSV1)-human telomerase reverse transcriptase
(hTERT)-GFP virus coupled with fluorescence-activated cell sorting
(FACS) and analysed their single nucleotide variant (SNV) profiles
through single-cell genome sequencing. The CTC behaviours were then
evaluated based on the SNVs found to be recurrently mutated in
different cells.
Materials and methods
Study procedure
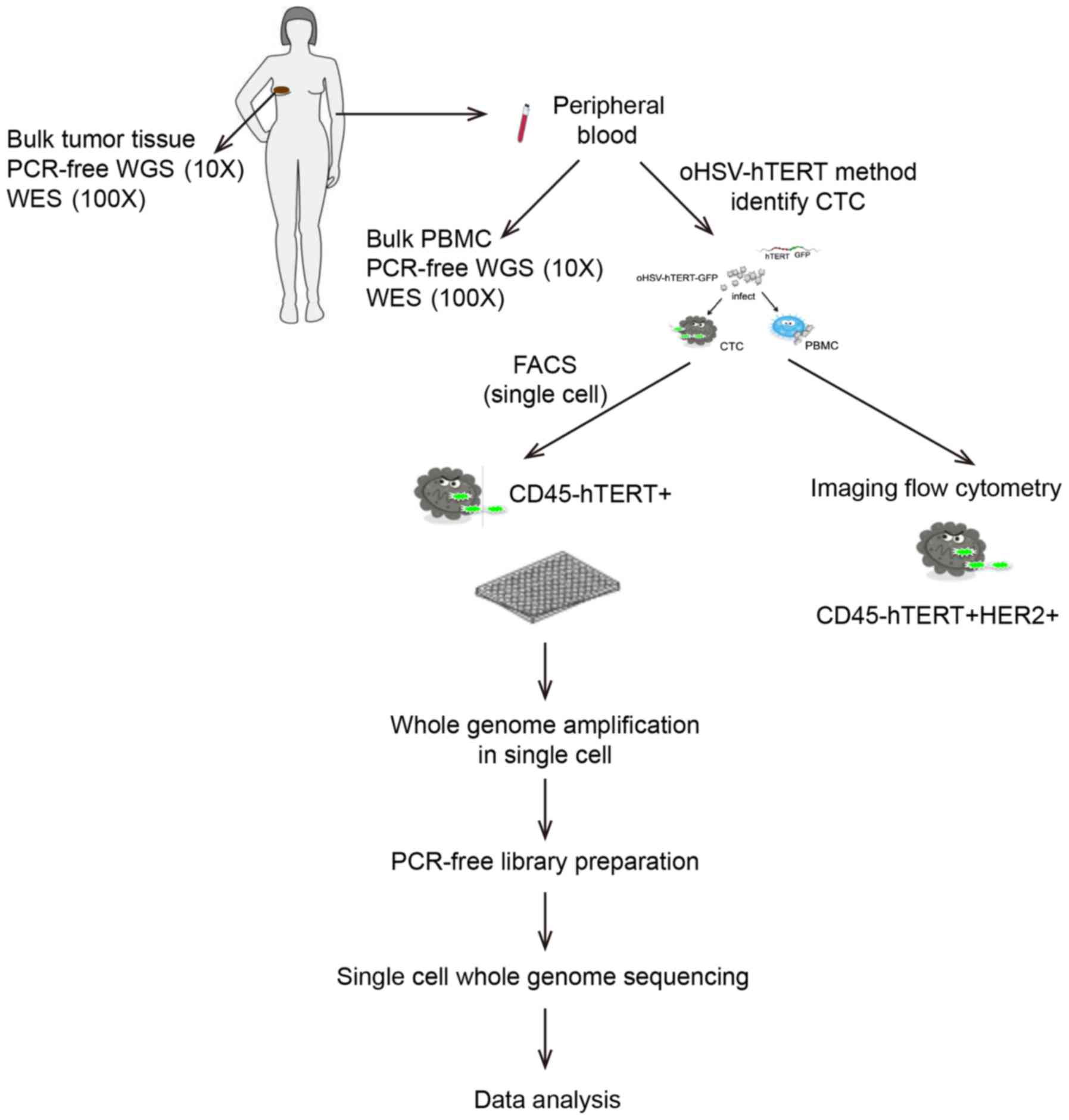
A flowchart of the study procedure is displayed in
Fig. 1.
Patients and specimens
The study involved 9 treatment-naïve patients who
were diagnosed with invasive breast carcinoma at the Cancer
Institute and Hospital of the Chinese Academy of Medical Sciences,
from November 2016 to July 2017. Their clinical information is
presented in Table I. A total of 8
ml of peripheral blood was collected from all patients, except
patient B54, into two K2EDTA vacuum tubes (BD Biosciences, Franklin
Lakes, NJ, USA; cat. no. 367844) prior to surgery, and the
corresponding resected tumour tissue samples were dissected into
small fragments and preserved in 3 ml of RNAlater (Invitrogen,
Carlsbad, CA, USA; cat. no. AM7021) immediately after the surgical
procedure. The blood and tissue samples were delivered on ice to
the laboratory within 1 h. A total of 4 ml of the peripheral blood
sample was used to sort CTCs. The remaining peripheral blood sample
in another tube was used to isolate germline DNA. The tumour tissue
fragments, which were derived from a typical region of the gross
specimen, were used for DNA extraction and histological
verification. In addition, only 4 ml of peripheral blood was
collected from patient B54 for imaging flow cytometry (patient B54
was not included in CTC sorting). The use of human samples and the
experimental procedures for this study were reviewed and approved
by the Ethics Committee of the Cancer Institute and Hospital of the
Chinese Academy of Medical Sciences, with the approval number
16–159/1238. The patients were informed, provided written informed
consent and thoroughly understood the research.
 | Table I.Patient characteristics and sample
information. |
Table I.
Patient characteristics and sample
information.
| Patient ID | CTC ID | Tissue ID | Age (years) | Sex | Pathological
typee | Molecular type | TNM | Countsi |
|---|
| B1 | B1CTC1 | B1Ta | 58 | Fd | BRCA, NST | Lum
B/HER2(−)f | T4N1M1 | 13 |
|
| B1CTC3 |
|
|
|
|
|
|
|
| B4 | B4CTC2 | B4T | 38 | F | BRCA, NST | Lum Ag | T1N2M0 | 27 |
|
| B4CTC3 |
|
|
|
|
|
|
|
|
| B4CTC4 |
|
|
|
|
|
|
|
| B5 | B5CTC3 | B5TA | 60 | F | BRCA, NST | Lum B/HER2(−) | T2N2M0 | 37 |
|
|
| B5TBb |
|
|
|
|
|
|
| B12 | B12CTC | B12T | 34 | F | BRCA, NST | Lum B/HER2(−) | T1N1M0 | 25 |
| B15 | B15CTC | B15T | 46 | F | BRCA, NST | Lum B/HER2(−) | T2mN1M0 | 51 |
| B16 | B16CTC | B16T | 48 | F | BRCA, NST | Lum B/HER2(+) | T1N0M0 | 13 |
| B20 | B20CTC | B20T | 42 | F | BRCA, NST | Lum A | T1N2M0 | 11 |
| B37 | B37CTC | B37T | 34 | F | BRCA, NST | HER2Eh | T2N3M0 | 5 |
|
|
| B37LNc |
|
|
|
|
|
|
| B54 | – | – | 25 | F | BRCA, NST | HER2E | T1N0M0 | – |
Isolating single circulating breast
tumour cells using oHSV1-hTERT-GFP
A total of 4 ml of fresh peripheral blood was
centrifuged for 20 min at 120 × g, and the platelet-rich plasma
phase was subsequently removed. The remaining layers, including the
leucocytes and RBCs, were incubated with erythrocyte lysis buffer
(Qiagen, Hilden, Germany; cat. no. 79217) for 10 min at room
temperature. After centrifugation at 400 × g for 5 min, the
supernatant was discarded, and the cell pellet was washed 2 times
with phosphate-buffered saline (PBS). After additional
centrifugation, the cells were resuspended in RPMI-1640 medium
(cat. no. SH30809.01B; HyClone Laboratories, Logan, UT, USA) and
infected with oHSV1-hTERT-GFP virus at an MOI=1 for 1 h at 37°C,
and the culture continued for another 23 h, and then the cells were
replenished in RPMI-1640 medium with 10% fetal bovine serum (FBS;
Gibco, Carlsbad, CA, USA; cat. no. 10099-141) (9). After staining the cells with 23 µl of
CD45 monoclonal antibody (HI30)-APC (cat. no. 17-0459-42;
eBioscience, San Diego, CA, USA), the cell suspension was evaluated
and sorted using flow cytometry (BD FACSJazz; BD Biosciences) in
single-cell mode. CD45−/hTERT+ cells were
defined as CTCs and sorted into strips of 8 tubes (cat. no.
