Introduction
Acute promyelocytic leukemia (APL) is distinguished
by a specific t(15;17) chromosomal translocation, contributing to
the expression of the oncoprotein PML-RARα, which counteracts
myeloid differentiation and facilitates APL-initiating cell
self-renewal (1,2). The dominant-negative effect of the
PML-RARα oncoprotein, which antagonizes the myeloid differentiation
process, can be reversed by pharmacological doses of
all-trans-retinoic acid (ATRA) (3). The combined therapy of ATRA and
arsenic trioxide (ATO), which eliminates APL by activating PML-RARα
degradation, demonstrates efficacy as an APL therapy and markedly
improves the prognosis of patients with APL (4). However, ATO and ATRA induce
irreversible resistance, and certain patients with APL ultimately
succumb to treatment-resistant diseases (5,6).
Therefore, the unifying mechanisms required for myeloid
differentiation and response to therapy in APL require further
investigation.
Long non-coding RNAs (lncRNAs), which are in excess
of 200 nucleotides, are involved in diverse biological processes
including epigenetic regulation, chromosome imprinting,
transcription, splicing, translation, cell-cycle control, and
differentiation (7). They have been
functionally coupled to cancer and cellular differentiation. The
investigation of lncRNA expression and function may contribute to
the current understanding of leukemogenesis, and the identification
of novel therapeutic targets and seminal posttranscriptional
factors associated with resistance to chemotherapy. Consequently,
the global aberrant expression of lncRNAs that occurs during
myeloid differentiation can assist in enhancing the efficacy of
current APL therapies and identifying novel therapeutic targets for
chemotherapies. lncRNAs have been considered as prognostic and
diagnostic molecular biomarkers for acute myeloid leukemia (AML)
(8,9). Previous investigations have
demonstrated that lncRNAs are crucial in myeloid differentiation
and APL therapy (10–12). HOTAIRM1, a myeloid-specific lncRNA,
has been defined as a key factor in ATRA-induced APL
differentiation (11,13). Furthermore, HOTAIRM1 can promote the
degradation of PML-RARα via a pathway associated with autophagy
during myeloid cell differentiation or ATRA-induced APL
differentiation (10). This
suggests that aberrant lncRNA expression may be underlying targets
for APL therapy and indicators for response to APL therapy. Several
lncRNAs have been identified in APL-associated myeloid
differentiation (10–13); however, additional key lncRNAs and
their functions in regulating myeloid maturation require
characterization in the APL-associated myeloid differentiation
transcriptome.
NB4 cells represent a suitable cell model to
investigate changes in lncRNA expression between APL cells and
their terminally differentiated counterparts. System analysis of
the transcriptome has been beneficial for the identification of
various pathways or cascades at the transcriptome level associated
with ATRA/ATO-induced cell differentiation (14). These are common in the absence of
the complicated and dynamic intracorporeal synergy between ATRA and
ATO in APL therapy, which may affect cell survival and response to
therapy. Although it has been suggested that lncRNAs are involved
in ATRA/ATO-induced extracorporeal APL differentiation, further
understanding of the lncRNA landscape in ATRA/ATO-induced
intracorporeal APL differentiation is required.
The aim of the present study was to examine lncRNA
profiles and regulatory functions in ATRA-based targeted therapy
for APL differentiation. The lncRNA and messenger RNA (mRNA)
profiles were compared in three post-induction bone marrow (BM)
samples from patients with APL and pre-induction (untreated)
matched controls via whole transcriptome microarray. Subsequently,
10 dysregulated lncRNAs were selected and verified by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis in another 27 APL BM samples. In addition, the functions
of dysregulated lncRNAs were predicted via their co-expressed
mRNAs. The findings identified the lncRNA landscape for myeloid
differentiation in APL and revealed potential mechanisms occurring
due to dysregulated lncRNA expression in ATRA-induced APL
differentiation; this may provide underlying targets for APL
therapy and lncRNA biomarkers for APL responses to therapy.
Materials and methods
Patient profiles
A total of 30 patients with APL who received
ATRA-based targeted therapy at the Second Affiliated Hospital and
Yuying Children's Hospital of Wenzhou Medical University (Wenzhou,
China) between January 2014 and December 2016 were recruited for
the present study. None of the patients received chemotherapy prior
to targeted therapy. Written informed consent was collected from
all the patients in conformity with the Declaration of Helsinki,
and the present study obtained permission from the Ethics Committee
of Wenzhou Medical University. The BM samples from the patients
with APL at diagnosis and corresponding BM samples from patients
with APL post-induction were collected in BD Vacutainer Heparin
tubes (BD Biosciences, San Diego, CA, USA). Patient diagnoses were
determined according to the 2016 World Health Organization criteria
(15). The patients with APL were
treated according to the International APL guidelines (15). Among the 60 samples, three paired
samples (comprising three post-induction samples and three
pre-induction samples) were used for lncRNA microarray analysis and
the other samples were used for RT-qPCR analysis. The primary
characteristics of the patients with APL are listed in Table I.
 | Table I.Primary characteristics of patients
with APL. |
Table I.
Primary characteristics of patients
with APL.
| Characteristic | n (%) |
|---|
| Patients | 30 |
| Sex |
|
Male | 18 (60.0) |
|
Female | 12 (40.0) |
| Age (years) |
| Median
(range) | 40 (19–67) |
| WHO
classification |
| APL
with t(15;17)(q22;q12); PML-RARa | 30 (100.0) |
|
Transcripts of PML-RARa | 30 (100.0) |
|
PML-RARa bcr1 | 18 (60.0) |
|
PML-RARa bcr2 | 1 (3.3) |
|
PML-RARa bcr3 | 11 (36.7) |
|
Received ATRA-based
therapy | 30 (100.0) |
| Paired
BM post-induction obtained | 30 (100.0) |
Sample collection and RNA
extraction
The mononuclear BM cells (MBMCs) were isolated from
~2 ml heparinized BM samples using density gradient medium
centrifugation (800 × g, for 20 min at room temperature).
Subsequently, 1×107 cells were resuspended in
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and temporarily stored at −80°C until
further analysis. Total RNA was extracted from the MBMCs according
to the manufacturer's protocol and dissolved in 100 µl
nuclease-free water. The RNA yield was measured using a NanoDrop
ND-2000 spectrophotometer from Thermo Fisher Scientific, Inc. and
the RNA integrity was assessed using an Agilent 2100 bioanalyzer
system (Agilent Technologies, Inc., Santa Clara, CA, USA). When the
28S:18S ratio was ascertained and the RNA integrity number (RIN) of
each sample was assigned; RNA samples with a 28S:18S ratio ≥0.7 and
RIN ≥7.0 were further analyzed.
lncRNA and mRNA microarray expression
profiling
RNA from each sample (~200 ng) was applied for
lncRNA and mRNA microarray analyses using Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm).
Gene expression was analyzed using an Affymetrix
GeneChip® Human Transcriptome Array 2.0 (Affymetrix,
Inc., Santa Clara, CA, USA). The microarray contained 67,539 probes
for 22,829 human lncRNAs and 44,710 human mRNAs, which were derived
from eight authoritative databases, including RefSeq, Ensembl,
UCSC, MGC, nocode, lncRNAdb, Broad Institute, TUCP catalogue and
Human Body Map lincRNAs. Additionally, the microarray contained
probes for small non-coding RNAs, but not microRNAs, and the
majority of these were small nuclear RNAs and small nucleolar RNAs.
The array experiments and computational analysis were performed
according to the manufacturer's protocol (Affymetrix, Inc.). The
raw data were extracted and standardized using the GeneChip Command
Console software 4.0 and Expression Console software 1.3.1 from
Affymetrix, Inc. Additional data processing was performed with
GeneSpring software 12.5 (Agilent Technologies, Inc.). Dysregulated
lncRNAs or mRNAs defined by an absolute value of fold change (FC)
≥2.0 and P≤0.05 (Student's t-test) were selected for further
analysis. The dysregulated mRNAs were categorized into different
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes
(KEGG) annotation groups. The lncRNA chip experiments were
performed in the laboratory at Shanghai OE Biotech Co., Ltd.
