Introduction
Acute myelogenous leukemia (AML) is a hematopoietic
cancer with a high mortality rate, and >50% of patients with AML
show no response to chemotherapy (1). Although individual therapeutic
modalities have been developed and applied to control AML, the
diversity of gene mutations and heterogeneity among cancer cells
has largely confounded efforts to achieve complete remission
(2,3). To overcome difficulties in identifying
effective drug targets, the focus in AML research has increasingly
turned from gene regulation to organelle-based therapy (4).
Mutant forms of the tyrosine kinase FMS-like
tyrosine kinase 3-Internal tandem duplication (FLT3-ITD),
encoded by an AML-associated gene, induce reactive oxygen species
(ROS) production and DNA damage, sequentially contributing to
genetic mutations and a poor prognosis (5). Mitochondria are responsible for
producing ~90% of intracellular ROS, and persistent ROS production
is associated with AML development (6). NADPH oxidase-2-mediated superoxide
production stimulates mitochondrial transfer from bone marrow
stromal cells to leukemic blast cells through AML-derived tunneling
nanotubes. Intact mitochondrial transfer causes higher oxidative
phosphorylation activity, which mediates the supplementation of
biosynthetic precursors and adenosine triphosphate (ATP), which
finally induces rapid proliferation of AML cells (7). In addition, depletion of autophagy
results in failure to remove dysfunctional mitochondria and causes
dysregulation of the glycolytic pathway, thereby reducing the
survival rate of patients with AML (8). Thus, regulation of
mitochondrial-related metabolic flow has potential as a treatment
strategy against AML or other cancer types characterized by somatic
mutations that affect mitochondrial metabolism.
L-Deprenyl is a highly selective and irreversible
inhibitor of monoamine oxidase B (MAO-B) (9) that prevents the deamination of
dopamine, thus maintaining the dopamine concentrations in the
synapses of the nigrostriatal pathway (10). This dopamine
concentration-maintaining action of L-deprenyl is the basis for its
use in the therapy of Parkinson's disease. As ROS are generated
through deamination, L-deprenyl also acts to decrease ROS levels in
neuronal cells, which alleviates the amount of dopaminergic
neuronal cell death from exo- and endogenous insults (11). The high concentration of L-deprenyl
is known to exert anticancer effects, exhibiting efficacy in
reducing breast tumor size and decreasing tumor cell viability.
There is also evidence that L-deprenyl decreases the activity of an
estrogen-receptor (ER)-dependent intracellular signaling pathway in
ER-positive human breast cancer (12). In addition to its effects on solid
tumors, L-deprenyl also reduces the number of monoblastic leukemia
cells by increasing production of norepinephrine, interferon-γ,
cluster of differentiation 8+ lymphocytes and natural
killer cells in the spleen (13).
However, whether an intracellular organelle-based mechanism
underlies the antitumor effect of L-deprenyl, and whether this
action is dependent on MAO-B inhibition, remains uncertain.
Moreover, the involvement of changes in mitochondrial respiration
in L-deprenyl-induced cytotoxicity has not been demonstrated. The
present study investigated mitochondrial respiration as a novel
target of L-deprenyl in AML cells, which undergo apoptotic cell
death in response to L-deprenyl.
Materials and methods
Animals
Male FLT3-ITD knock-in and wild-type (WT)
mice with a C57BL/6 background were purchased from Jackson
Laboratory (Bar Habor, ME, USA). All animal experiments were
conducted in the animal facility according to institutional
guidelines (standard operating procedure), and approved by the
Institutional Animal Care and Use Committee of Chungnam National
University Hospital (CNUH-015-A0007-2). A total of 6 mice (aged 8
weeks, weighed 20±2 g) were maintained in a controlled environment
(12-h light/dark cycle; temperature, 22°C; 55% humidity), and
provided with food (cat. no. AIN-76A; Research Diets Inc., New
Brunswick, NJ, USA) and water ad libitum.
Isolation of mouse bone marrow
mononuclear cells (BMMNCs)
Firstly, the mice were placed into a 1.5-liter
volume plastic chamber, then exposed to 100% CO2 at a
flow rate of 0.25 l/min. After the euthanasia, each end of the
femur and tibia were cut off. Warm phosphate-buffered saline (PBS;
20 mM, pH 7.4) was then injected into the marrow cavity. Fluids
were collected in 50-ml conical tubes containing 20 ml Lymphoprep
(Takeda Pharmaceuticals International GmbH, Zurich, Switzerland),
which is a density gradient medium recommended for the isolation of
MNCs. Centrifugation was performed at 800 × g for 20 min at 20°C.