4316567; Life Technologies, Carlsbad, CA, USA) containing 1X
ThermoPol Reaction Buffer (lysis buffer). In addition, to identify
the gate of the cell population of interest, we used a
single-stained or infected control (negative control, peripheral
blood of a healthy person; positive control, the hTERT-expressing
cancer cell line, SK-BR-3) (Fig.
2A-h-j).
Single-cell whole genomic
amplification (WGA)
Single-cell WGA was performed using the MALBAC
method (10), with a slight
pre-amplification step modification as a time-saving measure. The
prepared lysis buffer containing the sorted cells was vortexed,
spun at 6,000 × g for 10 sec and immediately chilled to 4°C,
instead of undergoing Qiagen Proteinase digestion. Subsequently,
linear pre-amplification and amplification were performed as
previously described (11). The
amplified DNA product was purified using the MinElute®
PCR Purification kit (Qiagen, Hilden, Germany; cat. no. 28004)
according to the manufacturer's instructions. The purified PCR
product was assessed using a Qubit® dsDNA HS Assay kit
in a Qubit® 3.0 Fluorometer (Life Technologies,
Carlsbad, CA, USA). The molecular weight of the DNA was evaluated
on 1% agarose gel. To reduce contamination, we performed the
WGA-associated experiments on a clean bench and all reagents,
except nucleotides and polymerases, were exposed to UV irradiation
for 30 min.
Isolation of DNA from bulk
specimens
DNA was extracted from breast-invasive carcinoma
tissues, and germline DNA was extracted from white blood cells
(WBCs) using the traditional phenol-chloroform method. The DNA was
quantified using Qubit 3.0 and high-molecular-weight DNA was
verified on 1% agarose gel.
PCR-free library preparation and whole
genome sequencing (WGS)
A PCR-free library is preferable to minimize the
base-composition bias in bulk samples and the already-existing PCR
amplification bias in single-cell samples. Thus, PCR-free libraries
were generated from 1.0 µg of genomic DNA from all bulk samples and
1.5 µg of amplified DNA from a single cell, based on their
ladder-like distribution base sizes, using an Illumina TruSeq DNA
PCR-Free LT Sample Prep kit (Illumina, San Diego, CA, USA; cat. no.
FC-121-3001) according to the manufacturer's instructions. The DNA
was sonicated into fragments with an average size of 350 bp,
followed by end-repair, A-tailing and ligation with full-length
adapter for Illumina sequencing but without further PCR
amplification. The final libraries were evaluated using an Agilent
2100 Bioanalyzer and quantified by real-time PCR. Libraries were
clustered on the cBot Cluster Generation System using a HiSeq X HD
PE Cluster kit (Illumina) and subsequently sequenced on the
Illumina HiSeq X Ten platform (paired ends, 150 bp).
Illumina target library preparation
and whole exome sequencing (WES)
All bulk DNA samples, cancer-originated DNA and
germline DNA were normalized to 1 µg and sheared to 150–200 bp. The
fragmented DNA was captured using an Agilent SureSelect Human All
Exon V6 kit (Agilent Technologies Inc., Santa Clara, CA, USA)
according to the manufacturer's instructions and the ends of the
enriched DNA fragments were repaired and ligated with adapters
using a Paired-End DNA Sample Prep kit (Illumina). The qualified
multi-regional target libraries were sequenced using an Illumina
HiSeq 4000 (paired ends, 150 bp).
Sequencing of data analysis
Adapter sequences and low-quality reads were removed
from the raw sequence data using Cutadapt (12) and sickle (http://github.com/najoshi/sickle/) software. After the
clean data were aligned to hg19 using the Burrows-Wheeler Aligner
(BWA) (13), SAM files were
produced. Picard tools (http://broadinstitute.github.io/picard/) were used to
transform the SAM files to a bam format and to sort and remove PCR
duplications. In addition with the help of the Genome Analysis
Toolkit (GATK) pipeline (14), the
original bam files were processed for local Indel realignment, base
quality estimation and recalibration for further analysis.
Somatic mutation calling was performed. For whole
exome data, VarScan2 (15) combined
with the JointSNVMix2 (16)
algorithm were used to identify somatic mutations and the
candidates were merged, while whole genomic data somatic mutation
calling used only the VarScan2 algorithm. To reduce false-positive
calls, the candidate calls were filtered using the following
criteria: i) zero mutant reads detected in the germline sample; ii)
at least 10X accumulated coverage for both the forward and reverse
reads of the tumour sample in the mutant alleles; and iii) the
nearest adjacent mutant base was at least 100 bp away in the same
sample. The filtered SNVs were annotated using Oncotator (17), and the variant allele fraction (VAF)
was calculated as follows: VAF = read counts that covered the
mutant position/all read counts that covered the position for data
from a single cell.
To determine the copy number, we used Ginkgo
(http://qb.cshl.edu/ginkgo), an
open-source web platform that specifically analyses single-cell
copy number variants (CNVs), and two R packages (HMMcopy and
DNAcopy), with hg19 as the reference genome.
Validating circulating breast tumour
cells using imaging flow cytometry
Slightly different from the oHSV1-hTERT-GFP-coupled
FACS method, the infected cells were stained with CD45 monoclonal
antibody (HI30)-eFluor 450 (cat. no. 48-0459-42; eBioscience, San
Diego, CA, USA) and human ErbB2/Her2 APC-conjugated antibody
(R&D Systems, Minneapolis, MN, USA; cat. no. FAB1129A) and the
cell suspension was subsequently imaged using an
ImageStream®X Mark II Imaging Flow cytometer (Merck
Millipore, Darmstadt, Germany).
Results
oHSV1-hTERT-GFP coupled with FACS is a
feasible method to detect and isolate CTCs
The efficacy and accuracy of the oHSV1-hTERT-GFP
method for detecting and isolating CTCs were validated using spike
and recovery experiments, as previously described, with a recovery
range between 75.5 and 87.19% (correlation r2=0.9909)
when 0, 10, 20, 40, 80, 160 and 320 BGC823 cells were spiked
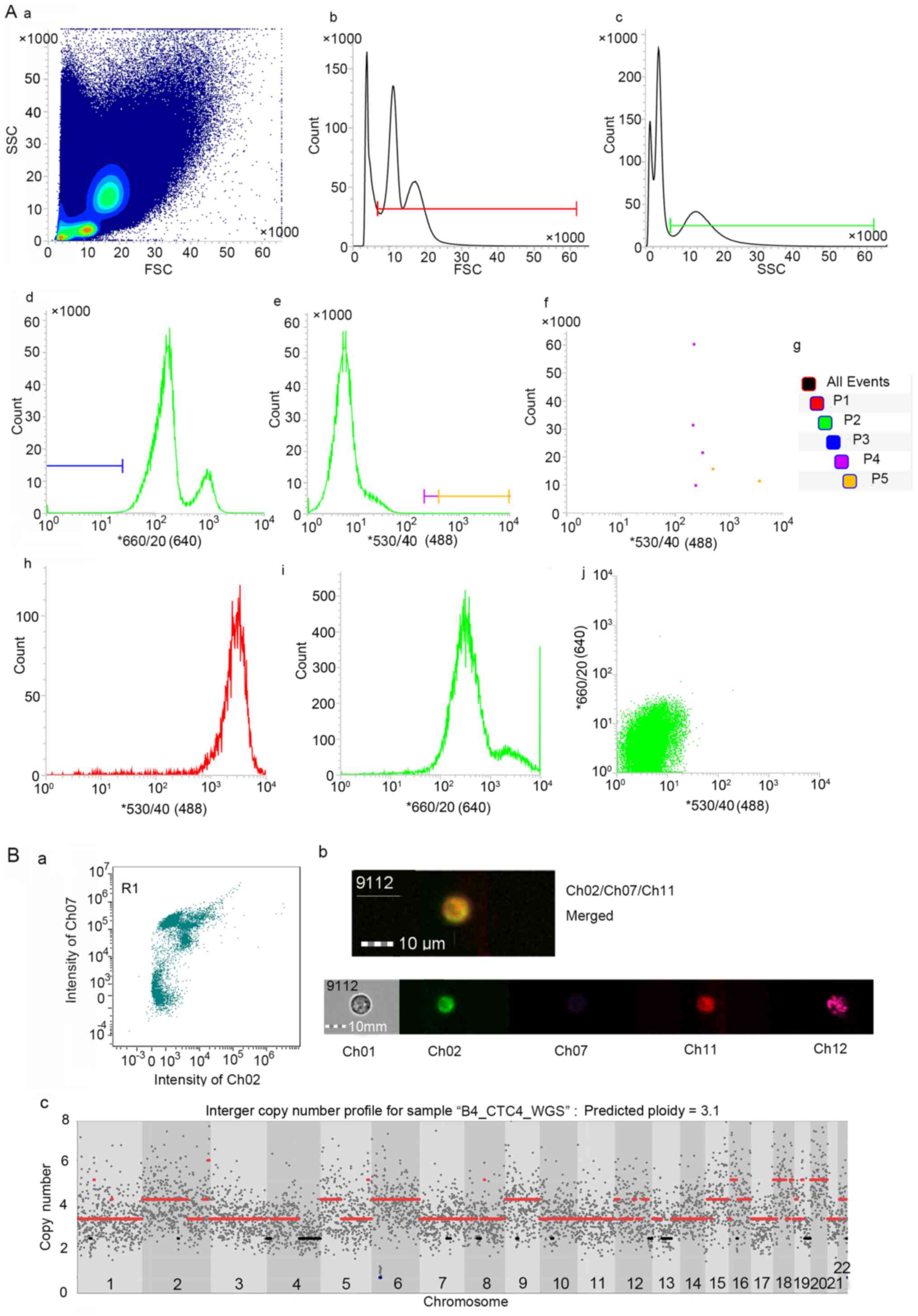
(9). The gates set to capture
viable CD45−/hTERT+ CTCs are displayed in
Fig. 2A. To further validate the
capacity for detecting circulating breast tumour cells, we
conducted imaging flow cytometry, and the observed
CD45−/hTERT+/ErbB2(Her2)+ cells
were defined as CTCs from breast cancer in patient B45 (Fig. 2B-a-b).
