Introduction
Hepatocellular carcinoma (HCC), the second most
frequent cause of cancer-associated mortality in men worldwide
(1), is notoriously refractory to
systemic chemotherapy (2). The
majority of patients with HCC are diagnosed when the disease is
already at an advanced stage. Sorafenib has been used as the
standard first-line systemic therapy for advanced HCC. However,
this promising treatment shows limited survival benefits with low
rates of tumor response (3,4). In certain patients with HCC, there is
an initial response to sorafenib, but eventually the disease
progresses (4), which indicates
that sorafenib resistance is common in HCC. Investigating the
underlying targets to overcome sorafenib resistance and enhancing
the response of patients to sorafenib are urgently required for
patients with HCC.
Sorafenib inhibits tumor cell proliferation not only
by inhibiting the Ras/Raf/mitogen-activated protein kinase
(MAPK)/extracellular signal-regulated kinase (ERK) signaling
pathway and inhibiting vascular endothelial growth factor receptor
and platelet-derived growth factor receptor (3), but also through activating the
phosphoinositide 3-kinase (PI3K)/Akt pathway, which regulates a
large number of molecules involved in all aspects of cancer
progression (5). cFLIP (Casper,
iFLICE, FLAME-1, CASH, CLARP, MRIT or usurpin) is an important
regulator against apoptosis (6).
The induction of apoptosis via death ligands and anticancer agents
can be abrogated by the ectopic expression of cFLIP variants
(7), indicating that the
overexpression of these proteins may cause resistance to multiple
anticancer drugs. Nuclear factor (NF)-κB and ERK signaling can be
activated continually by overexpressed cFLIPL binding to
adaptor proteins, including tumor necrosis factor receptor
(TNFR)-associated factor 1 (TRAF1) and 2 (TRAF2),
receptor-interacting protein 1 (RIP), and Raf-1 in each pathway
(8,9). cFLIPL also interacts with
Akt and enhances the anti-apoptotic functions of Akt (10,11) by
modulating the activity of glycogen synthase kinase 3β. The
cross-talk between the cFLIP, ERK and Akt pathways indicate that
the latent compensatory mechanism of cFLIP may contribute to
sorafenib resistance in HCC. However, the mechanisms that underlie
the role of cFLIP in sorafenib resistance remain to be fully
elucidated.
One of the most prominent modes of cytotoxic action
in several conventional chemotherapeutic drugs is the induction of
cells apoptosis. Certain drugs affect the mitochondrial pathway to
induce apoptosis, whereas other drugs, most notably the proteasome
inhibitors, act through the endoplasmic reticulum (ER) stress
(ERS)-mediated apoptotic pathway to induce cell death. In order to
survive, the ER can induce an alternate degradation system,
autophagy (12,13). It has been reported that
sorafenib-induced autophagy contributed to drug resistance
(14,15). As cFLIP is important in the protein
kinase RNA-like ER kinase (PERK)- and inositol-requiring enzyme-1
(IRE1)-mediated ER stress response and prevents procaspase-8
processing at the death-induced signaling complex, it may be a
potential target for overcoming sorafenib resistance. In the
present study, it was demonstrated that cFLIP is a potential target
for overcoming the acquired resistance to sorafenib in HCC. These
effects occurred partially through the reduction of ER
stress-related autophagy in HCC.
Materials and methods
Cell culture, antibodies and
reagents
Human Huh7 HCC cells were obtained from the Chinese
Academy of Sciences Cell Bank (Shanghai, China). The cells were
immediately expanded, and multiple aliquots were cryopreserved and
used within 3 months of resuscitation. The cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Hyclone Labotatories; GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (FBS; Biowest, Nuaillé, France). The antibodies
(Abs) against PERK, IRE1α, binding immunoglobulin protein (Bip),
Calnexin, ER oxidoreductin-1-like (Ero1-L)α, protein disulfide
isomerase (PDI), C/EBP homologous protein (Chop), ubiquitin
specific peptidase 2 (USP2), itchy E3 ubiquitin protein ligase
(ITCH), microtubule-associated protein 1 light chain 3 (LC3), P62,
Autophagy related 5 (Atg5), cFLIP, caspase-8, cleaved caspase-8,
caspase-9, caspase-3, cleaved caspase-3 and poly(ADP-ribose)
polymerase (PARP) were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Ab against caspase-12 was from Abcam
(Cambridge, MA, USA). The anti-β-actin Ab was from Sigma-Aldrich;
EMD Millipore (Billerica, MA, USA). Sorafenib, 3-methyladenine
(3-MA) and chloroquine phosphate (CQ) were from Selleck Chemicals
(Houston, TX, USA). 4-phenylbutyric acid (4-PBA) was purchased from
Sigma-Aldrich; EMD Millipore.
Sorafenib was dissolved in dimethyl sulfoxide to
prepare a stock solution of 20 µmol/l. CQ was dissolved in
phosphate-buffered saline (PBS) to prepare a stock solution of 50
mmol/l. 3-MA was dissolved in PBS at a stock concentration of 100
mmol/l by heating to 60–70°C immediately prior to use. The Annexin
V-FITC/propidium iodide (PI) apoptosis detection kit was from
Abcam. The MTT was purchased from Merck Millipore (Darmstadt,
Germany) dissolved in sterile PBS at a stock solution of 5 mg/l.
Lipofectamine 2000 was purchased from Thermo Fisher Scientific,
Inc. (Waltham, MA, USA).