As granulocytes and erythrocytes have a higher density than MNCs at
the osmotic pressure of Lymphoprep, during the centrifugation
process, granulocytes and erythrocytes sediment through the
Lymphoprep layer and the MNCs with lower densities remain at the
plasma Lymphoprep interface, which allows isolation of the BMMNCs.
Subsequent to centrifugation, the BMMNC layer was collected using a
Pasteur pipette, then washed with PBS and re-suspended in
Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 20% fetal bovine serum
(GE Healthcare Life Sciences, Logan, UT, USA) and
penicillin-streptomycin (Thermo Fisher Scientific, Inc.).
Cell line culture conditions
KG-1α human bone marrow-derived AML cells were
obtained from the American Type Culture Collection (Manassas, VA,
USA). KG-1α cells were maintained in Iscove's modified Dulbecco's
medium (Thermo-Fisher Scientific, Inc.) supplemented with 20% fetal
bovine serum (GE Healthcare Life Sciences) and
penicillin-streptomycin (Thermo Fisher Scientific Inc.) at 37°C in
a humidified 5% CO2 environment. HL-60 human
blood-derived acute promyelocytic leukemia cells were obtained from
the Korean Cell Line Bank (Seoul, South Korea). HL-60 cells were
maintained in Roswell Park Memorial Institute medium (Welgene Inc.,
Gyeongsan, South Korea) supplemented with 10% fetal bovine serum
(GE Healthcare Life Sciences) and penicillin-streptomycin (Thermo
Fisher Scientific, Inc.) at 37°C in a humidified 5% CO2
environment.
Cell viability assay
Cell viability was determined using Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., MD, USA). For
the 24-h experiment, BMMNCs and KG-1α cells were plated at
1×104 per well in 96-well tissue culture plates and
incubated at 37°C overnight. Following L-deprenyl (0.5–4 mM; cat.
no. M003; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and PBS
(control) treatment for 24 h, CCK-8 reagents, which allow sensitive
colorimetric determination of the number of viable cells, were
added to each well. In acute experiments, i.e., treatment with
L-deprenyl for 20 min, once KG-1α cells had been plated at
1×104 per well in 96-well tissue culture plates and
incubated at 37°C for overnight, CCK-8 reagents were added to the
KG-1α cells. After 40 min, L-deprenyl and PBS was added and the
plate was incubated for an additional 20 min. WST-8 is reduced by
dehydrogenase in cells to form an orange-colored formazan. The
amount of formazan and the number of living cells are in direct
proportion. Absorbance was measured at a wavelength of 450 nm using
a MultiSkan Ascent microplate spectrophotometer (Thermo Fisher
Scientific, Inc.).
Measurements of MAO activity
MAO activity of the whole brain tissues of WT mice,
the bone marrow cells of FLT3-ITD knock-in mice, and KG-1α
and HL-60 cells was determined using a commercially available kit
(Amplex Red Monoamine Oxidase Assay kit; cat. no. A12214; Thermo
Fisher Scientific, Inc.), following the manufacturer's protocols.
Following sacrifice and removal of the brain, whole brain tissue
was collected and then homogenized using a pellet pestle (cat. no.
Z359971; Sigma-Aldrich; Merck KGaA) with radioimmunoprecipitation
assay lysis (RIPA) buffer containing 10% phosphatase and protease
inhibitor cocktail (Roche Diagnostics, Basel, Switzerland), and
centrifuged at 14,000 × g for 15 min at 4°C. Bone marrow cells were
collected with 20 mM ice cold PBS (pH 7.4) from the femur and tibia
of FLT3-ITD knock-in mice. The bone marrow cells, KG-1α
cells and HL-60 cells were centrifuged at 14,000 × g for 5 min at
4°C and then pellets were placed in RIPA buffer containing 10%
phosphatase and protease inhibitor cocktail (Roche Diagnostics) and
centrifuged at 14,000 × g for 15 min at 4°C. Protein concentration
in the supernatant was determined using a Bradford-based assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA), and samples were
diluted to 500 µg per 2 ml total volume with reaction buffer.
Samples were pre-incubated for 30 min at room temperature with the
specific MAO-B inhibitor, pargyline hydrochloride (1 µM), and the
MAO-A inhibitor, clorgyline hydrochloride (1 µM). Subsequent to
incubation, samples were added to individual wells of a 96-well
microplate. The fluorimetric assay was initiated by adding 100 µl
of a reaction mixture containing Amplex Red reagent (400 µM),
horseradish peroxidase (HRP; 2 U/ml) and benzylamine (2 mM), a
specific substrate of MAO-B. Plates were incubated for 30 min at
room temperature, protected from light, and fluorescence was
measured at excitation and emission wavelengths of 550 and 590 nm,
respectively, using a microplate fluorometer (Berthold
Technologies, Bad Wildbad, Germany). H2O2 (10
µM) was used as a positive control, and reaction buffer alone was
used as a negative control.