(Shanghai, China).
RT-qPCR verification of 10
dysregulated lncRNAs
The lncRNA and mRNA expression were analyzed using
FastStart Universal SYBR-Green Master Mix (Rox) from Roche
Diagnostics (Indianapolis, IN, USA) and an ABI ViiA™ 7 Real-Time
PCR system from Applied Biosystems (Thermo Fisher Scientific,
Inc.). Briefly, the total RNA was transcribed into cDNA using a
Transcriptor First Strand cDNA Synthesis kit (Roche Diagnostics) as
per the manufacturer's protocol. PCR amplification was performed in
a 25 µl reaction containing 1 µl of cDNA template (~10 ng), 12.5 µl
of FastStart Universal SYBR-Green Master Mix (Rox), 10.5 µl of
nuclease-free water, and 0.5 µl of each pair of primers (Shanghai
GeneCore BioTechnologies Co., Ltd., Shanghai, China). The RT-qPCR
primers for the lncRNAs are listed in Table II. The reaction conditions were as
follows: 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec
and 60°C for 45 sec. All experiments were repeated three times in
parallel. The expression of lncRNAs was further standardized to the
GAPDH gene and calculated using the 2−ΔΔCq method
(16).
| Table II.Reverse transcription-quantitative
polymerase chain reaction primers for lncRNAs. |
Table II.
Reverse transcription-quantitative
polymerase chain reaction primers for lncRNAs.
| lncRNA primary
ID | Primer sequence
(5′-3′) | Product (bp) |
|---|
| NONHSAT121385 | Forward:
TACATGTTCCTGGTGAAAT | 108 |
|
| Reverse:
GGCATCAAGTATGTCTCT |
|
|
ENST00000469418 | Forward:
CAATTTACGGCTGGACGTTT | 128 |
|
| Reverse:
GAAAGGAATGCTGGGAAACA |
|
|
ENST00000424415 | Forward:
ATATTGAGATAGGAGGATGG | 107 |
|
| Reverse:
GGCTTCTTCTAGGATAAGT |
|
| NR_003186 | Forward:
TACATGTTCCTGGTGAAAT | 115 |
|
| Reverse:
ATCTTTGGGCATCAAGTA |
|
|
ENST00000536425 | Forward:
TTGAATAATCCTAAATTATACATAC | 78 |
|
| Reverse:
TCATAGTGACTAAATTGAATAAGTACCAAA |
|
| NONHSAT061249 | Forward:
AGGATCGCTTGAGATGCAGT | 110 |
|
| Reverse:
GCTACCGCTCTCAAGTTTGG |
|
|
TCONS_00017553-XLOC_008249 | Forward:
GTGTCTGTGTGTACAGAA | 187 |
|
| Reverse:
ACATTCCATACACACAAAC |
|
|
TCONS_l2_00030950-XLOC_l2_015963 | Forward:
GTTGGAAGATGAAGGAAC | 114 |
|
| Reverse:
ATCACTGTGTAAAGGACTA |
|
| NONHSAT076891 | Forward:
GGATCTCCCCTGTGTTCTCA | 146 |
|
| Reverse:
GACCAGGTAGTGGGGGAAGT |
|
|
TCONS_00022632-XLOC_010933 | Forward:
TCTTCCACGTAACAACCA | 124 |
|
| Reverse:
CTGACAGTGTCTTCCATA |
|
Microarray results analysis and
functional prediction of selected dysregulated lncRNAs
The identification of the overall functional
distributions for the dysregulated lncRNAs identified in the
experiment were performed as follows. Briefly, the co-expressed
mRNAs for each dysregulated lncRNA were first calculated, and
functional enrichment analysis for the set of co-expressed mRNAs
was then performed. The enriched functional terms served as the
predicted functional term for a given lncRNA. Furthermore, the
co-expressed mRNAs of lncRNAs were identified by calculating the
Pearson's correlation (P<0.05). The functional enrichment terms
for annotating the co-expressed mRNAs were determined using the
hypergeometric cumulative distribution function (17,18).
The top 200 reliable prediction associations between the lncRNAs
and the predicted functional terms were selected to reflect the
functional distribution of the dysregulated lncRNAs. The frequency
of each predicted functional term for these associations was
determined, following which the GO term and KEGG term with more
functional annotations were statistically identified (19).
Identification of cis-regulated mRNAs
for the dysregulated lncRNAs
The present study further examined how the
dysregulated lncRNAs may exert activities through cis-
and/or trans-regulated mRNAs. The regions of
cis-regulation were identified as follows: Gene locations
for different lncRNAs on the chromosome were determined; for each
dysregulated lncRNA, the mRNAs were identified as
cis-regulated mRNAs when the co-expressed mRNA loci were
within 300 kbp downstream or upstream of the given lncRNA and the
Pearson's correlation of lncRNA-mRNA expression was significant at
the P<0.05 level. When the mRNAs did not conform to the
cis-regulated mRNA rules, they were identified as possible
trans-regulated mRNAs.
Identification of transcription
factors associated with dysregulated lncRNAs
It has been documented that specific lncRNAs may be
involved in certain biological processes, including transcriptional
regulation, through key transcription factors (TFs) (17). Therefore, the TF/chromatin
regulation complexes that may have critical co-regulatory roles
with lncRNAs were identified (18,20).
Briefly, the set of co-expressed mRNAs for lncRNA and TF/chromatin
regulation complex target genes was determined, the enrichment
level of which was analyzed using the hypergeometric distribution.
Therefore, the TFs prominently associated with dysregulated lncRNAs
were finally determined. The lncRNA-TF network was established
using the hypergeometric distribution, and a graph showing the
associations between TFs and lncRNAs was drawn using Cytoscape
software (version 3.6.1; http://www.cytoscape.org/), an open source software
platform. In the network, the core TF with the highest degree of
expression was considered the centre of highest importance.
Results
General expression profiles of
dysregulated lncRNAs and mRNAs
To investigate the lncRNA landscape involved in
ATRA-induced APL differentiation, lncRNA and mRNA microarray
analyses were performed on BMMCs from patients with APL. The
microarray data were filtered through a volcano plot to determine
the dysregulated lncRNAs and mRNAs in BM samples from patients with
APL post-induction and corresponding BM samples from patients with
APL at diagnosis (Fig. 1A). The
lncRNA and mRNA expression data were clustered using Cluster 3.0
(Fig. 1B). Based on the similar
expression patterns, the samples were further classified into two
groups using dendrogram-based methods for clustering. It was found
that 825 lncRNAs were dysregulated between the patients with APL
post-induction and the matched controls at diagnosis, with 410
upregulated and 415 downregulated lncRNAs (Fig. 1C). Among the dysregulated lncRNAs,
NONHSAT076891 was upregulated the most, with an FC of 304.00,
whereas TCONS_00022632-XLOC_010933 was downregulated the most, with
an FC of 447.09. It was also found that 1,218 mRNAs were
dysregulated, with 660 upregulated and 558 downregulated mRNAs
(Fig. 1C). The most upregulated and
downregulated mRNAs were MPEG1 and CYTL1, with FCs of 257.13 and
1,169.37, respectively. The top 20 upregulated and downregulated
lncRNAs and mRNAs are listed in Table
III.
| Table III.Top 20 upregulated and downregulated
lncRNAs and mRNAs in patients with APL with induction therapy. |
Table III.