Copy number variations of CTCs
possibly confirm the capture and amplification method
A quality control was applied to the single-cell WGS
data for 11 CTCs from 8 patients, resulting in a mean coverage of
5.14±1.73 and an average global mapping rate of 84.59±6.5%. Both
were slightly lower than those for bulk sequencing (9.62±1.77 for
mean coverage and 93.29±3.62% for the mapping rate).
Genome instability is a hallmark of human cancers
(18). Amplifications of chr1q and
chr16p, commonly identified in breast cancer (19,20)
were discovered in 6 and 5 out of 11 CTCs, respectively.
Downregulation of human leukocyte antigen (HLA) class I expression
has been reported in a significant proportion of breast cancers
(21) and was found to be
significantly associated with a shorter disease-free interval
(22). The HLA super-locus in
chr6p21 was found to be lost in all 11 CTCs, indicating an escape
mechanism from cytotoxic T lymphocytes in the blood. In addition to
local gains and losses, whole-chromosome aneuploidy was also found
in B1CTC3 (with trisomies 10 and 18), B12CTC (with trisomies 1, 3
and 22), B4CTC2 (with trisomy 22), B4CTC3 (with trisomies 14 and
21) and B4CTC4 (with triploidy of the whole set of chromosomes).
The captured circulating aneuploid cells without leucocyte-common
antigen (CD45) indicated that our capture and amplification
procedure was satifactory.
SNVs accumulated sporadically between
CTCs and their matched primary tumours
As to the 11 CTCs analysed by single-cell
sequencing, on average, 299 high-confidence SNVs within exons in
each cell were found passing the filters mentioned in Materials and
methods. Compared with the single-cell sequencing data, the
abundance of SNVs (in exons) in bulk tissues was low (52 point
mutations on average for each primary or metastatic tissue), even
though we merged the mutations called from both WGS and WES data
(Table II). This result partially
reflected differences in low-abundance mutation mining competence
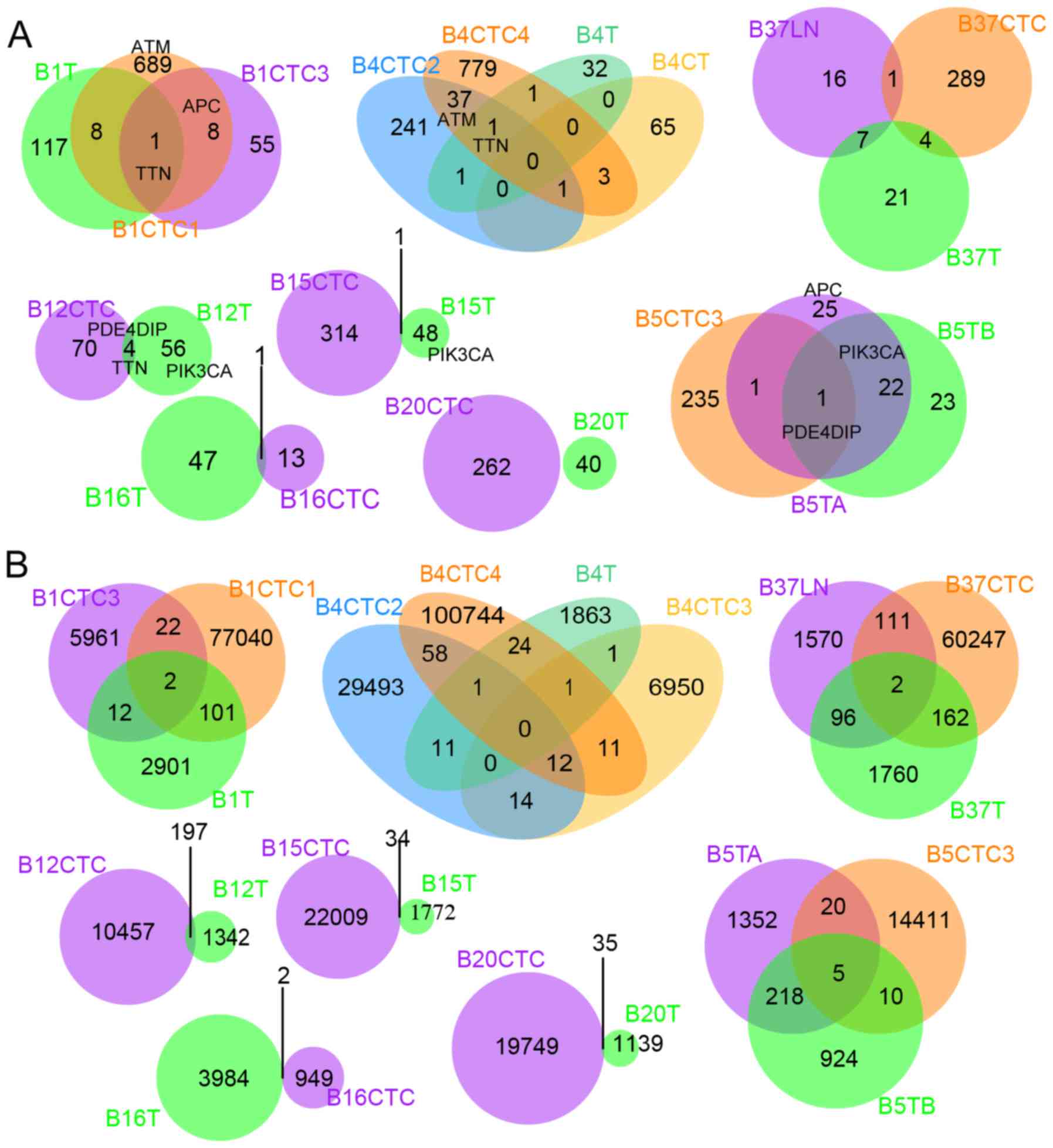
between single cells and bulk samples. Only 22 commonly mutated
genes were identified in CTCs and their matched primary or
metastatic tissues in the exome (Fig.
3A and Table III). However,
the shared mutation loci were much more prevalent genome wide
(Fig. 3B), indicating the
importance of genomic dark matter during the evolutionary process
of cancer cells. The heterogeneous mutation spectra of CTCs and
matched bulk cancers observed here is consistent with previous
studies (23–26).
| Table II.The point mutation counts of the exon
region and whole region of all samples. |
Table II.