Measurement of oxygen consumption rate
and extracellular acidification rate
Oxygen consumption rate (OCR) and extracellular
acidification rate (ECAR) were measured using a Seahorse Bioscience
XF24 Analyzer (Agilent Technologies, Inc., Santa Clara, CA, USA), a
24-well format system that automatically delivers drugs via
injection ports during the assay. Prior to plating KG-1α cells
(1×104 cells/well), the XF24 sensor cartridge was
initially activated by incubating with 1 ml calibrant solution for
8 h at 37°C under normal atmospheric CO2 conditions.
Following incubation for 24 h, the cells were treated with
L-deprenyl for 24 h or treated acutely by drug delivery via port A.
Cells were then washed with XF assay media, consisting of DMEM (pH
7.4) supplemented with 10 mM glucose, 1 mM sodium pyruvate and 2 mM
L-glutamine (without sodium bicarbonate), after which 450 µl XF24
assay media was added to each well. Following equilibration for 20
min, each well of the XF24 cartridge was sequentially injected with
the ATPase inhibitor oligomycin (2 µg/ml), the uncoupling agent
carbonyl cyanide 3-chlorophenylhydrazone (5 µM) and the
mitochondrial electron transport inhibitor rotenone (2 µM) (all
Sigma-Aldrich; Merck KGaA), and OCR and ECAR were measured in
real-time.
Western blot analysis
Subsequent to the KG-1α cells being washed with
ice-cold 20 mM PBS (pH 7.4), proteins were extracted using
radioimmunoprecipitation assay lysis buffer containing 10%
phosphatase and protease inhibitor cocktail (Roche Diagnostics).
Following centrifugation of the lysates at 14,000 × g for 15 min at
4°C, supernatants were collected and protein concentration was
determined using a Bradford-based assay. Proteins in cleared
lysates (10 µg/sample) were resolved by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis on 7% or 10.5% gels, and
then transferred to a nitrocellulose membrane. Non-specific binding
was blocked by incubating the membrane with Tris-buffered saline
plus Tween-20 [10 mM Tris-HCl (pH 7.6), 150 mM NaCl and 0.1%
Tween-20] containing 5% skimmed milk for 1 h. Thereafter, membranes
were firstly incubated at 4°C overnight with the following primary
antibodies: Anti-poly (ADP-ribose) polymerase 1 (PARP-1; rabbit
polyclonal; 1:1,000 dilution; cat. no. sc-7150; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), anti-actin (rabbit
polyclonal; 1:2,000 dilution; cat. no. sc-1616; Santa Cruz
Biotechnology, Inc.), anti-caspase-3 (rabbit polyclonal; 1:1,000
dilution; cat. no. 9662s; Cell Signaling Technology, Inc., Danvers,
MA, USA) and anti-cleaved caspase-3 (rabbit polyclonal; 1:500
dilution; cat. no. 9661; Cell Signaling Technology, Inc.). The
membranes were then incubated with the appropriate HRP-conjugated
anti-immunoglobulin G secondary antibody (1:2,000 dilution; cat.
no. 12-348; EMD Millipore, Billerica, MA, USA). Immunoreactive
proteins were detected using an enhanced chemiluminescence (ECL)
system (WEST-ZOL plus; Intron Biotechnology, Inc., Seongnam, South
Korea). The quantification of band intensity was normalized with
actin using Image J program (version 1.52c; National Institutes of
Health, Bethesda, MD, USA).
ATP
Total ATP content was measured using an ATPlite
assay in KG-1α cells (PerkinElmer, Inc., Waltham, MA, USA)
according to the manufacturer's protocols. The resulting
luminescence emitted by the ATP-dependent luciferase reaction was
measured using a Lumino Plate-reader (Thermo Labsystems, Santa
Rosa, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated in KG-1α cells using an eCube
Tissue RNA Mini kit (Ep52050; PhileKorea, Seoul, South Korea) and
cDNA was synthesized from 1 µg total RNA using Moloney murine
leukemia virus reverse transcriptase, 5X First-Strand Buffer,
dithiothreitol (DTT) (cat. no. 28025-021; Invitrogen; Thermo Fisher
Scientific, Inc.), deoxynucleotide triphosphates (cat. no. U15111;
Promega Corporation, Madison, WI, USA) and Oligo(dT)15
primer (cat. no. C1101; Promega Corporation). RT-qPCR was performed
on a 7500 Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using cDNA, SYBR Green PCR Master mix (iCycleriQ
Real-Time PCR Detection system; Bio-Rad Laboratories, Inc.), and
the following primer pairs (designed using Primer3 version 0.4.0;
Whitehead Institute, Cambridge, MA, USA and Howard Hughes Medical
Institute, Chevy Chase, MD, USA): Hexokinase-1 (HK1) forward,
5′-GGCCACGATGTAGTCACCTT-3′ and reverse, 5′-CACGTCCAGGTCAAATTCCT-3′;
phosphofructokinase-1 (PFK1) forward, 5′-AGAGGGTTTCGATGATGCTT-3′
and reverse, 5′-GTTGTAGGCAGCTCGGAGTC-3′; pyruvate dehydrogenase
(lipoamide) α1 (PDHA) forward, 5′-AGAACTTCTACGGGGGCAAT-3′ and
reverse, 5′-CGAATATCTGGCCCTGGTTA-3′; isocitrate dehydrogenase-2
(IDH2) forward, 5′-CTCATCAGGTTTGCCCAGAT-3′ and reverse,
5′-GTCCGTGGTGTTCAGGAAGT-3′; NADH dehydrogenase 1α-subcomplex
subunit 9 (NDUFA9) forward, 5′-CGAGACTGGGAAACCAAAAA-3′ and reverse,
5′-GCTTCCTTGGACAGTTGAGC-3′; and 18S rRNA forward,
5′-CTGGTTGATCCTGCCAGTAG-3′ and reverse, 5′-CGACCAAAGGAACCATAACT-3′.