Establishment of the
sorafenib-resistant HCC cell line
The cytotoxicity of sorafenib towards HCC cells was
initially detected by treating the cells with gradually increasing
concentrations of sorafenib in 96-well plates (104
cells/well) at 37°C, with cell proliferation measured at 24, 48 and
72 h, respectively. Into the medium of cells, the indicated
concentrations of sorafenib were added, which were just below their
respective IC50. The concentration of sorafenib was
slowly increased by 0.25 µmol/l every week. After 25–30 weeks, the
sorafenib-resistant cell line, termed Huh7-SR was obtained and was
continuously maintained by culture in the presence of
sorafenib.
MTT cytotoxicity assay
The cytotoxicity of sorafenib towards the cells was
determined using MTT, as previously described (16).
Annexin V/PI apoptosis assay
The cell death and apoptosis were evaluated by flow
cytometry using the Annexin V/PI binding kit (Abcam). Briefly,
following sorafenib treatment, the cells were trypsinized, stained
with Annexin V/PI, and then analyzed with a flow cytometer.
Transfection with small interfering
(si)RNA and cDNA
A double-stranded siRNA targeting human cFLIP
(5′-GCCUCAGAGCAUACCUGAATT-3′ and 5′-UCAUCUCGUACAUGACCACTT-3′) was
produced by Shanghai GenePharma Co., Ltd. (Shanghai, China) The
non-specific scrambled siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′ and
5′-ACGUGACACGUUCGGAGAATT-3′) served as a control. The USP2cDNA
clone plasmid (cat. no. RC200273) and empty vector plasmid (cat.
no. PS100001) were purchased from OriGene Technologies, Inc.
(Rockville, MD, USA). The transfection procedure was performed
according to the previously described protocol (16).
Western blot analysis
The protein extracts were obtained by suspending the
cells in RIPA lysis which containing protease inhibitor cocktail
(Pierce; Thermo Fisher Scientific, Inc.). The protein
concentrations were determined using a Bradford assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and 30 µg of cellular
proteins were electroblotted onto a PVDF membrane following
separation via 10% SDS polyacrylamide gel electrophoresis. The
immunoblot was blocked for 1 h with 5% milk at room temperature,
followed by incubation overnight at 4°C with 1:1,000 dilutions of
primary antibodies against PARP, caspase proteins, ERS proteins,
cFlip, p62, Atg5, LC3, ITCH, USP2 or β-actin. The blots were washed
twice with Tween-20/Tris- buffered saline (TTBS) prior to the
addition of a 1:1,000 dilution of HRP-conjugated secondary antibody
(cat. no. 7074; Cell Signaling Technology Inc.) for 1 h at room
temperature. The blots were washed again with TTBS, and developed
by enhanced chemiluminescence using Supersignal West Femto
chemiluminescent substrate (Pierce; Thermo Fisher Scientific,
Inc.). The band intensities were quantified using UN-SCAN-IT gel
analysis software (version 6; Silk Scientific, Inc., Orem, UT,
USA). The OD values for the target proteins were calculated as a
proportion of the OD value for β-actin. The western blot assays
were repeated three times (16).
Electronic microscopy
The cells were collected and fixed with 2.5%
glutaraldehyde solution for 1 h, then with 1% osmic acid for 1 h,
following which they were dehydrated with a graded series of
ethanol and embedded. Ultrathin sections (60–80 nm) were cut using
the LKB ultrotome with a diamond knife and double-stained with
uranium acetate and lead citrate. The cells were observed under a
transmission electronic microscope (HT7700; Hitachi, Ltd., Tokyo,
Japan).
Immunoprecipitation
The Huh7-R cells were transfected with the USP2
plasmid for 48 h, followed by sorafenib (5 µM) treatment for 6 h,
and the cells were washed three times with PBS. The pelleted cells
(300 × g for 5 min at room temperature) were then prepared and
analyzed by immunoprecipitation, as previously described (17).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical significance was determined using SPSS 19.0 for Windows
(IBM Corp., Armonk, NY, USA. Statistical comparison was performed
using Student's t-test (unpaired). P<0.05 was considered to
indicate a statistically significant difference.
Results
Sorafenib-resistant HCC cells are
refractory to sorafenib-induced growth inhibition and
apoptosis
Following incubation with 10 µM of sorafenib for 48
h, the cytotoxicity of Huh7-SR cells was 18.7%, respectively, which
was significantly lower than that of the Huh7 parent cells (54.6%;
Fig. 1A), as demonstrated by the
MTT assay. When the concentration of sorafenib reached 20 µM, the
cytotoxicity of the Huh7-SR cells was 45.9%, whereas almost
complete Huh7 parent cell death was observed (Fig. 1A). The apoptotic rates of the Huh7
cells were ~4.2-, 3.4- and 2.3-fold higher than those of the
Huh7-SR cells, following exposure to 5, 10 and 15 µM sorafenib,
respectively (Fig. 1B). The
apoptotic results were further supported by the expression of two
key apoptotic proteins, caspase-3 and PARP (Fig. 1C), showing that PARP and caspase 3
were significantly activated in the Huh7 cells following 10 µM
sorafenib treatment, compared with the Huh7-SR cells.
ER stress is involved in HCC cells
acquired resistance to sorafenib
To investigate the role of ERS in sorafenib
resistance, the Huh7 and Huh7-SR cells were incubated with 5, 10
and 20 µM sorafenib for 24 h, or with 10 µM sorafenib for 0, 3, 6,
12 or 24 h. The protein expression levels of PERK, IRE1α, PDI,
Calnexin, Bip and Chop, the key molecules involved in ER stress,
were detected. As shown in Fig 2A,
the expression levels of IRE1α, Ero1-Lα, and Bip were higher in the
Huh7-SR cells than those in the Huh7 cells. Following treatment
with different concentrations of sorafenib, these proteins remained
high in the Huh7-SR cells compared with the Huh7 cells. The
apoptosis-related protein Chop was significantly lower in the
Huh7-SR cells than in the Huh7 cells following sorafenib treatment.