Top 20 upregulated and downregulated
lncRNAs and mRNAs in patients with APL with induction therapy.
| Upregulated
lncRNAs | FC (abs) | Downregulated
lncRNAs | FC (abs) | Upregulated
mRNAs | FC (abs) | Downregulated
mRNAs | FC (abs) |
|---|
| NONHSAT076891 | 304.001 |
TCONS_00022632-XLOC_010933 | 447.093 | MPEG1 | 257.128 | CYTL1 | 1,169.366 |
| NONHSAT066562 | 301.188 |
ENST00000419668 | 145.949 | LRRK2 | 235.480 | HGF | 357.526 |
| NONHSAT104550 | 272.525 | NONHSAT032682 | 141.184 | PLBD1 | 223.158 | CPA3 | 206.801 |
| NONHSAT121385 | 86.674 |
TCONS_00022633-XLOC_010934 | 79.771 | P2RY13 | 220.328 | FLT3 | 119.563 |
| NR_003186 | 85.927 |
ENST00000429159 | 63.192 | CLEC7A | 165.126 | CFD | 82.293 |
| NR_003187 | 63.356 | NR_034105 | 52.276 | VCAN | 163.269 | KIT | 77.132 |
| NR_028324 | 53.106 | NR_003611 | 44.230 | FGL2 | 157.751 | PRSS57 | 75.924 |
|
TCONS_l2_00031106-XLOC_l2_016008 | 47.907 | NONHSAT021421 | 43.840 | RAB31 | 146.390 | CCNA1 | 75.130 |
| NR_039983 | 46.826 |
TCONS_00008717-XLOC_004231 | 43.062 | IFI30 | 138.929 | ITM2A | 73.741 |
|
ENST00000469418 | 42.355 |
ENST00000545120 | 40.173 | VNN2 | 101.935 | MAMDC2 | 40.283 |
|
TCONS_00028171-XLOC_013531 | 41.054 |
ENST00000579458 | 36.424 | S100A12 | 92.761 | STAB1 | 35.361 |
| NONHSAT144652 | 38.665 |
ENST00000535351 | 34.801 | C5AR1 | 81.921 | GTSF1 | 33.527 |
| NONHSAT060114 | 38.653 |
ENST00000423708 | 32.984 | SAMHD1 | 65.141 | RFX8 | 31.113 |
| NONHSAT097386 | 35.242 |
TCONS_l2_00004167-XLOC_l2_001953 | 30.059 | HLA-DRA | 63.702 | MMP2 | 26.319 |
| NONHSAT051747 | 32.846 |
ENST00000413504 | 24.286 | NCF2 | 59.800 | TDRD9 | 24.590 |
| NONHSAT150916 | 32.737 |
ENST00000451267 | 24.286 | GLT1D1 | 57.236 | UGT3A2 | 24.494 |
| NONHSAT120680 | 27.076 |
ENST00000426063 | 21.941 | STX11 | 55.971 | ZNF711 | 20.705 |
| NONHSAT098146 | 21.665 |
TCONS_l2_00003557-XLOC_l2_001971 | 21.569 | IGF2R | 54.459 | BLID | 16.816 |
|
TCONS_00013664-XLOC_006324 | 19.262 |
TCONS_l2_00003558-XLOC_l2_001972 | 19.349 | FLJ45445 | 53.106 | CALR | 16.644 |
| NONHSAT102906 | 17.430 |
ENST00000514473 | 19.153 | CEACAM1 | 50.279 | GABRE | 15.980 |
Verification of dysregulated
expression of lncRNAs and mRNAs
To verify the dysregulated expression of lncRNAs and
mRNAs associated with ATRA-induced APL differentiation, RT-qPCR
analysis was performed to examine the upregulation or
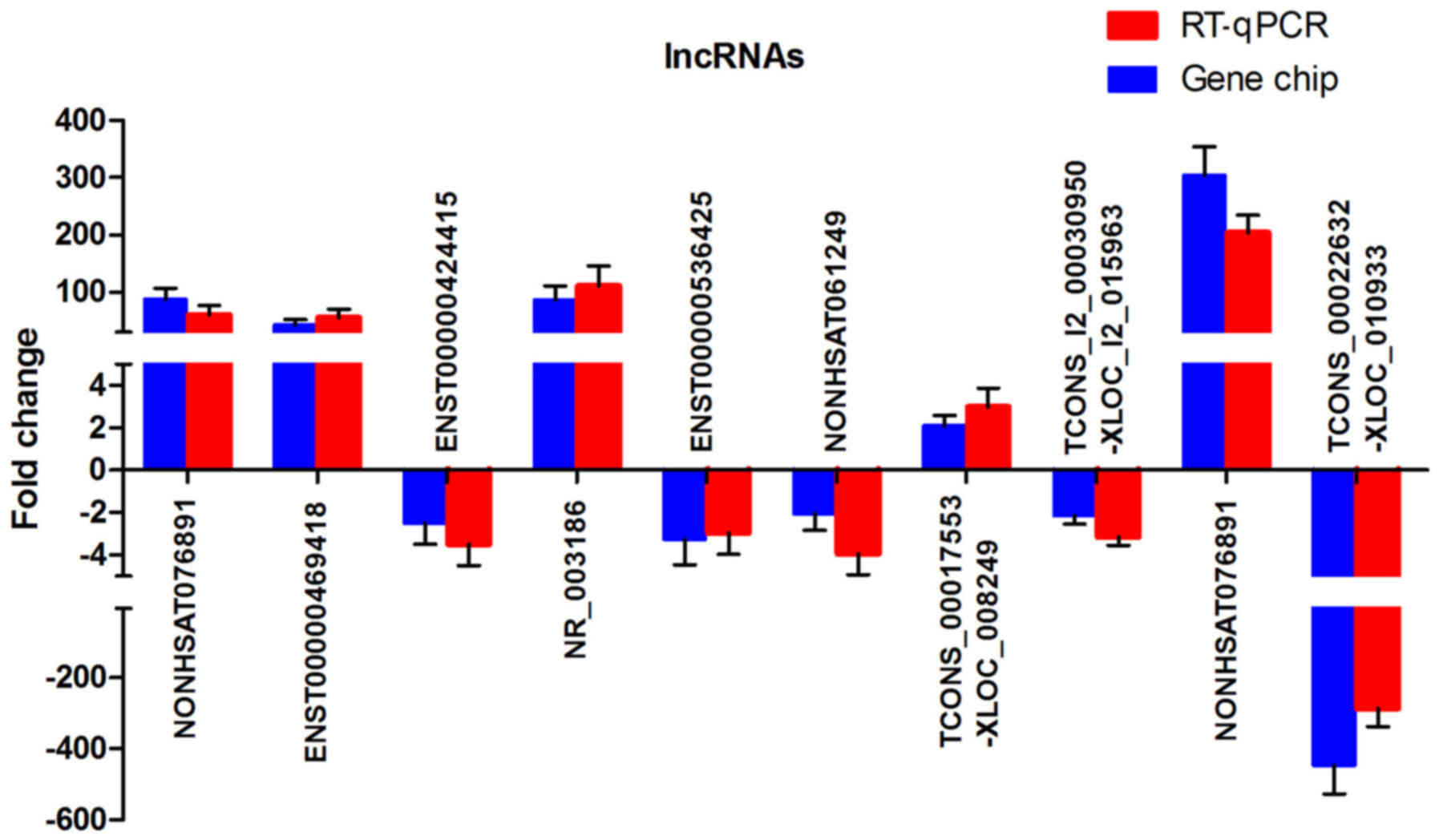
downregulation of the genes. A total of 10 dysregulated lncRNA
transcripts were selected for RT-qPCR analysis. These 10 lncRNAs
consisted of four randomly selected lncRNAs, including
NONHSAT121385, ENST00000469418, ENST00000424415, and NR_003186, and
six specifically selected lncRNAs, including ENST00000536425,
NONHSAT061249, TCONS_00017553-XLOC_008249,
TCONS_l2_00030950-XLOC_l2_015963, NONHSAT076891, and
TCONS_00022632-XLOC_010933. ENST00000536425 and NONHSAT061249 were
selected as a result of their predicted cis-regulating
potential. TCONS_00017553-XLOC_008249 and
TCONS_l2_00030950-XLOC_l2_015963 were selected due to their
predicted trans-regulating potential and their presence
among the top 100 lncRNA-TF pairs. NONHSAT076891 and
TCONS_00022632-XLOC_010933 were selected as they were the most
upregulated and downregulated, respectively (Table III). The RT-qPCR results were in
agreement with the results obtained from the microarray chip
analysis (Fig. 2). As shown in
Fig. 2, all selected lncRNAs were
dysregulated and exhibited the same trend of upregulation or
downregulation (P<0.05 for each lncRNA, Student's t-test).