The PCR amplification process was as follows: 5 min denaturation at
95°C, followed by 40 cycles at 95°C for 20 sec, 60°C for 15 sec and
72°C for 15 sec. Relative expression of target mRNAs was quantified
and normalized with respect to that of 18S rRNA, which was used as
an endogenous control, using the 2−ΔΔCq method (14).
Statistical analysis
GraphPad Prism (GraphPad Software, Inc., San Diego,
CA, USA) was used for all statistical analyses. All experiments
were performed 3–4 times, and results are presented as the mean ±
standard error of the mean. The significance of differences between
the groups was analyzed using one-tailed Student's t-test or
one-way analysis of variance with Dunnett's multiple comparison
test. P<0.05 was considered to indicate a statistically
significant difference. Individual P-values are indicated in figure
legends.
Results
L-Deprenyl induces cell death in
BMMNCs and KG-1α cells
A previous study reported that L-deprenyl inhibits
neurotoxin-induced apoptosis at low concentrations (10−9
to 10−13 M), but induces apoptosis at a high
concentration (10−3 M) in tissues of neuro-ectodermal
origin (11). Considering the focus
of the present study on inducing cytotoxic effects against AML
cells, high concentrations of L-deprenyl were used.
As FLT3-ITD mutation is one of the most
lethal mutations in AML (15) and
FLT3-ITD knock-in mice can develop myeloproliferative
disease (16), cell viability tests
were performed in isolated BMMNCs of FLT3-ITD knock-in and
WT mice. Following isolation from the bone marrow, cell viability
was measured in the BMMNCs using CCK-8 24 h after L-deprenyl (0.5–4
mM) treatment. L-Deprenyl treatment in isolated BMMNCs of
FLT3-ITD knock-in mice showed decreased cell viability in a
dose-dependent manner (Fig. 1A). To
measure the potency of L-deprenyl on immature leukocyte cells
specifically, the cell viability of isolated BMMNCs in
FLT3-ITD knock-in mice and WT mice were compared. As shown
in Table I, treatment with
L-deprenyl at a 2 mM concentration showed an almost two-fold potent
effect on isolated BMMNCs in FLT3-ITD knock-in mice compared
with that in WT mice.
 | Table I.Cytotoxic ratio of L-deprenyl in bone
marrow mononuclear cells of FLT3-ITD knock-in versus WT
mice. |
Table I.
Cytotoxic ratio of L-deprenyl in bone
marrow mononuclear cells of FLT3-ITD knock-in versus WT
mice.