Similarly, the expression of IRE1α, Ero1-Lα, and Bip were higher
following 10 µM sorafenib treatment for different times in the
Huh7-SR cells than those in the Huh7 cells. Chop was increased in a
time-dependent manner following sorafenib treatment in Huh7 cells,
but not in Huh7-SR cells (Fig. 2A).
Treatment with the ERS inhibitor 4-PBA + sorafenib inhibited the
expression of IRE1α and Ero1-Lα, and increased the expression of
Chop, compared with the sorafenib alone treatment group of Huh7-SR
cells (Fig. 2B). In addition, 4-PBA
increased sorafenib-induced Huh7 cell death (Fig. 2C). The results were supported by the
expression of apoptotic proteins, PARP, caspase-3, −8 and −9, which
were activated following 4-PBA + sorafenib treatment (Fig. 2D). However, ERS-induced
apoptosis-related caspase-12 was inhibited following 4-PBA
treatment.
 | Figure 2.ERS contributes to hepatocellular
carcinoma cell acquired resistance to sorafenib. (A) Expression of
unfolded protein response target genes IRE1a, Ero1-La, Bip, Chop,
PERK, PDI and Calnexin were analyzed by protein gel blots in Huh7
and Huh7-SR cells exposed to sorafenib at indicated concentrations
for 24 h (*P<0.05, **P<0.01 vs. corresponding untreated
cells) and at the indicated times (*P<0.05, **P<0.01 vs.
untreated Huh7 cells, #P<0.05 vs. untreated Huh7-SR
cells). Cells were incubated with indicated concentrations of
sorafenib for 48 h with or without 4-PBA treatment. (B) Cell
extracts were subjected to western blot analysis for ERS proteins.
*P<0.05, **P<0.01 vs. untreated cells, #P<0.05,
##P<0.01 vs. corresponding sorafenib-treated cells.
(C) Viability of Huh7-SR cells was assessed using a MTT assay. (D)
Cell extracts were subjected to western blot analysis for PARP and
caspase proteins. *P<0.05, **P<0.01 vs. untreated cells,
#P<0.05, ##P<0.01 vs. corresponding
sorafenib-treated cells. ERS, endoplasmic reticulum stress;
Ero1-Lα, ER oxidoreductin-1-like α; PDI, protein disulfide
isomerase; Bip, binding immunoglobulin protein; Chop, C/EBP
homologous protein; PERK, protein kinase RNA-like ER kinase; IRE1α,
inositol-requiring enzyme-1α, PDI, protein disulfide isomerase;
PARP, poly(ADP-ribose) polymerase; 4-PBA, 4-phenylbutyric acid; PI,
propidium iodide. |
ER stress increases autophagosome
formation in sorafenib-resistant HCC cells
P62, Atg 5 and LC3 are autophagy-related proteins.
As shown in Fig. 3A, P62 was
decreased, whereas Atg5 and the ratio of LC3-II/LC3-I were
increased prior to and following treatment with sorafenib in
Huh7-SR cells, compared with Huh7 cells. Co-incubation with the ERS
inhibitor (4-PBA) inhibited the sorafenib-induced increase of
LC3-II/LC3-I in the Huh7-SR cells (Fig.
3B). Electron microscopy showed the same results. Double
membrane vacuolar structures with morphological features of
autophagosomes and dilated ER lumens co-existed in the Huh7-SR
cells and sorafenib-treated cells, but not in the 4-PBA- and 4-PBA
+ sorafenib-treated cells (Fig.
3C). In addition, co-incubation with lysosomal protease
inhibitor (CQ) targeting the final steps of autophagic degradation,
and 3-MA, a PI3K inhibitor, enhanced sorafenib-induced Huh7-SR cell
apoptosis (Fig. 3D).