lncRNA and mRNA co-expression profiles
and lncRNA function prediction
Several hundred lncRNAs were co-expressed with
hundreds of mRNAs. For example, ENST00000419668 was co-expressed
with 515 mRNAs and TCONS_00022632-XLOC_010933 with 488 mRNAs. The
GO and KEGG pathway annotations of the co-expressed mRNAs were used
to predict the functions of the dysregulated lncRNAs. The lncRNAs
were clustered into hundreds of GO and KEGG terms with more
functional annotations. In the corresponding association between
the ‘lncRNA name’ and ‘functional prediction term’, the top 200
predicted associations were selected to reflect the functional
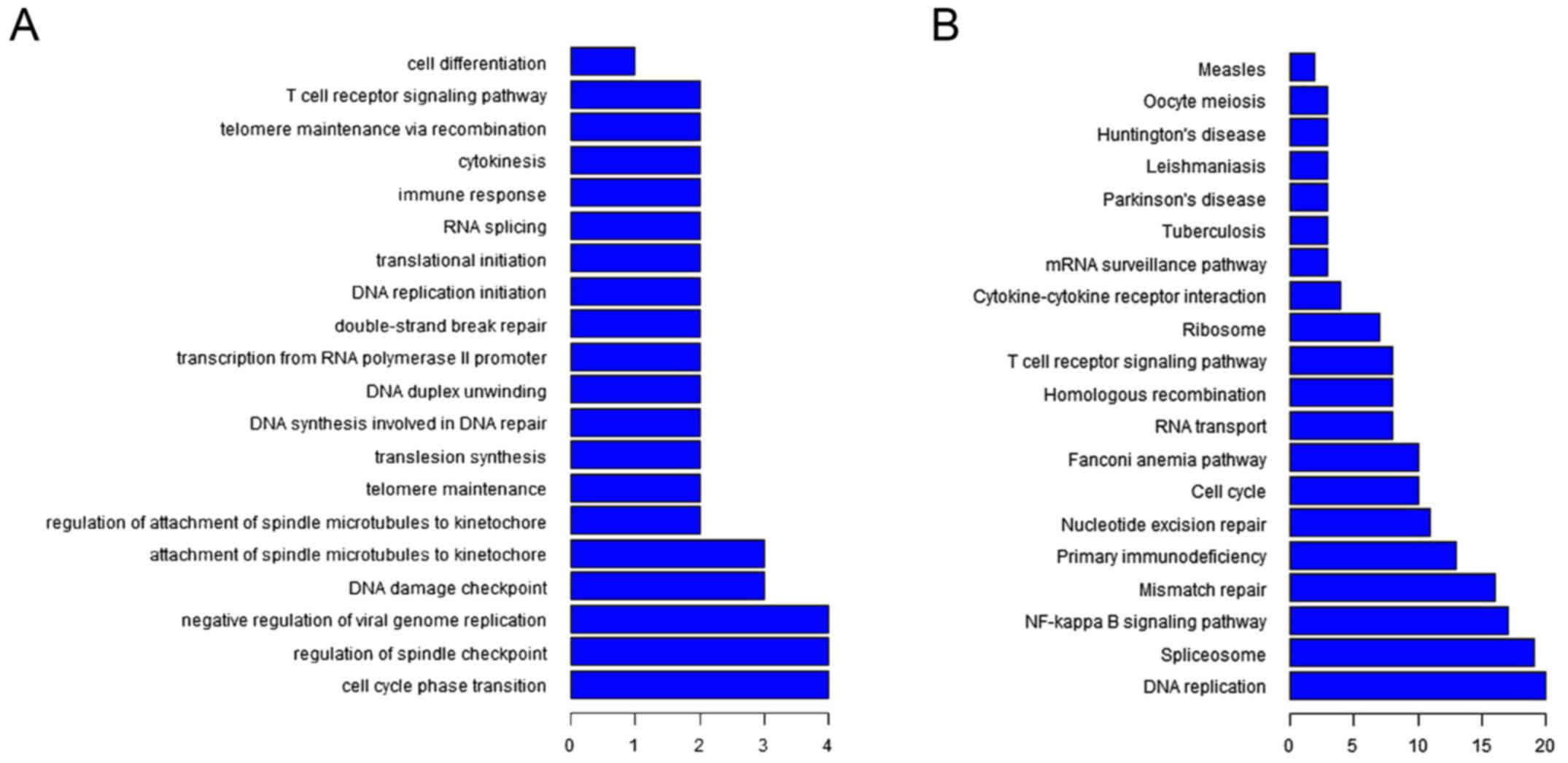
distribution of the dysregulated lncRNAs. Among the GO terms, the
most common biological processes for the dysregulated lncRNAs were
cell cycle phase transition, regulation of spindle checkpoint,
negative regulation of viral genome replication, DNA damage
checkpoint, and attachment of spindle microtubules to kinetochore
(Fig. 3A). The most common KEGG
terms were DNA replication, spliceosome, NF-κB signalling pathway,
mismatch repair, primary immunodeficiency, nucleotide excision
repair, and cell cycle (Fig. 3B).
Therefore, according to the enrichment, cell cycle phase transition
was the most enriched GO term and DNA replication was the most
enriched KEGG term (Fig. 3).
Analysis of ‘cis’ lncRNAs and their
adjacent co-expressed mRNAs
Evidence shows that a number of lncRNAs may
cis-regulate the transcription of themselves and their
adjacent mRNAs by recruiting remodelling factors to local chromatin
(21). To determine the potential
‘cis’ mRNAs associated with a specific lncRNA, the
co-expressed mRNAs 300 kb upstream and downstream of the
dysregulated lncRNAs were analyzed. In total, 48 lncRNAs and their
predictively cis-regulated mRNAs were identified using
accurate genomic mapping based on the criteria mentioned above. The
lncRNAs and their underlying cis-regulated mRNAs are listed
in Table IV. The results suggested
that lncRNA ENST00000536425 cis-regulates one mRNA, early
endosome antigen 1 (EEA1) (Fig.
4A), whereas lncRNA NONHSAT061249 cis-regulates two
mRNAs, including zinc finger protein 564 (ZNF564) and zinc finger
protein 44 (ZNF44) (Fig. 4B).