| Concentration of
L-deprenyl, mM | Toxicity ratio |
|---|
| 0 | 1.00 |
| 0.5 | 1.55 |
| 1 | 1.89 |
| 2 | 1.91 |
| 4 | 1.75 |
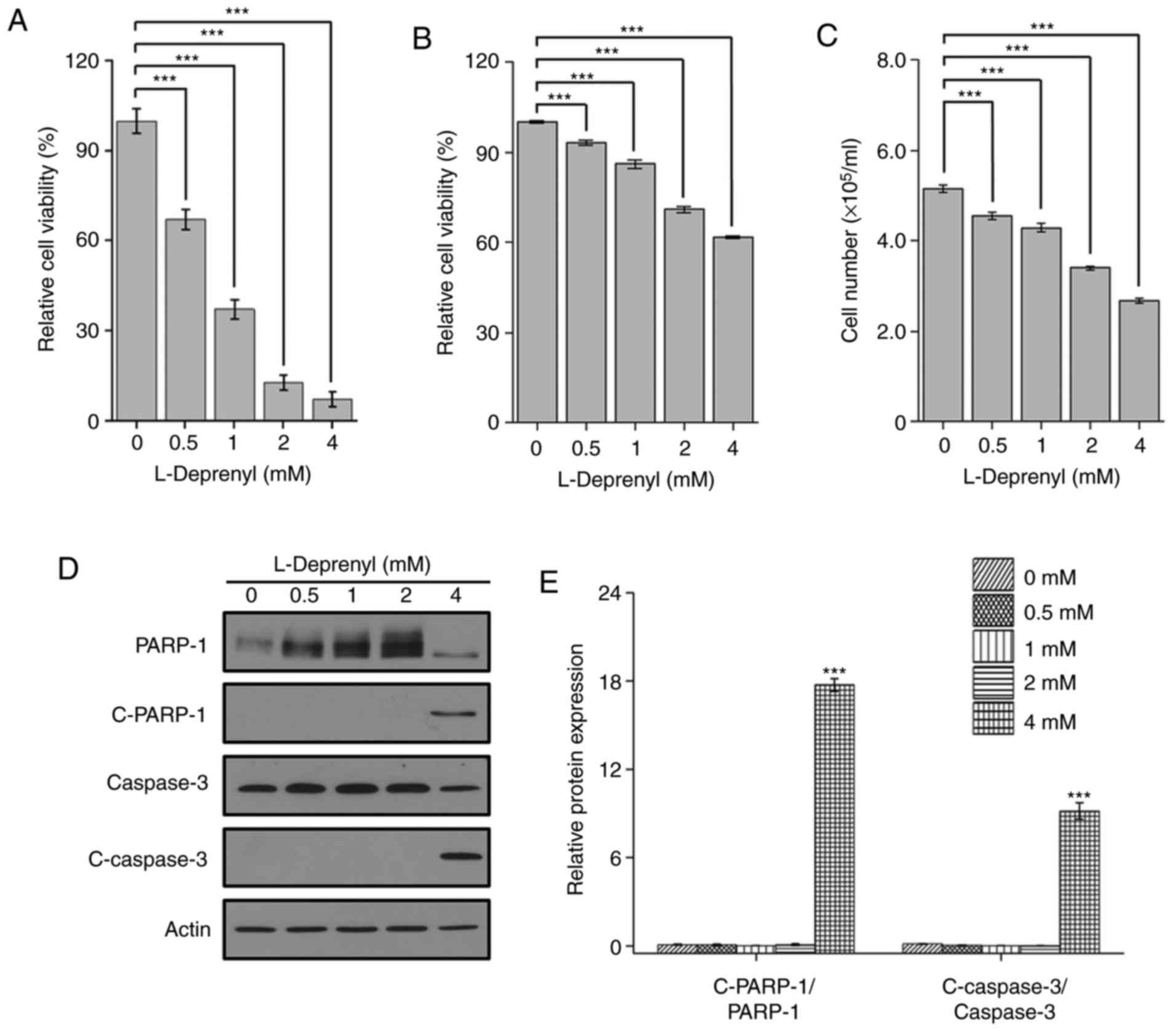
To identify the type of cell death induced by
L-deprenyl in leukemia cells, the cell viability was assessed in
KG-1α cells, one of the AML cell lines. Consistent with the result
in the BMMNCs, cell viability was reduced in the KG-1α cells in a
dose-dependent manner. The results were confirmed by CCK-8 assay
and by counting cell number using a hemocytometer (Fig. 1B and C). Moreover, an increase in
cleaved PARP-1 and caspase-3, markers of apoptosis, was detected by
western blotting following treatment with 4 mM L-deprenyl (Fig. 1D and E). These results suggest that
prolonged treatment with L-deprenyl induces apoptotic cell death in
BMMNCs and KG-1α cells.
Bone marrow cells and AML cell lines
lack MAO-A and B activity
Given that L-deprenyl selectively inhibits MAO-B,
the present study tested for the presence of MAO-B activity in bone
marrow cells and whole brain tissue, used as a positive control. As
expected, whole brain possessed MAO-B activity; notably, however,
bone marrow cells did not (Fig.
2A). Previous reports have indicated that cancer cells (glioma)
express high levels of MAO-B (17).
To determine whether bone marrow-derived cancer cells possess MAO
activity, MAO-A and MAO-B activity was assessed in the leukemia
KG-1α and HL-60 cell lines. Unexpectedly, there was no difference
found whether MAO-A and -B inhibitors were used or not, which means
that neither leukemic cell line showed MAO-A or -B activity
(Fig. 2B and C). These observations
suggest that the toxic effects of L-deprenyl on KG-1α cells are
independent of inhibition of MAO-B enzymatic activity.