The point mutation counts of the exon
region and whole region of all samples.
| Sample ID | Non-syn
SNVc | Stop-gain | Stop-loss | Syn
SNVd | Total mutation |
|---|
|
B1CTC1_WGSa | 493 | 26 | 0 | 222 | 77,165 |
| B1CTC3_WGS | 44 | 1 | 0 | 20 | 5,997 |
| B1T_WGS |
9 | 0 | 0 |
2 | 2,424 |
|
B1T_WESb | 80 | 6 | 0 | 31 | 590 |
| B4CTC2_WGS | 201 | 15 | 0 | 74 | 29,589 |
| B4CTC3_WGS | 54 | 3 | 0 | 16 | 6,989 |
| B4CTC4_WGS | 546 | 17 | 1 | 318 | 100,851 |
| B4T_WGS |
6 | 0 | 0 | 10 | 1,693 |
| B4T_WES | 15 | 0 | 0 |
5 | 192 |
| B5CTC3_WGS | 152 | 4 | 1 | 83 | 14,446 |
| B5TA_WGS | 10 | 0 | 0 |
4 | 1,303 |
| B5TA_WES | 21 | 2 | 0 | 13 | 281 |
| B5TB_WGS |
5 | 0 | 0 |
6 | 899 |
| B5TB_WES | 21 | 1 | 0 | 16 | 251 |
| B12CTC_WGS | 49 | 0 | 0 | 29 | 10,654 |
| B12T_WGS |
8 | 0 | 0 |
6 | 1,026 |
| B12T_WES | 16 | 0 | 0 | 30 | 499 |
| B15CTC_WGS | 229 | 5 | 0 | 89 | 22,043 |
| B15T_WGS | 10 | 1 | 0 |
4 | 1,383 |
| B15T_WES | 24 | 3 | 0 |
9 | 404 |
| B16CTC_WGS |
8 | 0 | 0 |
6 | 951 |
| B16T_WGS | 18 | 0 | 0 | 22 | 3,782 |
| B16T_WES |
8 | 0 | 0 |
4 | 195 |
| B20CTC_WGS | 173 | 16 | 1 | 83 | 19,784 |
| B20T_WGS |
5 | 0 | 0 |
2 | 911 |
| B20T_WES | 16 | 5 | 0 | 12 | 254 |
| B37CTC_WGS | 168 | 2 | 0 | 137 | 60,522 |
| B37T_WGS | 10 | 0 | 0 |
5 | 1,765 |
| B37T_WES |
9 | 2 | 0 |
6 | 251 |
| B37LN_WGS |
7 | 0 | 0 |
4 | 1,607 |
| B37LN_WES |
5 | 0 | 0 |
8 | 163 |
| Table III.Mutation genes appeared in both CTCs
and their matched primary or metastatic tissues and the concurring
mutant between CTC-shared SNVs and bulk SNVs. |
Table III.
Mutation genes appeared in both CTCs
and their matched primary or metastatic tissues and the concurring
mutant between CTC-shared SNVs and bulk SNVs.
| Category | Gene |
|---|
| B1 | XIRP2, TTN, PKHD1,
MUC17, C12orf29, UTP20, AHNAK2, MYO1E, TLN2, XIRP2 |
| B4 | TTN, CDCA2,
TGA9 |
| B5 | PDE4DIP,
RFPL4A |
| B12 | TTN, PDE4DIP, CUBN,
PRH1 |
| B15 | FAM184A |
| B16 | DSPP |
| B37 | OXCT2, OR8D1,
LILRA6, POTEH, FAM46A |
| Co-mutant between
CTC-shared | TTN, APC, MDN1,
MUC17, UTP20, AHNAK2, MYO1E, FBN1, DNAH17, KMT2C, |
| SNVs and bulk
SNVs | NBPF8, LRP1B,
XIRP2, KLHL41, CUBN, PZP, NAV3, CIT, PDE4DIP, ARHGEF5, FAM186A,
NBPF10, COL6A6, NUMA1, DDRGK1, COL24A1, MANSC1, MUC12, TOP2B,
ZFHX4 |
Characteristics of CTC-shared SNVs in
the exome
To prevent bias and errors from the WGA, 394
concurring mutation genes (hereafter referred to as CTC-shared
SNVs), which were identified in at least two CTCs, were used for
further analysis (Fig. 4A).
Intersecting the CTC-shared SNVs with the SNVs in the exome
(hereafter, exome SNVs) of the bulk tissues, 30 CTC-bulk shared
mutation genes were identified, including the well-established
tumour suppressor gene APC and the putative tumour suppressor gene
LRP1B (Fig. 4B and Table III). The CTC-shared SNVs comprised
some prevalent mutated genes that have been reported in the Cancer
Genome Atlas (TCGA, https://cancergenome.nih.gov/) database, including
USH2A, TTN, MUC16 (CA125) and other members of the mucin family
(i.e., MUC17 and MUC3A). In particular, mutations with high
frequencies in metastatic samples reported in TCGA, such as
mutations in LRP1B, were frequently observed among the CTCs
(Fig. 4C) (27). We further compared the allele
frequencies of seven relatively prominent genes between our samples
and another published database, the Catalogue of Somatic Mutations
in Cancer (COSMIC) (28). The
frequencies of PIK3CA, a mutation driver in breast cancer (29), were higher in the bulk sample than
the COSMIC record, and the mutation rate of TTN exceeded 10% in all
three datasets (Fig. 4D).
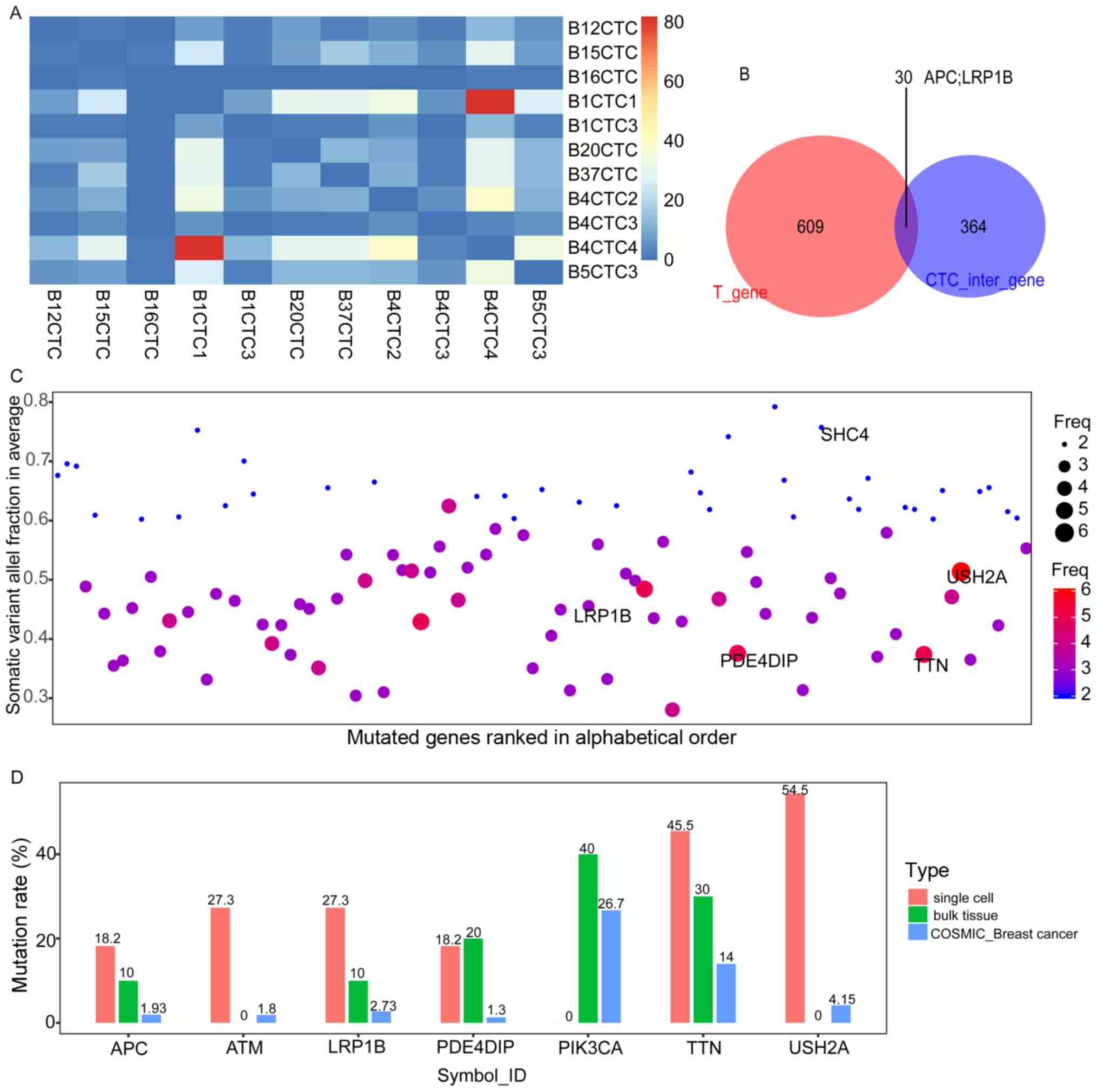 | Figure 4.The CTCs-shared SNVs. (A) The heat
map of the number of CTCs-shared SNVs contributed by each cell. The
majority of CTCs-shared SNVs are offered by B1CTC1 and B4CTC4, and
a few overlapping SNVs in B16CTC and other single cells. The
diagonal of this heat map represents the overlap of the same cells,
which were set to zero. (B) Intersecting the CTC-shared SNVs with
the exome SNVs of the bulk tissues, 30 CTC-bulk shared mutation
genes were identified, including the tumour suppressor gene APC and
the putative tumour suppressor gene LRP1B. (C) The somatic variant
allele fraction (VAF) distribution and mutation frequency in the
CTCs-shared SNVs. The CTCs-shared SNVs demonstrated here are
covered with more than ten reads without any mutations in the