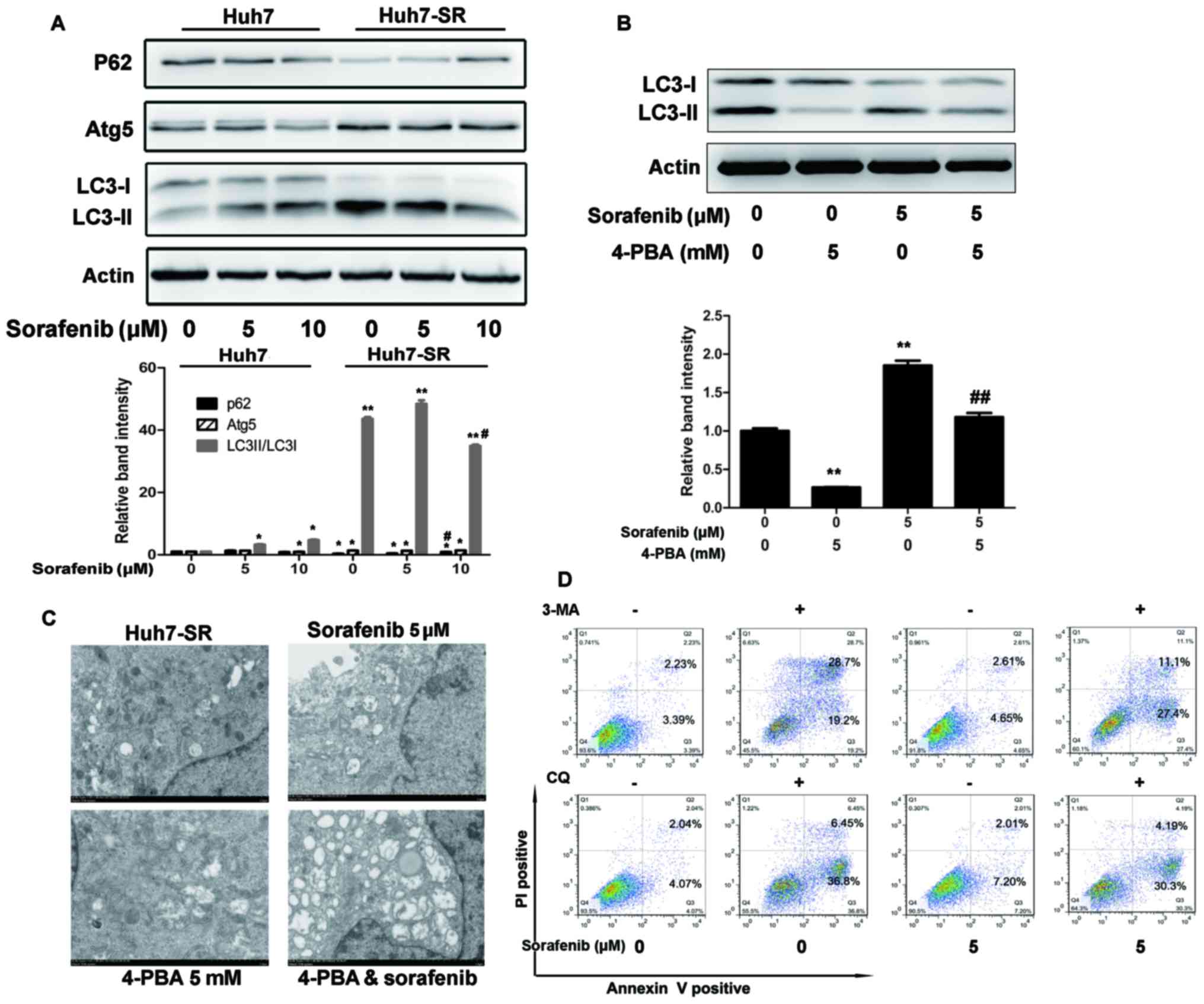 | Figure 3.ER stress increases autophagosome
formation in sorafenib-resistant HCC cells. (A) Lysates of Huh7 and
Huh7-SR cells incubated with 0, 5 or 10 µM of sorafenib for 48 h
were immunoblotted to detect the expression of autophagy-associated
proteins. *P<0.05, **P<0.01 vs. untreated Huh7 cells,
#P<0.05 vs. untreated Huh7-SR cells. (B) Huh7-SR
cells were exposed for 48 h to sorafenib at 5 µM in the presence or
absence of 4-PBA (5 mmol/l). Lysates of cells were immunoblotted to
detect the expression of LC3-I and II. **P<0.01 vs. untreated
cells, ##P<0.01 vs. sorafenib-treated cells. (C)
Electron microscopy of Huh7-SR cells exposed to sorafenib with or
without 4-PBA for 12 h. Typical autophagosome multivesicular,
body-like vesicles and multilamellar structures were observed next
to the dilated ER (magnification, ×5,000). (D) Huh7-SR cells were
exposed for 48 h to sorafenib at 5 µM in the presence or absence of
3-MA or CQ. Cells were stained with PI/Annexin V and then analyzed
by flow cytometry. ER, endoplasmic reticulum; LC3,
microtubule-associated protein 1 light chain 3; Atg5, autophagy
related 5; 4-PBA, 4-phenylbutyric acid; PI, propidium iodide. |
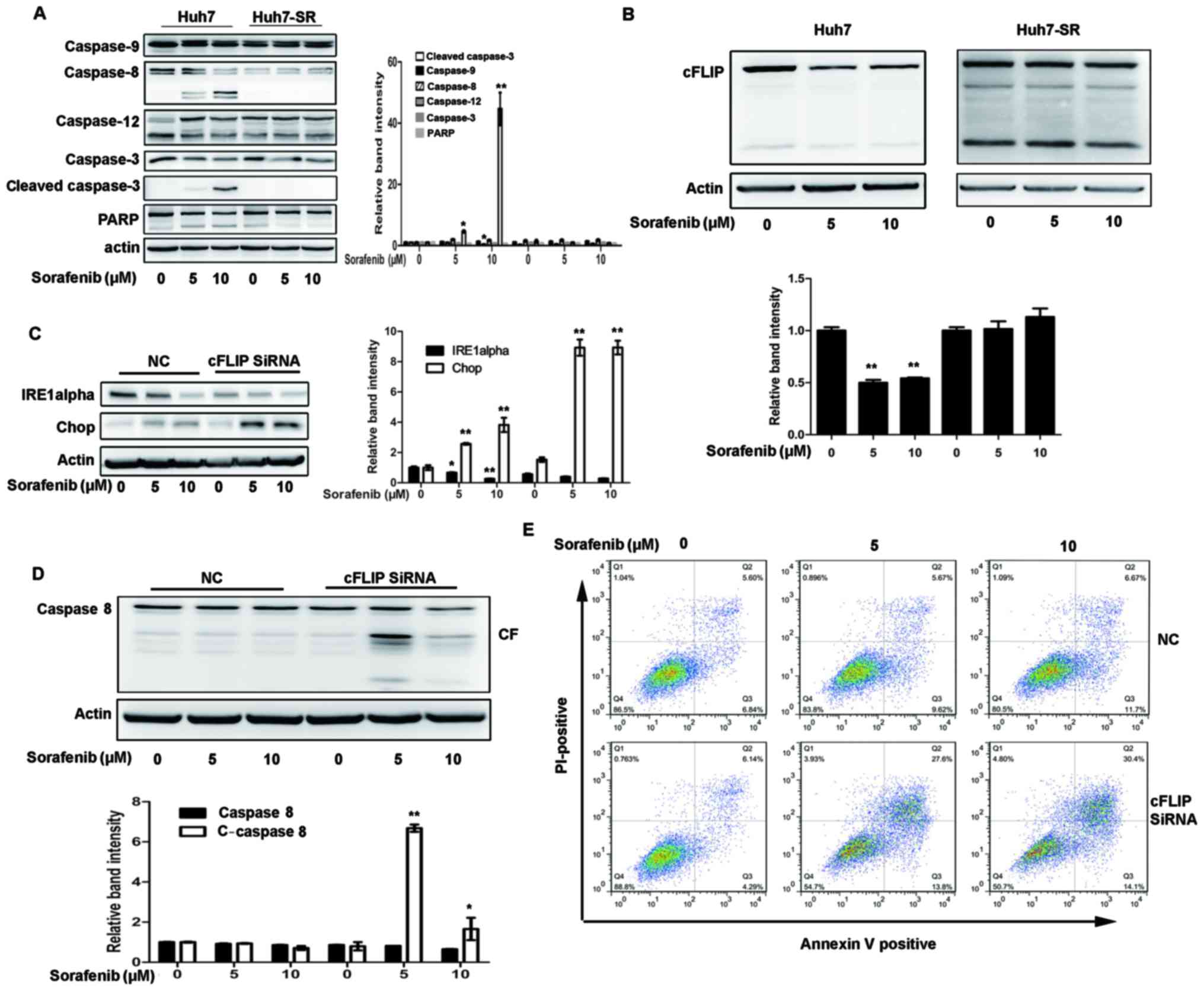
High expression of cFLIP is required
for sorafenib resistance to HCC cells
cFLIP is one of the specific inhibitors of caspase-8
(18). As shown in Fig. 4A, the level of cleaved caspase 8 was
decreased in the Huh7-SR cells following sorafenib treatment,
compared with that in the Huh7 cells. It was also found that cFLIP
was continuously increased in the Huh7-SR cells following sorafenib
treatment (Fig. 4B). Whether the
knockdown of cFLIP by siRNAs can increase sorafenib-induced Huh7-SR
and cytotoxicity and apoptosis was then examined. As shown in
Fig. 4C, transfection of the
Huh7-SR cells with cFLIP siRNA markedly reduced the expression of
IRE1a prior to and following sorafenib treatment, and increased the