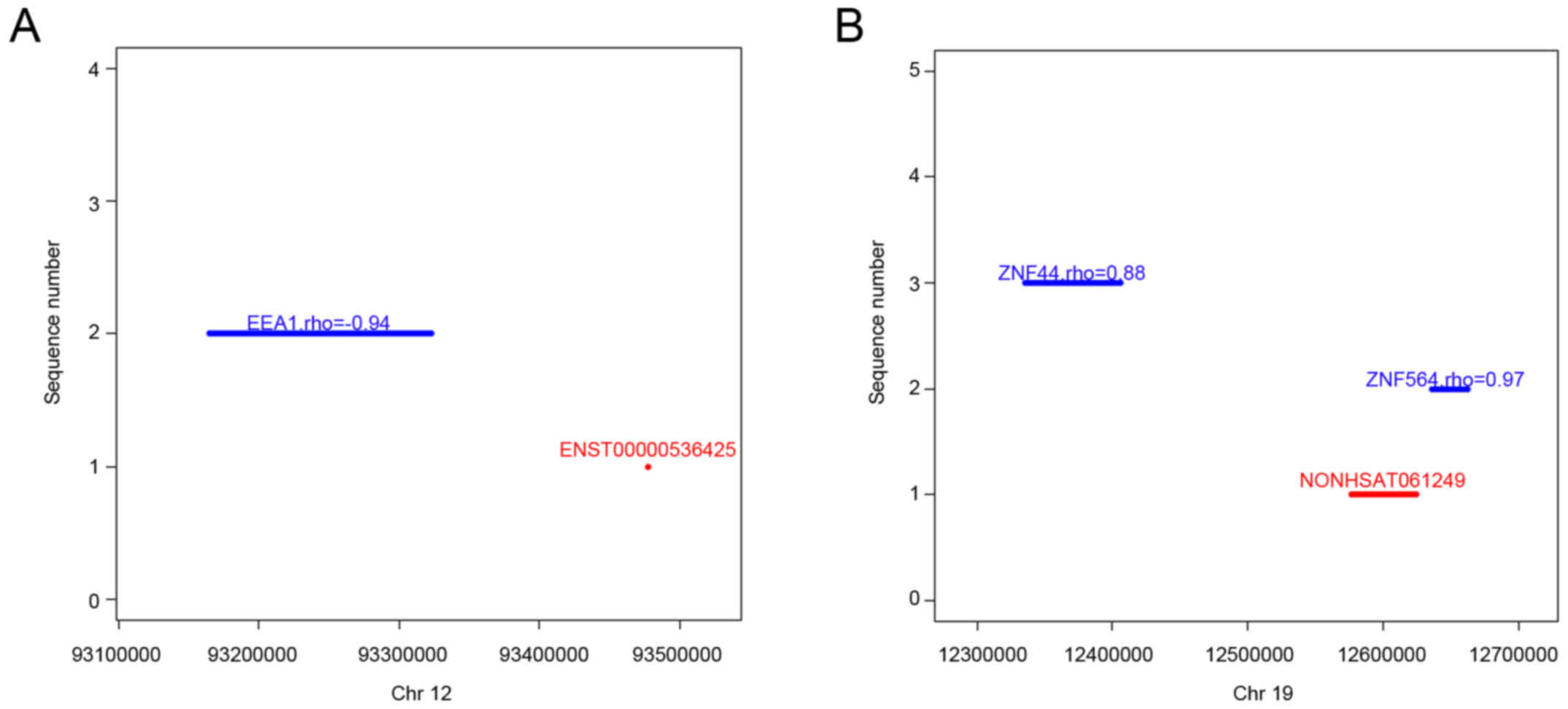 | Figure 4.Representative lncRNAs, their
‘cis’ mRNAs, and their positions on chromosomes. (A)
Co-expression of ENST00000536425 and downstream EEA1 mRNA on
chromosome 12. (B) Co-expression of NONHSAT061249 and downstream
ZNF564 or ZNF44 mRNA on chromosome 19. The abscissa shows the
genomic position, the red line/point indicates lncRNA genomic
position, the blue line indicates the mRNA location, the rho value
represents the correlation coefficients between the lncRNA and its
‘cis’ mRNA. lncRNAs, long non-coding RNAs; mRNAs, messenger
RNAs; EEA1, early endosome antigen 1; ZNF564, zinc finger protein
564; ZNF44, zinc finger protein 44. |
| Table IV.All lncRNA transcripts and their
potentially cis-regulated mRNA transcripts. |
Table IV.
All lncRNA transcripts and their
potentially cis-regulated mRNA transcripts.
| Chrom | Strand | Correlation | lncRNA primary
ID | txStart | txEnd | mRNA primary
ID | txStart | txEnd |
|---|
| 10 | − | −0.846333103 |
ENST00000437232 | 17256238 | 17271983 | CUBN | 16865963 | 17171830 |
| 10 | − | −0.889863566 |
ENST00000439671 | 94428529 | 94429500 | IDE | 94211441 | 94333852 |
| 10 | − | −0.827414528 |
ENST00000453853 | 46798370 | 46809021 | SYT15 | 46952762 | 46971400 |
| 15 | + | −0.844858112 |
ENST00000499528 | 64663186 | 64667537 | TRIP4 | 64679952 | 64747502 |
| 5 | + | −0.821398914 |
ENST00000512856 | 96120481 | 96121703 | LNPEP | 96271098 | 96373219 |
| 11 | − | −0.931044512 |
ENST00000525500 | 62357121 | 62357959 | UBXN1 | 62443970 | 62446562 |
| 11 | − | 0.904455214 |
ENST00000525500 | 62357121 | 62357959 | HNRNPUL2 | 62480097 | 62494857 |
| 11 | − | −0.832660059 |
ENST00000525500 | 62357121 | 62357959 | AHNAK | 62201016 | 62314332 |
| 12 | − | −0.936840914 |
ENST00000536425 | 93477374 | 93477451 | EEA1 | 93164413 | 93323107 |
| 14 | − | 0.933330841 |
ENST00000555403 | 50793244 | 50794627 | L2HGDH | 50704281 | 50779266 |
| 14 | + | 0.908030062 |
ENST00000555460 | 22886826 | 22887645 | TRAV38-1 | 22739821 | 22740446 |
| 14 | + | 0.871856814 |
ENST00000555460 | 22886826 | 22887645 | TRAJ61 | 22944306 | 22944365 |
| 14 | + | 0.825846372 |
ENST00000555460 | 22886826 | 22887645 | TRAV30 | 22636293 | 22636884 |
| 1 | − | −0.833908257 | NONHSAT005886 | 145372088 | 145373798 | POLR3GL | 145456236 | 145470388 |
| 1 | + | 0.83645797 | NONHSAT007706 | 173865073 | 173871776 | DARS2 | 173793641 | 173827684 |
| 11 | − | −0.901219661 | NONHSAT021765 | 62334412 | 62340225 | HNRNPUL2 | 62480097 | 62494857 |
| 11 | − | 0.874686208 | NONHSAT021765 | 62334412 | 62340225 | UBXN1 | 62443970 | 62446562 |
| 11 | − | −0.844180413 | NONHSAT025134 | 130434325 | 130628495 | ADAMTS8 | 130274818 | 130298888 |
| 12 | − | 0.834550722 | NONHSAT028485 | 54059021 | 54062993 | CALCOCO1 | 54104902 | 54121529 |
| 12 | − | 0.904345693 | NONHSAT031503 | 122826057 | 122839031 | ZCCHC8 | 122956146 | 122985543 |
| 14 | + | −0.837693156 | NONHSAT035752 | 22564302 | 22564896 | TRAV41 | 22788568 | 22789123 |
| 14 | + | −0.820822874 | NONHSAT035752 | 22564302 | 22564896 | TRAV12-1 | 22309321 | 22309956 |
| 16 | − | −0.864461252 | NONHSAT051747 | 223162 | 223620 | AXIN1 | 337440 | 402676 |
| 19 | − | 0.971374656 | NONHSAT061249 | 12576359 | 12624647 | ZNF564 | 12636184 | 12662356 |
| 19 | − | 0.880808619 | NONHSAT061249 | 12576359 | 12624647 | ZNF44 | 12335501 | 12405714 |
| 5 | − | −0.889390591 | NONHSAT101116 | 39105326 | 39105857 | RICTOR | 38938021 | 39074510 |
| 5 | − | 0.827904836 | NONHSAT101123 | 39383148 | 39393457 | DAB2 | 39371776 | 39462402 |
| 5 | + | −0.83173824 | NONHSAT102906 | 96071868 | 96077284 | LNPEP | 96271098 | 96373219 |
| 5 | − | −0.921818421 | NONHSAT104550 | 149782674 | 149782885 | RBM22 | 150070352 | 150080669 |
| 7 | + | 0.833858641 | NONHSAT119665 | 27135713 | 27139877 |
OTTHUMG00000065294 | 27420181 | 27420825 |
| X | + | 0.82095135 | NONHSAT137231 | 54841718 | 54842362 | PAGE2B | 55101496 | 55105342 |
| 19 | − | 0.83717369 | NONHSAT180547 | 52359056 | 52391189 | SIGLEC14 | 52114781 | 52150151 |