normal tissue, with VAF ≥0.6 or frequency ≥3. The vertical axis
shows the average somatic VAF within a gene. The horizontal axis
depicts the mutated genes (official_symbol_ID) in alphabetical
order. The diameter or colour of the circle represents the observed
mutation frequency in the 11 cells. (D) The frequency of several
prominent mutations in our data and COSMIC data. The vertical axis
presents the prevalence of the mutations in each group, including
the 11 cells, 10 bulk tissues, and the COSMIC breast cancer
samples. The horizontal axis depicts APC, ATM (DNA-damage response
gene that regulates the tumour suppressor proteins p53 and BRCA1
and some checkpoint or DNA repair proteins), LRP1B (LDL
receptor-related protein 1 B, which is ranked in the top 3 mutated
genes in at least one cancer type, particularly in the metastatic
samples of TCGA), PDE4DIP, PIK3CA, TTN and USH2A (with mutants
occurring in 6 cells, GO annotation relates to collagen binding and
myosin binding). |
Verification of the breast-originating
identity of CTC-shared SNVs
Although the relationship between SNVs and the cDNA
library is obscure, it is believed that SNVs can alter the
expression level of mutated genes in several ways. For example,
some SNVs located in the promoter or 3′UTR of genes, can influence
expression levels by altering the regulatory influence of the
transcription factors on the promoter region or some regulatory
non-coding RNAs on their mRNA targets. To explore the general
classification of CTC-shared SNVs, the SNV-bearing genes were
identified in the Cancer Genome Anatomy Project library (CGAP,
https://cgap.nci.nih.gov/), particularly the EST
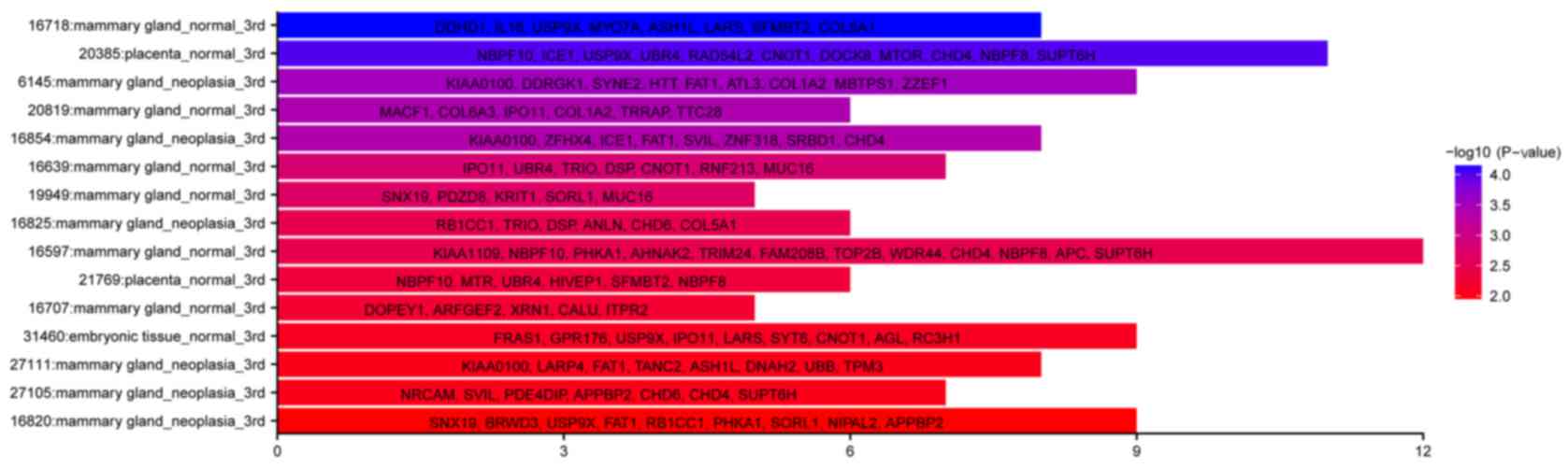
database (dbEST) with the tissue tool. In total, 285 annotated
genes were included in the dbEST, and these could be divided into
three major categories including mammary gland_normal, mammary
gland_neoplasia and placenta_normal (Fisher's exact test,
P<0.05; Fig. 5). The former two
categories uncovered the origin of these CTCs. As to the category
referring to normal placenta, it may represent a cluster of genes
with hormone-associated, invasive or low-immunogenicity functions,
which are known as features of the trophoblast model of cancer
(30).
CTC-shared SNVs and their
CTC-associated functions
To further understand the potential biological
functions of CTC-shared SNVs, KEGG pathway (http://www.kegg.jp/) and GO (http://www.geneontology.org/) biological process
analyses were used to perform enrichment analysis on the genes
bearing the 394 CTC-shared SNVs. As displayed in Tables IV and V, the data indicated that these genes
alter the following functions of the CTCs: i) intravasation
competency through ECM remodelling and collagen catabolism; ii)
increased migration or motility dependent on cilia motility, actin
cytoskeleton organization and other myocardium-like movement,
indicating that captured viable CTCs undergo EMT; iii) initiation
of cell-cell interactions, such as integrin-mediated cell adhesion;
iv) alterations to energy metabolism, including lipoprotein
transport, glycan processing and ABC transporters; v) platelet or
coagulation system activation, affecting wound healing; and vi)
dysfunctional mitosis, exhibited through DNA repair deficiency
(Fanconi anaemia pathway enriched in the KEGG pathway analysis)
(31) and dysfunction in DNA
double-strand break repair by non-homologous end-joining (mutant
genes enriched in GO: histone H3-K36 dimethylation, H3K36me2)
(32).
| Table IV.CTCs-shared SNVs involved in KEGG
pathway. |
Table IV.
CTCs-shared SNVs involved in KEGG
pathway.
| Category | Term | Count | P-value |
|---|
| KEGG_PATHWAY | hsa04512:
ECM-receptor interaction | 14 | 9.21E-09 |
| KEGG_PATHWAY | hsa04510: Focal
adhesion | 17 | 1.98E-06 |
| KEGG_PATHWAY | hsa04974: Protein
digestion and absorption | 11 | 6.71E-06 |
| KEGG_PATHWAY | hsa04151: PI3K-Akt
signaling pathway | 16 | 0.002665514 |
| KEGG_PATHWAY | hsa05410:
Hypertrophic cardiomyopathy (HCM) | 6 | 0.017366873 |
| KEGG_PATHWAY | hsa05414: Dilated
cardiomyopathy | 6 | 0.02318041 |
| KEGG_PATHWAY | hsa02010: ABC
transporters | 4 | 0.053062934 |
| KEGG_PATHWAY | hsa04260: Cardiac
muscle contraction | 5 | 0.05722822 |
| KEGG_PATHWAY | hsa03460: Fanconi
anemia pathway | 4 | 0.082951282 |
| Table V.CTCs-shared SNVs confer biology
process on free-floating tumour cells (GO biology process). |
Table V.