expression of Chop following sorafenib treatment (Fig. 4C). Sorafenib-induced caspase-8
activation was also significantly increased in the Huh7-SR cells
transfected with cFLIP siRNA (Fig.
4D). Sorafenib-induced Huh7-SR cell apoptosis was effectively
increased in cells transfected with cFLIP siRNA (Fig. 4E), compared with control siRNA
transfection.
Suppression of USP2 contributes to the
deubiquitination of cFLIP in sofafenib-resistant cells
A major post-transcriptional mechanism that controls
protein levels in cells is ubiquitin-dependent proteasomal turnover
(19). To determine whether the
proteasome is involved in sorafenib-induced cFLIP decay, Huh7 cells
were incubated with MG132, a specific proteasome inhibitor. As
shown in Fig. 5A, pretreatment with
MG132 significantly prevented sorafenib-induced cFLIP degradation
in the Huh7 cells (Fig. 5A).
Accordingly, it was found that the ubiquitin of cFLIP was higher in
Huh7 cells than in Huh7-SR cells following sorafenib treatment
(Fig. 5B). It has been reported
that the knockdown of USP2, a de-ubiquitinating enzyme, can protect
hepatocytes from tumor necrosis factor-α-induced apoptosis by
decreasing the cellular levels of the ubiquitinligas ITCH, a
negative regulator of cFLIP, and the subsequent turnover of cFLIP
(20). Therefore, the present study
determined the expression of USP2 and ITCH in Huh7 and Huh7-SR
cells following sorafenib treatment. The expression of USP2 was
decreased in the Huh7-SR cells following sorafenib treatment,
compared with that in the Huh7 cells (Fig. 5C). The expression of ITCH in the
Huh7-SR cells was also lower than that in the Huh7 cells (Fig. 5C). The overexpression of USP2
enhanced the expression of ITCH and decreased the expression of
cFLIP in the Huh7-SR cells treated with sorafenib (Fig. 5D). These results suggested that the
downregulation of USP2 following long term exposure to sorafenib
may contribute to decreased proteasomal degradation of cFLIP.