L-Deprenyl suppresses mitochondrial
respiration without affecting extracellular acidification in KG-1α
cells
Previous studies have shown that high-dose
L-deprenyl treatment causes toxic effects on cancer cells and
mammary tumors through production of ROS (12,13,18).
Since the main sites of ROS generation are mitochondrial oxidative
phosphorylation complex I and III, the mitochondrial OCR was
measured using an XF24 analyzer (19). Treatment with L-deprenyl for 24 h
inhibited mitochondrial respiration in KG-1α cells (Fig. 3A and B). As depletion of
mitochondrial ATP production is abruptly compensated for by
upregulation of the glycolytic pathway so as to maintain
intracellular ATP content (20),
the ECAR, which reflects intracellular lactate production, was also
measured (Fig. 3C and D).
Unexpectedly, although ECAR showed a decreasing tendency, this
difference did not reach statistical significance.
 | Figure 3.L-Deprenyl suppresses mitochondrial
respiration without affecting glycolysis in KG-1α cells. (A) OCR in
KG-1α cells pretreated with L-deprenyl for 24 h. Oligomycin (2
µg/ml), CCCP (5 µM) and rotenone (2 µM) were sequentially injected
into cells. (B) The AUC of OCR, calculated using the third to fifth
time points from the graphs in (A). (C) ECAR of KG-1α cells after
L-deprenyl pretreatment for 24 h. Oligomycin (2 µg/ml), CCCP (5 µM)
and rotenone (2 µM) were sequentially injected onto cells. (D) The
AUC of ECAR, calculated using the third to fifth measurement points
in graphs in (C) Data are presented as the mean ± standard error of
the mean (error bars). Each OCR and ECAR measurement point (rate)
consisted of a drug injection for 2 min, wait time for 2 min, and
detection of oxygen and hydrogen for 3 min. *P<0.05 vs.
untreated controls. OLI, oligomycin; ROT, rotenone; CCCP, carbonyl
cyanide 3-chlorophenylhydrazone; OCR, oxygen consumption rate;
ECAR, extracellular acidification rate; AUC, area under the curve;
NS, not significant. |
Acute treatment with L-deprenyl
suppresses mitochondrial respiration
L-Deprenyl is rapidly absorbed into the body and
reaches its highest plasma concentration within 30–120 min
(21). To investigate the effects
of acute L-deprenyl treatment on KG-1α cells, the cell viability
was first assessed following treatment with L-deprenyl for 30 min.
It was found that cell viability was not changed over this time
frame following treatment with 0.5 to 4 mM L-deprenyl (Fig. 4A). Next, mitochondrial OCR was
assessed using an XF24 analyzer following injection of L-deprenyl
into 24-well XF cell plates. In contrast to the absence of an acute
effect on viability, L-deprenyl decreased mitochondrial
respiration, every 7 min, by 21 to 32% in a concentration-dependent
manner (Fig. 4B and C). In general,
inhibition of mitochondrial respiration is accompanied by
upregulation of the glycolytic pathway, which serves to maintain
intracellular ATP homeostasis. As expected, L-deprenyl increased
ECAR at 0.5 mM (Fig. 4D and E);
however, at higher concentrations, it decreased ECAR and led to
depletion of total ATP content (Fig.
4F). Taken together with the effects of the 24-h treatment with
L-deprenyl, these findings suggest that high concentration and
prolonged treatment with L-deprenyl cause deterioration in the
intracellular energy supply in KG-1α cells and lead to apoptotic
cell death.