| 22 | − | 0.859220203 | NONHSAT192618 | 18710726 | 18711888 | CLTCL1 | 18900087 | 19279239 |
| 1 | + | −0.82131915 | NR_003133 | 89873238 | 89890493 | GBP6 | 89829436 | 89853719 |
| 6 | + | −0.865567866 | NR_003288 | 160514114 | 160517244 | SLC22A1 | 160542805 | 160579750 |
| 19 | + | −0.90635912 | NR_024247 | 1285890 | 1378430 | DAZAP1 | 1407584 | 1435683 |
| 4 | + | 0.882358648 | NR_036694 | 88812995 | 88815167 | DMP1 | 88571454 | 88585513 |
| 7 | + | −0.879682301 |
TCONS_00012852-XLOC_006277 | 141870970 | 141923474 | TRBV4-2 | 142045252 | 142045816 |
| 7 | + | −0.852206355 |
TCONS_00012852-XLOC_006277 | 141870970 | 141923474 | TRBV2 | 142000747 | 142211011 |
| 10 | − | −0.823174131 |
TCONS_00017969-XLOC_008805 | 46798370 | 46809021 | SYT15 | 46952762 | 46971400 |
| 1 | − | −0.880035509 |
TCONS_l2_00002788-XLOC_l2_001347 | 228155359 | 228158853 | C1orf148 | 228351787 | 228353213 |
| 12 | + | −0.842080292 |
TCONS_l2_00005685-XLOC_l2_002994 | 53547053 | 53548417 | ESPL1 | 53662083 | 53778657 |
| 12 | + | 0.933722808 |
TCONS_l2_00005705-XLOC_l2_003009 | 56904734 | 56906923 | IL23A | 56732663 | 56734194 |
| 15 | + | −0.853220916 |
TCONS_l2_00008528-XLOC_l2_004602 | 30846790 | 30848689 | ARHGAP11B | 30916697 | 30931023 |
| 17 | + | −0.948957318 |
TCONS_l2_00011527-XLOC_l2_005686 | 18330175 | 18333940 | TRIM16L | 18601311 | 18639432 |
| 17 | + | 0.815854971 |
TCONS_l2_00011527-XLOC_l2_005686 | 18330175 | 18333940 | LGALS9C | 18380098 | 18398259 |
| 19 | + | 0.840205468 |
TCONS_l2_00012696-XLOC_l2_006815 | 54278778 | 54279108 | MIR516A2 | 54264387 | 54264476 |
| 2 | + | −0.866656281 |
TCONS_l2_00014331-XLOC_l2_007832 | 242626503 | 242633704 | ING5 | 242641450 | 242668896 |
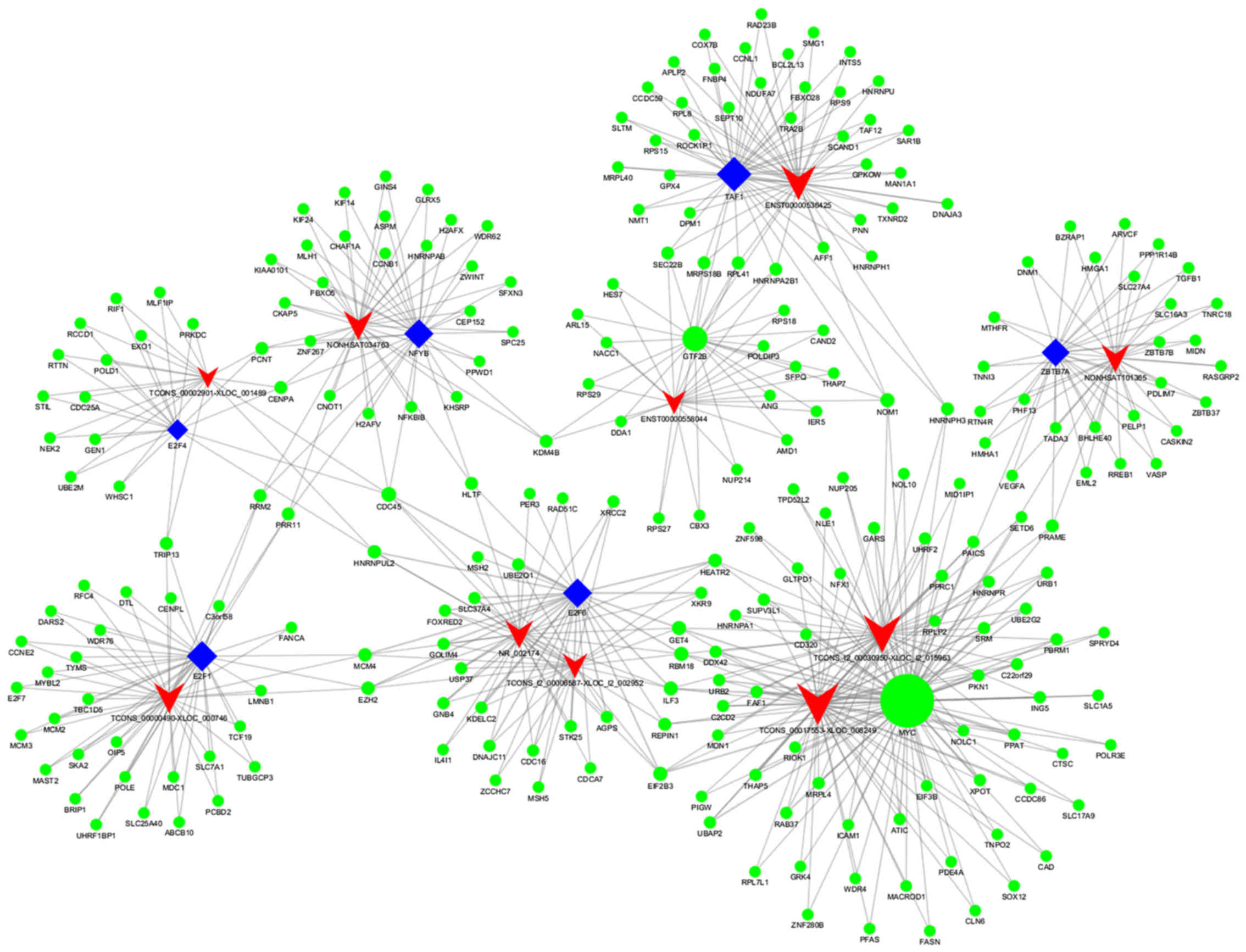
Dysregulated lncRNA ‘trans’ mechanism
and construction of the TF-lncRNA-target gene network
As numerous dysregulated lncRNAs were involved in
mRNA regulation, a ‘TF-lncRNA’ network is likely to be large and
complex. Consequently, the top 100 associations were selected to
generate a core TF-lncRNA network, visualized by hypergeometric
distribution analysis (Fig. 5). The
core TF-lncRNA network map for patients with APL post-induction,
vs. matched controls at diagnosis is shown in Fig. 5A. The majority of potential
trans-regulation lncRNAs were found to be involved in
pathways regulated by three TFs, including E2F transcription factor
1 (E2F1), E2F transcription factor 6 (E2F6), and early B cell
factor 1 (EBF1) (Fig. 5B). In the
core network of lncRNA-TF pairs, E2F1, E2F6, and EBF1 regulated the
expression of 16 lncRNAs, 13 lncRNAs and 10 lncRNAs,
respectively.
Based on the lncRNA co-expression results, the
target genes were incorporated into a ‘TF-lncRNA’ network to
determine the TF-lncRNA-target network. Due to the large and
complex networks, the top 10 associations were selected to produce
a core TF-lncRNA-target network map (Fig. 6). The core TF-lncRNA-target gene
association for patients with APL post-induction therapy, vs.
pre-induction therapy is shown in Fig.
6, and includes 10 dysregulated lncRNAs
(TCONS_l2_00006587-XLOC_l2_002952, ENST00000536425, NR_002174,
TCONS_00002901-XLOC_001489, ENST00000558044, NONHSAT101365,
TCONS_00017553-XLOC_008249, TCONS_00000490-XLOC_000746,
NONHSAT034763, and TCONS_l2_00030950-XLOC_l2_015963), 247 target
genes, and eight TFs (TAF1, GTF2B, E2F4, ZBTB7A, E2F1, NFYB, MYC,
and E2F6) in the core map. As shown in Fig. 6, the core TF MYC association
regulated two lncRNAs (TCONS_00017553-XLOC_008249 and
TCONS_l2_00030950-XLOC_l2_015963) and 97 target genes. As observed
for ‘MYC-TCONS_00017553-XLOC_008249-RIOK1’ in this map, target
genes, including RIOK1, were co-expressed for
TCONS_00017553-XLOC_008249. As observed for ‘MYC-
TCONS_l2_00030950-XLOC_l2_015963-RPLP2’ in the map, target genes,
including RPLP2, were co-expressed for
TCONS_l2_00030950-XLOC_l2_015963-RPLP2. Therefore, these maps
provided vital information on the lncRNAs, TFs and target
genes.