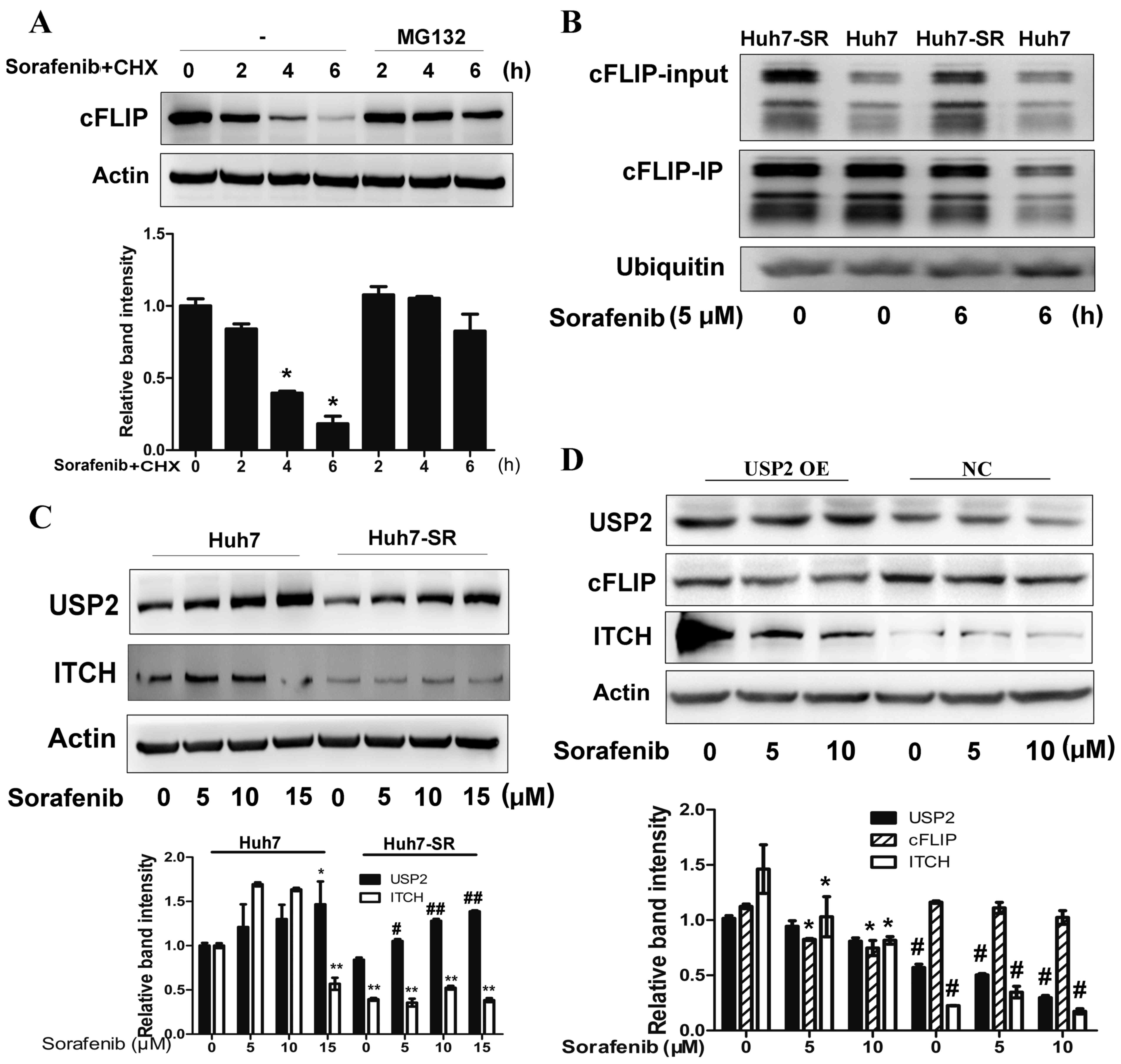 | Figure 5.Suppression of USP2 contributes to
deubiquitination of cFLIP following long term sorafenib
exposure-induced Huh7 SR cells. (A) Huh7 cells were treated with or
without the proteasome inhibitor MG-132 30 min prior to addition of
sorafenib (10 µM) and CHX (100 µg/ml). At the indicated times, cell
lysates were prepared and analyzed by immunoblotting with
antibodies targeting cFLIP and actin. *P<0.01 vs. corresponding
untreated cells. (B) Huh7 and Huh7-SR cells were incubated with
sorafenib. At the indicated times, the lysed cells were
immunoprecipitated (IP) with anti-cFLIP antibodies, gel separated,
and immunoblotted with anti-ubiquitin antibodies. (C) Huh7 and
Huh7-SR cells were treated with 0, 5, 10 or 15 µM of sorafenib for
24 h and analyzed for USP2 and ITCH protein via western blot
analysis. *P<0.05, **P<0.01 vs. untreated Huh7 cells,
#P<0.05, ##P<0.01 vs. untreated Huh7-SR
cells. (D) Huh7-SR cells were transfected with USP2 plasmid, and
the lysed cells were analyzed for cFLIP and ITCH protein via
western blot analysis. *P<0.01 vs. corresponding untreated
cells, #P<0.01 vs. corresponding cells with USP2
overexpression. USP2, ubiquitin specific peptidase 2; ITCH, itchy
E3 ubiquitin protein ligase; OE, overexpression; NC, negative
control. |
Discussion
Sorafenib remains unique in the treatment of HCC,
particularly for late HCC (21).
Investigating potential targets is urgently required to reverse
sorafenib resistance to HCC. In the present study, a high
expression of cFLIP was found following long-term exposure to
sorafenib. It was demonstrated that a high expression of cFLIP was
involved in the acquired sorafenib resistance through increasing
autophagy from a cytoprotective ERS in HCC cells. It was also found
that sorafenib inhibited the activity of caspase-8 via the
upregulation of cFLIP and abated sorafenib-induced apoptosis. cFLIP
may be an effective target for reversing sorafenib resistance in
HCC.
The results indicated that sorafenib induced
parental HCC cell death and apoptosis in a dose-dependent manner.
However, following long-term exposure, the sorafenib treated HCC
cells exhibited markedly reduced death and apoptotic rates
(Fig. 1). This was due to the
acquired resistance of HCC cells towards long-term sorafenib
treatment. The mechanism of sorafenib-induced cell resistance
requires further investigation.