CTCs-shared SNVs confer biology
process on free-floating tumour cells (GO biology process).
| Term_ID | Term | P-value |
|---|
|
GO:0060285a | Cilium-dependent
cell motility | 1.30E-06 |
|
GO:0060048a | Cardiac muscle
contraction | 2.59E-04 |
|
GO:0002027a | Regulation of heart
rate | 4.66E-04 |
|
GO:0030198b | Extracellular
matrix organization | 6.02E-04 |
|
GO:0030049a | Muscle filament
sliding | 9.09E-04 |
|
GO:0030574b | Collagen catabolic
process | 0.001734612 |
|
GO:0006941a | Striated muscle
contraction | 0.001956422 |
|
GO:0007155c | Cell adhesion | 0.002348521 |
| GO:0007507 | Heart
development | 0.003843656 |
|
GO:0007010a | Cytoskeleton
organization | 0.005175679 |
|
GO:0010880a | Regulation of
release of sequestered calcium ion into cytosol by sarcoplasmic
reticulum | 0.005183546 |
| GO:0031119 | tRNA pseudouridine
synthesis | 0.005677556 |
|
GO:0045214a | Sarcomere
organization | 0.019748792 |
| GO:0045666 | Positive regulation
of neuron differentiation | 0.020028672 |
|
GO:0006936a | Muscle
contraction | 0.020559102 |
|
GO:0042060d | Wound healing | 0.022090779 |
| GO:0031581 | Hemidesmosome
assembly | 0.023078156 |
| GO:0051480 | Regulation of
cytosolic calcium ion concentration | 0.025660253 |
|
GO:0030239a | Myofibril
assembly | 0.026918471 |
| GO:0007512 | Adult heart
development | 0.026918471 |
| GO:0060402 | Calcium ion
transport into cytosol | 0.030995997 |
| GO:0071044 | Histone mRNA
catabolic process | 0.030995997 |
|
GO:0016266e | O-glycan
processing | 0.031968827 |
|
GO:0030335a | Positive regulation
of cell migration | 0.032042051 |
| GO:0010951 | Negative regulation
of endopeptidase activity | 0.034801689 |
|
GO:0033627c | Cell adhesion
mediated by integrin | 0.035299764 |
|
GO:0042953e | Lipoprotein
transport | 0.035299764 |
| GO:0034728 | Nucleosome
organization | 0.039619843 |
|
GO:0030036a | Actin cytoskeleton
organization | 0.046670842 |
|
GO:0002026a | Regulation of the
force of heart contraction | 0.054569593 |
|
GO:0010881a | Regulation of
cardiac muscle contraction by regulation of the release of
sequestered calcium ion | 0.054569593 |
|
GO:0097676f | Histone H3-K36
dimethylation | 0.058838886 |
| GO:0002331 | Pre-B cell allelic
exclusion | 0.058838886 |
| GO:0032439 | Endosome
localization | 0.058838886 |
| GO:0000472 | Endonucleolytic
cleavage to generate mature 5′-end of SSU-rRNA from (SSU-rRNA, 5.8S
rRNA, LSU-rRNA) | 0.058838886 |
| GO:0007030 | Golgi
organization | 0.060936288 |
|
GO:0007067f | Mitotic nuclear
division | 0.06220872 |
|
GO:0022617b | Extracellular
matrix disassembly | 0.065918416 |
| GO:0006816 | Calcium ion
transport | 0.065918416 |
| GO:0090287 | Regulation of
cellular response to growth factor stimulus | 0.077674441 |
|
GO:0007018a | Microtubule-based
movement | 0.079266502 |
| GO:0035023 | Regulation of Rho
protein signal transduction | 0.079266502 |
| GO:0001701 | In utero embryonic
development | 0.082288621 |
| GO:0060070 | Canonical Wnt
signaling pathway | 0.084954447 |
| GO:0006887 | Exocytosis | 0.084954447 |
| GO:0022604 | Regulation of cell
morphogenesis | 0.088600004 |
| GO:0008544 | Epidermis
development | 0.09083611 |
|
GO:0003356a | Regulation of
cilium beat frequency | 0.096134138 |
| GO:0002634 | Regulation of
germinal center formation | 0.096134138 |
| GO:0042415 | Norepinephrine
metabolic process | 0.096134138 |
Discussion
More than a decade has passed since the first trial
enumerated CTCs through EpCAM-based detection to estimate the
prognosis of patients with breast cancer (3). In addition, following the validation
of their prognostic power in breast cancer, CTC counts have been
correlated to poor prognosis in other high-morbidity cancers,
including lung (33), prostate
(34) and colorectal cancer
(35). In addition,
single-nucleotide mutation characteristics have been reported by
whole genome sequencing in kidney tumours (23), prostate (36) and lung cancer (24). Although invasive breast carcinoma
(no special type) is known to derive from the ductal epithelium and
prognosis prediction through EpCAM-phenotype CTCs has been long
validated (37), the molecular
characterization of these EpCAM-based free-floating tumour cells
had, until recently, only been performed using whole genomes
(20). However, the present study
is the first to profile the mutant characteristics of hTERT-based
CTCs in the whole genome. In addition, the status and phenotypes of
circulating breast cancer cells are diverse and include stem-like
cells (38), dormant states
(39,40) and dynamic EMT changes (6,41).
Thus, it is imperative to determine the molecular characteristics
of specific cell populations, as they exhibit a great deal of
genomic disparity among them that can lead to the initiation of
metastasis or can act as a decisive factor in treatment failure.
Unfortunately, molecular profiling has been hindered by the rarity
and heterogeneity of CTCs, coupled with unsophisticated technology
for their enrichment, isolation and genomic amplification. Compared
with several studies that have analysed some typical mutations
(TP53, PIK3CA, KRAS, RB1 and ErbB2) in CTCs and bulk tissues
(8,42,43),
which simply represent the tip of the iceberg, this study is the
first to examine the whole genome of hTERT-positive CTCs. In
addition, this is the first study to profile SNVs in viable
hTERT-positive CTCs, rather than EpCAM-CTCs, to define CTC
behaviours.
In contrast with the reported bulk sequencing data,
one mutant gene (SHC4/RaLP/SHCD) was highlighted in our single-CTC
data. Recent studies concerning SHC4 have focused on metastatic
melanoma and reproductive organ cancer (particularly prostate and
ovarian cancers involving the BRCA1 pathway) and have identified
several plausible roles for SHC4, such as regulating apoptosis
(44), stem cell differentiation
(45), cell invasion and migration
(44). It appears, therefore, that
although considered a ‘low-probability event’, when the specific
cell population acquired the essential mutation during the random
accumulation process, they began to detach and survive in the
bloodstream.
We extracted 394 CTC-shared SNVs from all the
identified CTC mutations, and these may confer primary tumour cells
with the following alterations. During the metastasis initiation
phase, the selected population interacts with ECM receptors
(hsa04512), followed by extracellular matrix organization
(GO:0030198) via collagen catabolism (GO:0030574). Similarly, the
results from single-cell RNA sequencing data in a pancreatic cancer
experimental animal model revealed that the aberrant expression of
stromal ECM genes by EM-CTCs are liable to aid spread into the
bloodstream (46). Indeed, the
five-step model for proteolytic cell migration has been suggested
for many years. This model proposes that the elongated cell
morphology and dynamic membrane protrusions that are tailored for
ECM degradation promote cell detachment from one spot (47,48).
Specifically, von Nandelstadh et al (48) confirmed that the actin-associated
protein paladin functions in actin-based pseudopods (cell
cytoskeleton alteration) to drive invasion through ECM degradation.
Thus, the ever-increasing motility (GO:0060285, cilium-dependent
cell motility; GO:0030036, actin cytoskeleton organization) and ECM
degradation of CTCs may co-occur in CTC behaviours, similar to our
data. The motility-enhanced EM-CTCs not only tend to penetrate into
vessels, but also are prone to escape immune attack through
autophagy when encountering T cells (49). Therefore, the increased mobility of
CTCs ensures both detachment and survival.
To ensure survival in the circulation, the detached
tumour cells are equipped not only with increased motility but also
enhanced focal adhesion (KEGG, hsa04510), cell adhesion
(GO:0007155), and cell adhesion mediated through integrins
(GO:0033627). Cell adhesion can be classified as follows: CTC-CTC
interactions; CTC-non-CTC interactions, which include all cell
types except CTCs and various molecules, such as hormones,
cytokines and growth factors in the bloodstream; and CTC-CTC
interactions bridged by non-CTCs in the circulatory system. In
contrast with isolated, floating CTCs, CTCs and their binding
partners favour resistance to anoikis, and the counterparts of cell
adhesion, wound healing (GO:0042060) and its child term, blood
coagulation (GO:0007596), play significant roles in survival.
Collectively, the terms associated with cell adhesion and wound
healing highlight a novel theory. A secure shelter of CTCs is built
when neutrophil extracellular traps (NETs) sequester CTCs via
β1-integrin-mediated interactions (50) and act as a scaffold to recruit
platelets and coagulation factors (51). Activating platelets and the
coagulation system predisposes the tumour to dissemination
(52).