Discussion
In the present study, the expression patterns of
genome-wide lncRNAs were first evaluated in BM samples from
patients with APL post-induction and corresponding BM samples from
patients with APL at diagnosis using microarray analysis. Their
potential functions were then examined by analysing their
co-expressed mRNAs. The experimental results showed that 825
lncRNAs and 1,218 mRNAs were dysregulated. Furthermore, 10 selected
dysregulated lncRNAs were validated by RT-qPCR analysis. Several
hundred lncRNAs were co-expressed with hundreds of mRNAs, and a
number of these may contribute to ATRA/ATO-induced intracorporeal
myeloid differentiation by affecting these mRNAs in trans
and/or in cis. The present study not only clarified the
contributions of lncRNAs to myeloid differentiation in APL and/or
intracorporeal therapy response, but also elucidated possible
dysregulated lncRNA expression mechanisms associated with
ATRA-induced APL differentiation.
APL has shifted from a complex problem in the past
into a paradigm with successful targeted therapies, and a series of
published randomized clinical trials in patients with APL have all
demonstrated efficacy on the frontline of ATRA/ATO association. The
identification of a novel class of lncRNAs has attracted attention
and may have encouraged investigations focused on characterizing
lncRNAs associated with myeloid differentiation and responses to
APL therapy. For example, several lncRNAs, including NEAT1 and
HOTAIRM1, are indispensable during APL differentiation induced by
ATRA (10,12,22).
However, numerous lncRNAs and their roles in APL differentiation
remain to be fully elucidated. A systematic analysis of the
ATRA/ATO-induced intracorporeal myeloid differentiation
transcriptome may reflect the complicated and dynamic
intracorporeal synergy between ATRA and ATO in patients with APL.
lncRNA and mRNA analysis in BM from patients with APL is expected
to reflect real changes.
To examine global lncRNA and mRNA profiling in the
present study, the Human Transcriptome Array 2.0 was used to screen
the dysregulated lncRNAs in three patients with APL post- and
pre-induction therapy. The results showed that 825 lncRNAs and
1,218 mRNAs were dysregulated, including 410 upregulated lncRNAs,
415 downregulated lncRNAs, 660 upregulated mRNAs, and 558
downregulated mRNAs, suggesting that these dysregulated lncRNAs may
be involved in ATRA-induced myeloid differentiation. Consequently,
the present study may assist in determining whether these
dysregulated lncRNAs and mRNAs can be applied for the early
assessment of APL therapy response or efficacy. The present study
also cross-validated the dysregulated gene results from the
GeneChip with the results of RT-qPCR analysis; this comparison
showed consistency between the microarrays and RT-qPCR results,
which supports further predictions.
An increasing number of lncRNAs have been recognized
as critical factors in gene programs to control cell
differentiation and function, and serving as scaffolds or decoys at
the transcriptional and translational levels (23). Although the number of novel and
known lncRNAs is increasing exponentially, only a small subset of
these have functional annotations, and the most commonly used
method to predict lncRNA function is by investigating their
co-expressed mRNAs and associated biological pathways. In the
present study, it was found that thousands of mRNAs are
co-expressed with the dysregulated lncRNAs. Through functional
prediction with the co-expressed mRNAs, the present study
identified lncRNAs involved in DNA replication associated with cell
cycle phase transition as the most affected by ATRA-induced myeloid
differentiation. When stabilized and inhibited via cell cycle phase
transitions, lncRNAs function as a vital factor for regulating cell
cycle during the course of myeloid maturation in NB4 APL cells
(13). Therefore, lncRNAs may be a
potential modulator of DNA replication, in addition to certain
regulatory TFs.
Compared with mRNAs, lncRNAs have the inherent
characteristic of cis-regulation (24) and can cis-regulate their
adjacent mRNAs (25). The
epigenetic upregulation of lncRNAs is associated with
cis-downregulation of a functional gene cluster in leukemia
(26). In the present study, 48
lncRNAs were found to potentially promote cis-regulation of
their adjacent mRNAs. Although the majority of the identified
lncRNAs have not been characterized, two sets of lncRNA-mRNAs,
including ENST00000536425 and EEA1 mRNA and NONHSAT061249
and ZNF564 and ZNF44 mRNAs, were identified. A
previous study revealed that EEA1 serves as an identifying
marker of early endosomes for neddylation of type II receptor, and
aberrant neddylation results in the development of leukemia
(27). The biological processes
associated with ZNF564 and ZNF44 are involved in
transcription and transcription regulation, respectively. These
findings suggest that lncRNA-mRNA networks may contribute to the
regulation of cell responses to ATRA-induced APL
differentiation.
Certain lncRNAs have been demonstrated to be
involved in cis-regulation; however, the majority of
characterized lncRNAs are functionally trans-regulating
(18,24). The present study predicted the
trans-regulatory functions of lncRNAs through TFs. In the
central network of lncRNA-TF pairs, the potential
trans-regulatory lncRNAs were mainly trans-regulated
by E2F1, E2F6, and EBF1. E2F1 and E2F6 are TFs that contribute to
controlling cell cycle. Several studies have associated the
activity of E2F with cell-cycle control (28,29).
The E2F1-C/EBPa feedback loop regulates the expression of the
oncogene tribbles 2, which is important for AML cell proliferation
control (30). E2F6 may
transcriptionally regulate cell-cycle G1/S genes via recruiting
BRG1 (29). EBF1, a
TF that is critical for normal and malignant B-lymphocyte
development, controls DNA repair in a dose-dependent manner, which
may explain the reason for the frequent loss of the EBF1
gene in leukemia (31). In the
present study, the dysregulated lncRNAs involved in the pathways
mainly regulated by E2F1, E2F6, and EBF1 were candidate
participants in ATRA-induced myeloid differentiation. Consequently,
the ‘trans’ analysis provides a method to interpret the
functions of lncRNAs and their biological processes in APL
therapy.
The lncRNA-TF analysis identified novel lncRNAs and
three TFs for enriched dysregulated mRNAs that contributed to APL
treatment. The results of the ‘cis’ and ‘trans’
analyses provided essential clues on the modular regulation of
lncRNAs. The data obtained may promote future investigations on
therapeutic mechanisms in APL. However, the present study had
several limitations. First, the sample size was small; thus,
investigations with larger sample sizes are required. Second, as
the functions of several of the identified lncRNAs have not been
annotated, it was only possible to predict the functions of lncRNAs
through network and pathway analyses with their co-expressed mRNAs.
Therefore, the biological functions of these lncRNAs require
further validation.
In conclusion, the present study offered insight
into the genome-wide patterns of lncRNA expression during the
course of ATRA-based APL therapy. A set of dysregulated lncRNAs
were identified in patients with APL who received ATRA-based
therapy compared with untreated matched controls. Several lncRNAs
may be involved in biological pathways associated with ATRA-induced
myeloid differentiation through the cis and/or trans
regulation of mRNAs. Furthermore, targeting aberrantly activated
pathways in APL cells may offer strategies to circumvent or
mitigate disease. The present study provides a foundation for
future investigations on lncRNAs associated with ATRA-based APL
therapy as therapeutic and diagnostic targets by supplying
candidate genes. Additionally, the results provide a platform for
systematically evaluating a large number of lncRNAs and mRNAs to
identify pathways critical for APL elimination using primary APL
patient specimens prior to and following targeted therapy.
Acknowledgements
We thank Professor Yan Li and Professor Yongqing
Tong (Department of Clinical Laboratory, Renmin Hospital of Wuhan
University) for their selfless assistance. We thank Shanghai OE
Biotech Co., Ltd. (Shanghai, China) for providing lncRNA chip
analyses and American Journal Experts (AJE) for providing language
help.
Funding
The present study was supported by the Medical and
Health Research Science and Technology Plan Project of Zhejiang
Province (grant nos. 2017KY112 and 2018KY523), the Public Welfare
Science and Technology Plan Project of Wenzhou City (grant no.
Y20170201), the National Nature Science Foundation of China (grant
no. 81701426) and the PhD Research Launching Fund Project of the
Second Affiliated Hospital of Wenzhou Medical University (grant no.
FEY001).