The data showed that ERS was more active in Huh7-SR
cells than in Huh7 cells, which was reflected in the high
expression of IRE1a, Ero1-La and Bip (Fig. 2A). The ERS inhibitor, 4-PBA,
markedly reduced the expression of IRE1a and Ero1-La, increased the
expression of Chop, and subsequently enhanced sorafenib-induced
cell death (Fig. 2B and C). This
can be explained by the following: i) ERS is important in sorafenib
resistance in HCC. It has been shown that ERS contributes to drug
resistance and that drug resistance can be reversed by the
inhibition of ERS (22–24); ii) ER stress induces high regulation
of IRE1 signals, critical for HCC cell protective effects. 4-PBA
increased the sorafenib induced-activation of PARP, caspase-3, −8
and −9, but not caspase-12, which was associated with ERS-induced
apoptosis (Fig. 2D). This was
explained by the decrease in ERS by 4-PBA leading to inhibited
ER-stress-associated apoptosis, and sorafenib resistance being
independent of ERS-associated apoptosis. ERS has pleiotropic
effects on tumor cells, involving pro-survival or pro-apoptotic
signals (14,25–27).
The cell death or apoptosis was alleviated by ERS-related
autophagy. Inhibiting autophagy by using either pharmacological
inhibitors or RNA interference of essential autophagy genes
potentiates sorafenib-induced apoptosis in HCC cells (14). In the present study, autophagy was
enhanced following sorafenib treatment (Fig. 3A). The ERS inhibitor, 4-PBA,
effectively inhibited the activation of autophagy in Huh7-SR cells
(Fig. 3B and C). The autophagy
inhibitors, 3-MA and CQ, effectively enhanced sorafenib-induced
cell apoptosis (Fig. 3D), which
indicated that the ERS-induced protective role against cell death
of Huh7-SR cells occurred in an autophagy-dependent manner.
No significant differences were found in caspase-12
and caspase-9 between the Huh7-SR cells and Huh7 cells following
sorafenib treatment, unlike caspase-8. This can be explained by the
following: i) ERS-induced cell apoptosis may be not in involved in
sorafenib-induced resistance; ii) cFLIP, a caspase-8-like protein
that lacks a catalytic site and inhibits caspase 8-mediated
apoptosis, may be involved in sorafenib resistance. cFlip is a
well-characterized anti-apoptotic regulator. It can inhibit death
receptor-induced apoptosis through antagonizing caspase-8
activation at the stage of TNFR-complex II formation (28,29).
The data obtained in the present study indicated that the
expression of cFLIP remained higher in Huh7-SR cells following
sorafenib treatment, compared with that in Huh7 cells. The
silencing of cFLIP effectively enhanced the activity of caspase-8
and sorafenib-induced apoptosis of Huh7-SR cells, which was
accompanied by decreased IRE1a and increased Chop. This can be
explained by the following: i) high expression of cFLIP reduces
sorafenib-induced apoptosis by inhibiting of caspase-8 in
sorafenib-resistant cells; ii) high expression of cFLIP is involved
in the resistance of HCC cells to sorafenib by inducing
ERS-associated autophagy.
It has been reported that the phosphorylation and
activation of the E3 ubiquitin ligase Itch specifically
ubiquitinates cFLIP and induces its proteasomal degradation
(30). Furthermore, the artificial
knockdown of de-ubiquitinating enzyme USP2 decreased actinomycin
D/TNFa-induced hepatocyte apoptosis in vitro, which was
correlated with increased levels of cFlip and a concomitant
decrease in levels of the ubiquitinligase Itch (20). In the present study, it found that
treatment with specific proteasome inhibitor, MG132, completely
prevented sorafenib-induced cFLIP degradation in the Huh7 cells,
and reduced ubiquitinated cFLIP was found in the Huh7-SR cells.
This can be explained by the following: Decreased proteasomal
degradation of cFLIP contributes to the high expression of cFLIP
following long-term exposure to sorafenib. Reduced USP2 may be
responsible for elevated levels of cFLIP and reduced ITCH. The data
in the present study also showed that the overexpression of USP2
prevented the elevated levels of cFLIP. Therefore, USP2 depletion
in Huh7-SR cells enhanced the expression of cytoprotective cFlip,
degradation of Itch, and resistance against sorafenib-induced
apoptosis.
In conclusion, cFLIP was identified as an underlying
target that may reverse sorafenib resistance in HCC. These effects
occur partially through the reduction of ERS-related autophagy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Scientific Foundation of China (grant no. 81473487 to JD;
grant no. 81430101 to CL) and the Shanghai Natural Scientific
Foundation (grant no. 14ZR1408400 to JD).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
CL and JD conceived and designed the study.
Acquisition of data, including establishment of sorafenib-resistant
cells, MTT, flow cytometry, immunoprecipitation, western blot
analyses were performed by DL, YF, JL, WL and XL. Analysis and
interpretation of data, including statistical analysis,
biostatistics, computational analysis was performed by DL, JD and
BC. JD and DL wrote, reviewed and/or revised the manuscript. CL
supervised the study. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu AX: Systemic treatment of
hepatocellular carcinoma: Dawn of a new era? Ann Surg Oncol.