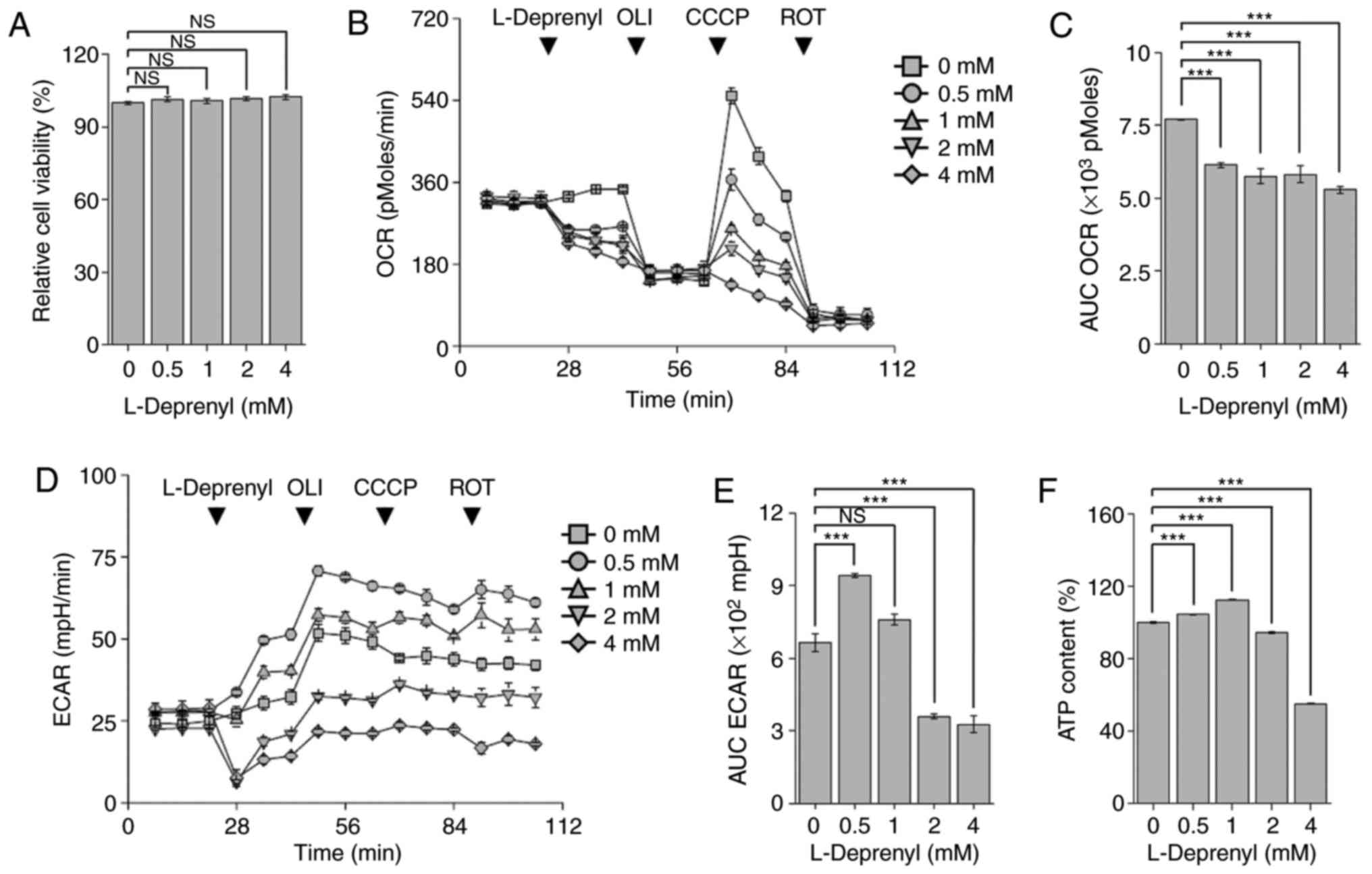 | Figure 4.Acute treatment with L-deprenyl
suppresses mitochondrial respiration and ATP content. (A) Viability
of KG-1α cells following treatment with L-deprenyl for 20 min. (B)
Real-time measurement of OCR in KG-1α cells. L-Deprenyl was
injected via port A of the XF24 analyzer. The four arrows indicate
injection times of L-deprenyl (0, 0.5, 1, 2 and 4 mM) and OCR drugs
[oligomycin (2 µg/ml), CCCP (5 µM) and rotenone (2 µM)]. (C) The
AUC of OCR, calculated from the fourth to sixth time points from
graphs in (A). (D) Real-time measurement of ECAR in KG-1α cells.
L-Deprenyl, oligomycin, CCCP and rotenone were injected in order
from port A to D. Every arrow indicates injection time points of
the four drugs. (E) The AUC of ECAR, calculated using the fourth to
sixth measurement points in graphs in (C). (F) ATP content in KG-1α
cells following treatment with L-deprenyl for 20 min. Data are
presented as the mean ± standard error of the mean (error bars)
(***P<0.001 vs. untreated controls; NS, not significant). Each
OCR and ECAR measurement point (rate) consisted of a drug injection
for 2 min, wait time for 2 min, and detection of oxygen and
hydrogen for 3 min. ATP, adenosine triphosphate; OLI, oligomycin;
ROT, rotenone; CCCP, carbonyl cyanide 3-chlorophenylhydrazone; OCR,
oxygen consumption rate; ECAR, extracellular acidification rate;
AUC, area under the curve; NS, not significant. |
Acute treatment with L-deprenyl alters expression
levels of mRNAs for glycolysis- and tricarboxylic cycle-related
genes in KG-1α. To investigate the physiological relevance of
changes caused by acute treatment with L-deprenyl at the molecular
level, changes in the expression of genes encoding proteins
involved in glycolysis and the tricarboxylic acid cycle were
examined. KG-1α cells were treated with L-deprenyl for 20 min, and
levels of HK1, PFK1, PDHA, IDH2 and NDUFA9 mRNAs were measured by
RT-qPCR. Although there was no change in mRNA levels of HK1
(Fig. 5A), consistent with the ECAR
results, a high concentration (4 mM) of L-deprenyl reduced the
level of PFK1 mRNA (Fig. 5B) and a
low concentration (0.5 mM) of L-deprenyl induced a strong increase
in PDHA mRNA levels (Fig. 5C).