Additional distinguishing behaviours of CTCs are the
dysfunction of energy metabolism, ABC transporters (KEGG, hsa02010)
and lipoprotein transport (GO:0042953). High-fat diets increase the
frequency and reduce the time to occurrence of breast cancer
(53). LRP1B, which encodes a
member of the low-density lipoprotein (LDL) receptor family,
affects LDL receptor activity (54)
and acquired chemotherapeutic resistance (55), with mutations detected in three
CTCs.
The enumerated CTC behaviours facilitate metastasis
initiation, and survival is rooted in genomic alterations,
including DNA repair dysfunction (KEGG, hsa03460; GO:0097676) and
mitotic nuclear division resulting in aneuploidy (GO:0007067).
Genomic (DNA repair-associated genes) and epigenomic (histone
methylation, particularly H3K36me2) alterations regulate DNA repair
to destabilize the genome and produce a series of CTC-shared
SNVs.
Although the CTC behaviours deduced from the 394
CTC-shared SNVs are supported by previous studies, there are no
sufficiently amplified DNA samples to validate the SNVs detected
through next-generation sequencing via Sanger sequencing since more
DNA is needed to prepare PCR-free libraries than traditional
sequencing libraries. However, the PCR-free library reduced the
bias from PCR. Reflecting the limitations of the unsophisticated
isolation and amplification technologies, the same sorting and
amplification work flow deficiencies were observed in the present
study, and only a few CTCs were successfully amplified and
sequenced from each patient, which is far from covering all the
heterogeneous CTCs. There were two reasons for only 1–3 CTCs being
sorted for one patient in the present study. Firstly, to balance
the sorted speed and efficiency in our flow cytometry (BD FACSJazz;
BD Biosciences), we sorted the peripheral blood until 5 to 10 CTCs
collected, and counted the CTC number in the rest. However, in most
cases, less than 5 CTCs could be captured successfully into a PCR
tube for one patient, even there were dozens of CTCs counted by
FACS. The limitations of the sorting accuracy of the flow cytometry
are the main reason. Secondly, the amount of DNA needed to prepare
the PCR-free library are much more than that for the traditional
library. Not all single cell DNA amplificated by the MALBAC method
could pass the quality control for PCR-free library construction.
Recently, one protocol combining immunomagnetic enrichment and
fluorescence-activated cell sorting (IE/FACS) increased the yield
of CTCs (56). As for the isotype
control in flow cytometry, more and more researchers abandoned the
isotype control as it indicated that such controls may be
unreliable to be used as gating controls or to determine background
signal (57). The application of
Fc-blocking reagent is recommended instead. In addition, during our
CTC isolated period, 10% FBS was added into the medium as the
Fc-blocking reagent. Thus, considering the convenience and the
blood consumption, we did not use the isotype control to detect the
specificity of the CD45 antibody in the group of these breast
cancer patients.
The genomic data in the present study provided clues
to the behaviour of the hTERT-positive metastasis initiation cells
in peripheral blood. There are many interesting events to be
investigated in the future, for example, the function of the
high-frequency mutant SHC4/RaLP/SHCD in CTCs, the relationship of
the mutant of LRP1B and their distinguished metabolic way.
Uncovering the specific properties of CTCs as the major drivers of
metastasis initiation and the pool of treatment resistance and
tumour recurrence is urgently needed.
Acknowledgements
We thank the National Center for Protein Sciences
Beijing (Peking University) and Ms Fei Wang for her technical
assistance with the imaging flow cytometry. We thank the National
Cancer Center/Cancer Hospital (CAMS and PUMC) and Professor Shujun
Cheng.
Glossary
Abbreviations
Abbreviations:
|
APC
|
allophycocyanin
|
|
CTC
|
circulating tumour cell
|
|
CNV
|
copy number variant
|
|
ECM
|
extracellular matrix
|
|
EM-CTC
|
epithelial and mesenchymal
biphenotypic CTC
|
|
ErbB2
|
v-erb-b2 avian erythroblastic
leukaemia viral
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
hTERT
|
human telomerase reverse
transcriptase
|
|
Her2
|
oncogene homologue 2
|
|
MALBAC
|
multiple annealing and looping-based
amplification cycles
|
|
oHSV
|
oncolytic herpes simplex virus
|
|
SNV
|
single nucleotide variant
|
|
WGA
|
whole genome amplification
|
|
WGS
|
whole genome sequencing
|
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giuliano M, Giordano A, Jackson S, De
Giorgi U, Mego M, Cohen EN, Gao H, Anfossi S, Handy BC, Ueno NT, et
al: Circulating tumor cells as early predictors of metastatic
spread in breast cancer patients with limited metastatic
dissemination. Breast Cancer Res. 16:4402014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Magbanua MJ, Carey LA, DeLuca A, Hwang J,
Scott JH, Rimawi MF, Mayer EL, Marcom PK, Liu MC, Esteva FJ, et al:
Circulating tumor cell analysis in metastatic triple-negative
breast cancers. Clin Cancer Res. 21:1098–1105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klein CA: Selection and adaptation during
metastatic cancer progression. Nature. 501:365–372. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deng G, Krishnakumar S, Powell AA, Zhang
H, Mindrinos MN, Telli ML, Davis RW and Jeffrey SS: Single cell
mutational analysis of PIK3CA in circulating tumor cells and
metastases in breast cancer reveals heterogeneity, discordance, and
mutation persistence in cultured disseminated tumor cells from bone
marrow. Bmc Cancer. 14:4562014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu M, Bardia A, Wittner BS, Stott SL, Smas
ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al:
Circulating breast tumor cells exhibit dynamic changes in
epithelial and mesenchymal composition. Science. 339:580–584. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng T, Wang Y, Seiler M and Hu Z:
Functional characterization of breast cancer using pathway
profiles. Bmc Med Genomics. 7:452014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Polzer B, Medoro G, Pasch S, Fontana F,
Zorzino L, Pestka A, Andergassen U, Meier-Stiegen F, Czyz ZT,
Alberter B, et al: Molecular profiling of single circulating tumor
cells with diagnostic intention. EMBO Mol Med. 6:1371–1386. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang W, Bao L, Yang S, Qian Z, Dong M,
Yin L, Zhao Q, Ge K, Deng Z, Zhang J, et al: Tumor-selective
replication herpes simplex virus-based technology significantly
improves clinical detection and prognostication of viable
circulating tumor cells. Oncotarget. 7:39768–39783. 2016.PubMed/NCBI
|
|
10
|
Zong C, Lu S, Chapman AR and Xie XS:
Genome-wide detection of single-nucleotide and copy-number
variations of a single human cell. Science. 338:1622–1626. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Bourcy CF, De Vlaminck I, Kanbar JN,
Wang J, Gawad C and Quake SR: A quantitative comparison of
single-cell whole genome amplification methods. PLoS One.
9:e1055852014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martin M: Cutadapt removes adapter
sequences from high-throughput sequencing reads. Embnet J.
17:10–12. 2011. View Article : Google Scholar
|
|
13
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roth A, Ding J, Morin R, Crisan A, Ha G,
Giuliany R, Bashashati A, Hirst M, Turashvili G, Oloumi A, et al:
JointSNVMix: A probabilistic model for accurate detection of
somatic mutations in normal/tumour paired next-generation
sequencing data. Bioinformatics. 28:907–913. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramos AH, Lichtenstein L, Gupta M,
Lawrence MS, Pugh TJ, Saksena G, Meyerson M and Getz G: Oncotator:
Cancer variant annotation tool. Hum Mutat. 36:E2423–E2429. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Silva GO, He X, Parker JS, Gatza ML, Carey
LA, Hou JP, Moulder SL, Marcom PK, Ma J, Rosen JM and Perou CM:
Cross-species DNA copy number analyses identifies multiple 1q21-q23
subtype-specific driver genes for breast cancer. Breast Cancer Res
Treat. 152:347–356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gulbahce N, Magbanua MJM, Chin R, Agarwal
MR, Luo X, Liu J, Hayden DM, Mao Q, Ciotlos S, Li Z, et al:
Quantitative whole genome sequencing of circulating tumor cells
enables personalized combination therapy of metastatic cancer.