17:1247–1256. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, De Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhai B and Sun XY: Mechanisms of
resistance to sorafenib and the corresponding strategies in
hepatocellular carcinoma. World J Hepatol. 5:345–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Micheau O: Cellular FLICE-inhibitory
protein: An attractive therapeutic target? Expert Opin Ther
Targets. 7:559–573. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Poukkula M, Kaunisto A, Denessiouk K,
Katajamäki T, Johnson MS, Sistonen L and Eriksson JE: Rapid
turnover of c-FLIPshort is determined by its unique C-terminal
tail. J Biol Chem. 280:27345–27355. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ueffing N, Singh KK, Christians A, Thorns
C, Feller AC, Nagl F, Fend F, Heikaus S, Marx A, Zotz RB, et al: A
single nucleotide polymorphism determines protein isoform
production of the human c-FLIP protein. Blood. 114:572–579. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chaudhary PM, Eby MT, Jasmin A, Kumar A,
Liu L and Hood L: Activation of the NF-kappaB pathway by caspase 8
and its homologs. Oncogene. 19:4451–4460. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iyer AK, Azad N, Talbot S, Stehlik C, Lu
B, Wang L and Rojanasakul Y: Antioxidant c-FLIP inhibits Fas
ligand-induced NF-kappaB activation in a phosphatidylinositol
3-kinase/Akt-dependent manner. J Immunol. 187:3256–3266. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quintavalle C, Incoronato M, Puca L, Zanca
C, Romano G, Garofalo M, Iaboni M, Croce CM and Condorelli G:
c-FLIPL enhances anti-apoptotic Akt functions by modulation of
Gsk3beta activity. Cell Death Differ. 24:11342017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ding WX and Yin XM: Sorting, recognition
and activation of the misfolded protein degradation pathways
through macroautophagy and the proteasome. Autophagy. 4:141–150.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fujita E, Kouroku Y, Isoai A, Kumagai H,
Misutani A, Matsuda C, Hayashi YK and Momoi T: Two endoplasmic
reticulum-associated degradation (ERAD) systems for the novel
variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and
autophagy/lysosome ERAD(II). Hum Mol Genet. 16:618–629. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke
AW, Wang XY, Dai Z, Peng YF, Gu CY, et al: Targeting autophagy
enhances sorafenib lethality for hepatocellular carcinoma via ER
stress-related apoptosis. Autophagy. 7:1159–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhai B, Hu F, Jiang X, Zhao D, Liu B, Pan
S, Dong X, Tan G and Wei Z: Inhibition of Akt reverses the acquired
resistance to sorafenib by switching protective autophagy to
autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther.
13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du J, Wu J, Fu X, Tse AK, Li T, Su T and
Yu ZL: Icariside II overcomes TRAIL resistance of melanoma cells
through ROS-mediated downregulation of STAT3/cFLIP signaling.
Oncotarget. 7:52218–52229. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Salim K, Fenton T, Bacha J,
Urien-Rodriguez H, Bonnert T, Skynner HA, Watts E, Kerby J, Heald
A, Beer M, et al: Oligomerization of G-protein-coupled receptors
shown by selective co-immunoprecipitation. J Biol Chem.
277:15482–15485. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krammer PH: CD95's deadly mission in the
immune system. Nature. 407:789–795. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ciechanover A and Schwartz AL: The
ubiquitin system: Pathogenesis of human diseases and drug
targeting. Biochim Biophys Acta. 1695:3–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haimerl F, Erhardt A, Sass G and Tiegs G:
Down-regulation of the de-ubiquitinating enzyme ubiquitin-specific
protease 2 contributes to tumor necrosis factor-alpha-induced
hepatocyte survival. J Biol Chem. 284:495–504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berasain C: Hepatocellular carcinoma and
sorafenib: Too many resistance mechanisms? Gut. 62:1674–1675. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang CC, Yang F, Thorne RF, Zhu BK,
Hersey P and Zhang XD: Human melanoma cells under endoplasmic
reticulum stress acquire resistance to microtubule-targeting drugs
through XBP-1-mediated activation of Akt. Neoplasia. 11:436–447.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fan L, Sun G, Ma T, Zhong F, Lei Y, Li X
and Wei W: Melatonin reverses tunicamycin-induced endoplasmic
reticulum stress in human hepatocellular carcinoma cells and
improves cytotoxic response to doxorubicin by increasing CHOP and
decreasing survivin. J Pineal Res. 55:184–194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fan L, Song B, Sun G, Ma T, Zhong F and
Wei W: Endoplasmic reticulum stress-induced resistance to
doxorubicin is reversed by paeonol treatment in human
hepatocellular carcinoma cells. PloS One. 8:e626272013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kato H and Nishitoh H: Stress responses
from the endoplasmic reticulum in cancer. Front Oncol. 5:932015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu F, Han J, Zhai B, Ming X, Zhuang L, Liu
Y, Pan S and Liu T: Blocking autophagy enhances the apoptosis
effect of bufalin on human hepatocellular carcinoma cells through
endoplasmic reticulum stress and JNK activation. Apoptosis.
19:210–223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang WA, Groenendyk J and Michalak M:
Endoplasmic reticulum stress associated responses in cancer.
Biochim Biophys Acta. 1843:2143–2149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Micheau O, Lens S, Gaide O, Alevizopoulos
K and Tschopp J: NF-kappaB signals induce the expression of c-FLIP.
Mol Cell Biol. 21:5299–5305. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song JH, Tse MC, Bellail A, Phuphanich S,
Khuri F, Kneteman NM and Hao C: Lipid rafts and nonrafts mediate
tumor necrosis factor related apoptosis-inducing ligand induced
apoptotic and nonapoptotic signals in non small cell lung carcinoma
cells. Cancer Res. 67:6946–6955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang L, Kamata H, Solinas G, Luo JL,
Maeda S, Venuprasad K, Liu YC and Karin M: The E3 ubiquitin ligase
itch couples JNK activation to TNFalpha-induced cell death by
inducing c-FLIP(L) turnover. Cell. 124:601–613. 2006. View Article : Google Scholar : PubMed/NCBI
|