Availability of data and materials
The datasets used during the present study are
available from the corresponding authors upon reasonable
request.
Authors' contributions
ZGC, JCR and XQZ designed and supervised the
project. YYB and JJY provided the samples and clinical information.
JY and XLG performed the experiments. YYB and XQZ provided
instructions for the experiments. ZGC and JY analyzed the data and
wrote the manuscript. ZGC and XLG edited the manuscript. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Written informed consent was collected from all the
patients in conformity with the Declaration of Helsinki, and the
present study obtained permission from the Ethics Committee of
Wenzhou Medical University.
Patient consent for publication
Written informed consent was collected from all the
patients.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
APL
|
acute promyelocytic leukemia
|
|
lncRNAs
|
long non-coding RNAs
|
|
ATRA
|
all-trans-retinoic acid
|
|
mRNA
|
messenger RNA
|
|
BM
|
bone marrow
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
ATO
|
arsenic trioxide
|
|
AML
|
acute myeloid leukemia
|
|
MBMCs
|
mononuclear bone marrow cells
|
|
TFs
|
transcription factors
|
|
FC
|
fold change
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GO
|
Gene Ontology
|
|
EEA1
|
early endosome antigen 1
|
|
ZNF564
|
zinc finger protein 564
|
|
ZNF44
|
zinc finger protein 44
|
|
E2F1
|
E2F transcription factor 1
|
|
EBF1
|
early B cell factor 1
|
|
E2F6
|
E2F transcription factor 6
|
References
|
1
|
Ablain J, Rice K, Soilihi H, de Reynies A,
Minucci S and de Thé H: Activation of a promyelocytic
leukemia-tumor protein 53 axis underlies acute promyelocytic
leukemia cure. Nat Med. 20:167–174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li K, Wang F, Cao WB, Lv XX, Hua F, Cui B,
Yu JJ, Zhang XW, Shang S, Liu SS, et al: TRIB3 promotes APL
progression through stabilization of the oncoprotein PML-RARα and
inhibition of p53-mediated senescence. Cancer Cell. 31:697–710
e697. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang ZY and Chen Z: Acute promyelocytic
leukemia: From highly fatal to highly curable. Blood.
111:2505–2515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lo-Coco F, Avvisati G, Vignetti M, Thiede
C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona
E, et al: Retinoic acid and arsenic trioxide for acute
promyelocytic leukemia. N Engl J Med. 369:111–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lehmann-Che J, Bally C and de Thé H:
Resistance to therapy in acute promyelocytic leukemia. N Engl J
Med. 371:1170–1172. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu HH, Qin YZ and Huang XJ: Resistance to
arsenic therapy in acute promyelocytic leukemia. N Engl J Med.
370:1864–1866. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan JQ, Zhang YQ, Wang JH, Xu P and Wang
W: lncRNA co-expression network model for the prognostic analysis
of acute myeloid leukemia. Int J Mol Med. 39:663–671. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao L, Xiao PF, Tao YF, Hu SY, Lu J, Zhao
WL, Li ZH, Wang NN, Wang J, Feng X, et al: Microarray profiling of
bone marrow long non-coding RNA expression in Chinese pediatric
acute myeloid leukemia patients. Oncol Rep. 35:757–770. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen ZH, Wang WT, Huang W, Fang K, Sun YM,
Liu SR, Luo XQ and Chen YQ: The lncRNA HOTAIRM1 regulates the
degradation of PML-RARA oncoprotein and myeloid cell
differentiation by enhancing the autophagy pathway. Cell Death
Differ. 24:212–224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei S, Zhao M, Wang X, Li Y and Wang K:
PU.1 controls the expression of long noncoding RNA HOTAIRM1 during
granulocytic differentiation. J Hematol Oncol. 9:442016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeng C, Xu Y, Xu L, Yu X, Cheng J, Yang L,
Chen S and Li Y: Inhibition of long non-coding RNA NEAT1 impairs
myeloid differentiation in acute promyelocytic leukemia cells. BMC
Cancer. 14:6932014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X, Weissman SM and Newburger PE:
Long intergenic non-coding RNA HOTAIRM1 regulates cell cycle
progression during myeloid maturation in NB4 human promyelocytic
leukemia cells. RNA Biol. 11:777–787. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng PZ, Wang KK, Zhang QY, Huang QH, Du
YZ, Zhang QH, Xiao DK, Shen SH, Imbeaud S, Eveno E, et al: Systems
analysis of transcriptome and proteome in retinoic acid/arsenic
trioxide-induced cell differentiation/apoptosis of promyelocytic
leukemia. Proc Natl Acad Sci USA. 102:7653–7658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sanz MA, Grimwade D, Tallman MS, Lowenberg
B, Fenaux P, Estey EH, Naoe T, Lengfelder E, Büchner T, Döhner H,
et al: Management of acute promyelocytic leukemia: Recommendations
from an expert panel on behalf of the European LeukemiaNet. Blood.
113:1875–1891. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guttman M, Amit I, Garber M, French C, Lin
MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al:
Chromatin signature reveals over a thousand highly conserved large
non-coding RNAs in mammals. Nature. 458:223–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guttman M and Rinn JL: Modular regulatory
principles of large non-coding RNAs. Nature. 482:339–346. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gerstein MB, Kundaje A, Hariharan M, Landt
SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et
al: Architecture of the human regulatory network derived from
ENCODE data. Nature. 489:91–100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khalil AM, Guttman M, Huarte M, Garber M,
Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van
Oudenaarden A, et al: Many human large intergenic noncoding RNAs
associate with chromatin-modifying complexes and affect gene
expression. Proc Natl Acad Sci USA. 106:11667–11672. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guenzl PM and Barlow DP: Macro lncRNAs: A
new layer of cis-regulatory information in the mammalian genome.
RNA Biol. 9:731–741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Fu L, Sun A, Tang D, Xu Y, Li Z,
Chen M and Zhang G: C/EBPβ contributes to transcriptional
activation of long non-coding RNA NEAT1 during APL cell
differentiation. Biochem Biophys Res Commun. 499:99–104. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alvarez-Dominguez JR and Lodish HF:
Emerging mechanisms of long noncoding RNA function during normal
and malignant hematopoiesis. Blood. 130:1965–1975. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JT: Epigenetic regulation by long
noncoding RNAs. Science. 338:1435–1439. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ørom UA, Derrien T, Beringer M, Gumireddy
K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q,
et al: Long noncoding RNAs with enhancer-like function in human
cells. Cell. 143:46–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Garding A, Bhattacharya N, Claus R, Ruppel
M, Tschuch C, Filarsky K, Idler I, Zucknick M, Caudron-Herger M,
Oakes C, et al: Epigenetic upregulation of lncRNAs at 13q14.3 in
leukemia is linked to the In Cis downregulation of a gene cluster
that targets NF-κB. PLoS Genet. 9:e10033732013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zuo W, Huang F, Chiang YJ, Li M, Du J,
Ding Y, Zhang T, Lee HW, Jeong LS, Chen Y, et al: c-Cbl-mediated
neddylation antagonizes ubiquitination and degradation of the TGF-β
type II receptor. Mol Cell. 49:499–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Attwooll C, Lazzerini Denchi E and Helin
K: The E2F family: Specific functions and overlapping interests.
EMBO J. 23:4709–4716. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Leung JY and Nevins JR: E2F6 associates
with BRG1 in transcriptional regulation. PLoS One. 7:e479672012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rishi L, Hannon M, Salomè M, Hasemann M,
Frank AK, Campos J, Timoney J, O'Connor C, Cahill MR, Porse B and
Keeshan K: Regulation of Trib2 by an E2F1-C/EBPα feedback loop in
AML cell proliferation. Blood. 123:2389–2400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Prasad MA, Ungerbäck J, Åhsberg J,
Somasundaram R, Strid T, Larsson M, Månsson R, De Paepe A,
Lilljebjörn H, Fioretos T, et al: Ebf1 heterozygosity results in
increased DNA damage in pro-B cells and their synergistic
transformation by Pax5 haploinsufficiency. Blood. 125:4052–4059.
2015. View Article : Google Scholar : PubMed/NCBI
|