Cancer Res. 77:4530–4541. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Palmisano GL, Pistillo MP, Capanni P, Pera
C, Nicolò G, Salvi S, Perdelli L, Pasciucco G and Ferrara GB:
Investigation of HLA class I downregulation in breast cancer by
RT-PCR. Hum Immunol. 62:133–139. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kaneko K, Ishigami S, Kijima Y, Funasako
Y, Hirata M, Okumura H, Shinchi H, Koriyama C, Ueno S, Yoshinaka H
and Natsugoe S: Clinical implication of HLA class I expression in
breast cancer. BMC Cancer. 11:4542011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu X, Hou Y, Yin X, Bao L, Tang A, Song L,
Li F, Tsang S, Wu K, Wu H, et al: Single-cell exome sequencing
reveals single-nucleotide mutation characteristics of a kidney
tumor. Cell. 148:886–895. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z,
Zong C, Bai H, Chapman AR, Zhao J, et al: Reproducible copy number
variation patterns among single circulating tumor cells of lung
cancer patients. Proc Natl Acad Sci USA. 110:21083–21088. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Waters J, Leung ML, Unruh A, Roh
W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, et al: Clonal
evolution in breast cancer revealed by single nucleus genome
sequencing. Nature. 512:155–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao Y, Ni X, Guo H, Su Z, Ba Y, Tong Z,
Guo Z, Yao X, Chen X, Yin J, et al: Single-cell sequencing
deciphers a convergent evolution of copy number alterations from
primary to circulating tumour cells. Genome Res. 27:1312–1322.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim N, Hong Y, Kwon D and Yoon S: Somatic
mutaome profile in human cancer tissues. Genomics Inform.
11:239–244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the catalogue of somatic
mutations in cancer. Nucleic Acids Res. 39:D945–D950. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ross CA: The Trophoblast Model of Cancer.
Nutr Cancer. 67:61–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alix-Panabieres C, Cayrefourcq L, Mazard
T, Maudelonde T, Assenat E and Assou S: Molecular portrait of
metastasis-competent circulating tumor cells in colon cancer
reveals the crucial role of genes regulating energy metabolism and
DNA repair. Clin Chem. 63:700–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fnu S, Williamson EA, De Haro LP,
Brenneman M, Wray J, Shaheen M, Radhakrishnan K, Lee SH, Nickoloff
JA and Hromas R: Methylation of histone H3 lysine 36 enhances DNA
repair by nonhomologous end-joining. Proc Natl Acad Sci USA.
108:540–545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang J, Wang HT and Li BG: Prognostic
significance of circulating tumor cells in small-cell lung cancer
patients: A meta-analysis. Asian Pac J Cancer Prev. 15:8429–8433.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Goldkorn A, Ely B, Quinn DI, Tangen CM,
Fink LM, Xu T, Twardowski P, Van Veldhuizen PJ, Agarwal N, Carducci
MA, et al: Circulating tumor cell counts are prognostic of overall
survival in SWOG S0421: A phase III trial of docetaxel with or
without atrasentan for metastatic castration-resistant prostate
cancer. J Clin Oncol. 32:1136–1142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Romiti A, Raffa S, Di Rocco R, Roberto M,
Milano A, Zullo A, Leone L, Ranieri D, Mazzetta F, Medda E, et al:
Circulating tumor cells count predicts survival in colorectal
cancer patients. J Gastrointestin Liver Dis. 23:279–284.
2014.PubMed/NCBI
|
|
36
|
Lohr JG, Adalsteinsson VA, Cibulskis K,
Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ,
Shalek AK, Satija R, et al: Whole-exome sequencing of circulating
tumor cells provides a window into metastatic prostate cancer. Nat
Biotechnol. 32:479–484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weissenstein U, Schumann A, Reif M, Link
S, Toffol-Schmidt UD and Heusser P: Detection of circulating tumor
cells in blood of metastatic breast cancer patients using a
combination of cytokeratin and EpCAM antibodies. BMC Cancer.
12:2062012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Papadaki MA, Kallergi G, Zafeiriou Z,
Manouras L, Theodoropoulos PA, Mavroudis D, Georgoulias V and
Agelaki S: Co-expression of putative stemness and
epithelial-to-mesenchymal transition markers on single circulating
tumour cells from patients with early and metastatic breast cancer.
BMC Cancer. 14:6512014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kanwar N, Hu P, Bedard P, Clemons M,
McCready D and Done SJ: Identification of genomic signatures in
circulating tumor cells from breast cancer. Int J Cancer.
137:332–344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Meng S, Tripathy D, Frenkel EP, Shete S,
Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, et
al: Circulating tumor cells in patients with breast cancer
dormancy. Clin Cancer Res. 10:8152–8162. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aktas B, Tewes M, Fehm T, Hauch S, Kimmig
R and Kasimir-Bauer S: Stem cell and epithelial-mesenchymal
transition markers are frequently overexpressed in circulating
tumor cells of metastatic breast cancer patients. Breast Cancer
Res. 11:R462009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bingham C, Fernandez SV, Fittipaldi P,
Dempsey PW, Ruth KJ, Cristofanilli M and Alpaugh Katherine R:
Mutational studies on single circulating tumor cells isolated from
the blood of inflammatory breast cancer patients. Breast Cancer Res
Treat. 163:219–230. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mu Z, Benali-Furet N, Uzan G, Znaty A, Ye
Z, Paolillo C, Wang C, Austin L, Rossi G, Fortina P, et al:
Detection and characterization of circulating tumor associated
cells in metastatic breast cancer. Int J Mol Sci. 17:E16652016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fagiani E, Giardina G, Luzi L, Cesaroni M,
Quarto M, Capra M, Germano G, Bono M, Capillo M, Pelicci P and
Lanfrancone L: RaLP, a new member of the Src homology and collagen
family, regulates cell migration and tumor growth of metastatic
melanomas. Cancer Res. 67:3064–3073. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Turco MY, Furia L, Dietze A, Diaz
Fernandez L, Ronzoni S, Sciullo A, Simeone A, Constam D, Faretta M
and Lanfrancone L: Cellular heterogeneity during embryonic stem
cell differentiation to epiblast stem cells is revealed by the
ShcD/RaLP adaptor protein. Stem Cells. 30:2423–2436. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ting DT, Wittner BS, Ligorio M, Jordan
Vincent N, Shah AM, Miyamoto DT, Aceto N, Bersani F, Brannigan BW,
Xega K, et al: Single-cell RNA sequencing identifies extracellular
matrix gene expression by pancreatic circulating tumor cells. Cell
Rep. 8:1905–1918. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Friedl P and Wolf K: Proteolytic
interstitial cell migration: A five-step process. Cancer Metastasis
Rev. 28:129–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
von Nandelstadh P, Gucciardo E, Lohi J, Li
R, Sugiyama N, Carpen O and Lehti K: Actin-associated protein
palladin promotes tumor cell invasion by linking extracellular
matrix degradation to cell cytoskeleton. Mol Biol Cell.
25:2556–2570. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Akalay I, Janji B, Hasmim M, Noman MZ,
André F, De Cremoux P, Bertheau P, Badoual C, Vielh P, Larsen AK,
et al: Epithelial-to-mesenchymal transition and autophagy induction
in breast carcinoma promote escape from T-cell-mediated lysis.
Cancer Res. 73:2418–2427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Najmeh S, Cools-Lartigue J, Rayes RF,
Gowing S, Vourtzoumis P, Bourdeau F, Giannias B, Berube J, Rousseau
S, Ferri LE and Spicer JD: Neutrophil extracellular traps sequester
circulating tumor cells via β1-integrin mediated interactions. Int
J Cancer. 140:2321–2330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Demers M, Krause DS, Schatzberg D,
Martinod K, Voorhees JR, Fuchs TA, Scadden DT and Wagner DD: Cancer
predispose neutrophils to release extracellular DNA traps that
contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA.
109:13076–13081. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Goubran HA, Burnouf T, Radosevic M and
El-Ekiaby M: The platelet-cancer loop. Eur J Intern Med.
24:393–400. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hulka BS and Stark AT: Breast cancer:
Cause and prevention. Lancet. 346:883–887. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
May P, Woldt E, Matz RL and Boucher P: The
LDL receptor-related protein (LRP) family: An old family of
proteins with new physiological functions. Ann Med. 39:219–228.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cowin PA, George J, Fereday S, Loehrer E,
Van Loo P, Cullinane C, Etemadmoghadam D, Ftouni S, Galletta L,
Anglesio MS, et al: LRP1B deletion in high-grade serous ovarian
cancers is associated with acquired chemotherapy resistance to
liposomal doxorubicin. Cancer Res. 72:4060–4073. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Magbanua MJ and Park JW: Isolation of
circulating tumor cells by immunomagnetic enrichment and
fluorescence-activated cell sorting (IE/FACS) for molecular
profiling. Methods. 64:114–118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Andersen MN, Al-Karradi SN, Kragstrup TW
and Hokland M: Elimination of erroneous results in flow cytometry
caused by antibody binding to Fc receptors on human monocytes and
macrophages. Cytometry A. 89:1001–1009. 2016. View Article : Google Scholar : PubMed/NCBI
|