However, no concentration of L-deprenyl treatment changed the mRNA
levels of IDH2 and NDUFA9 (Fig. 5D and
E). Collectively, these results indicate that, although
L-deprenyl improves glycolysis at a low concentration, thereby
compensating for decreased ATP content, high concentrations of
L-deprenyl reduce mitochondrial respiration and glycolysis, and
decrease ATP content, leading to cell death in the chronic
phase.
 | Figure 5.Expression levels of glycolysis- and
TCA cycle-related genes with acute treatment of L-deprenyl in
KG-1α. (A-E) mRNA levels of genes encoding enzymes involved in
glycolysis and the TCA cycle. KG-1α cells were treated with
L-deprenyl for 20 min, after which (A) HK1, (B) PFK1, (C) PDHA, (D)
IDH2 and (E) NDUFA9 mRNAs were quantified by reverse
transcription-quantitative polymerase chain reaction. Data are
presented as the mean ± standard error of the mean (error bars)
(*P<0.05 vs. untreated controls). NS, not significant; TCA,
tricarboxylic acid; HK1, hexokinase-1; PFK1, phosphofructokinase-1;
PDHA, pyruvate dehydrogenase (lipoamide) α1; IDH2, isocitrate
dehydrogenase-2; NDUFA9, NADH dehydrogenase 1α-subcomplex subunit
9. |
Discussion
AML cells have a larger mitochondrial content than
leukocytes and require mitochondrial ATP for survival and
proliferation (22). The complexity
of AML development involving variable genetic mutations creates
challenges for achieving complete remission through
genetic-targeting strategies, shifting the focus towards
development of metabolic regulators. The higher content of
mitochondria, the intracellular energy supply centers, in AML cells
has driven the development of mitochondrial-targeting drugs. The
results of the present study suggest the L-deprenyl reduces AML
proliferation through inhibition of mitochondrial respiration.
Efforts to develop mitochondria-inhibiting drugs
that arrest AML progression have employed molecular and genetic
targeting strategies. For example, inhibition of fatty acid
oxidation in the mitochondrial matrix by etomoxir and inhibition of
glutaminase-1 by
bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide have
been studied for their potential to induce metabolic changes that
suppress the energy supply required for AML proliferation (23,24).
In addition, AG-221 and AGI-6780 target mutant IDH2, thereby
inhibiting 2-hydroxyglutarate production and inducing the
differentiation of AML cells (25,26).
However, the clinical trials necessary to demonstrate the safety of
these mitochondrial-targeting agents involve considerable time and
expense. By contrast, L-deprenyl is already widely used to treat
Parkinson's disease patients and could be rapidly enrolled as a
novel therapeutic agent for AML as a mitochondrial
respiration-targeting drug.
It has been reported that treatment with high-dose
L-deprenyl induces toxicity against monoblastic leukemia and
pituitary and mammary cancer (13,27–29).
In addition, treatment of mono-mac-6 cells with a high
concentration of L-deprenyl reduces cell proliferation and
metastasis (13). It has also been
shown that decreasing norepinephrine and dopamine, metabolites of
MAO, by treatment with L-deprenyl reduces tumor size in vivo
(27–29). However, these previous reports did
not identify the molecular- and organelle-based mechanism
underlying the resulting apoptotic cell death. One possible
mitochondria-associated mechanism by which L-deprenyl could exert
these effects would be through modulation of the stability of
mitochondrial membrane potential (MMP) (30,31).
Low concentrations of L-deprenyl stabilize MMP in neuronal cells by
increasing expression of the B-cell lymphoma 2 gene (32). As high concentrations of L-deprenyl
have cytotoxic effects on the integrity of cellular homeostasis
mechanisms in cancer cells, inhibition of mitochondrial respiration
could be one mechanism underlying L-deprenyl-induced apoptotic cell
death (13). The changes in ATP
production caused by inhibition of mitochondrial respiration are
consistent with the observed reduction in key glycolysis-regulating
enzymes, and depletion of ATP content without compensatory
glycolysis can lead to apoptotic cell death (33). Moreover, The R(−) amphetamine and
R(−) methamphetamine produced by L-deprenyl increase the production
of ROS, which impose oxidative stress on the mitochondria. As a
result, mitochondria release apoptotic proteins, including
cytochrome c and apoptosis inducing factor, which lead to
apoptotic cell death (18).
Definitive confirmation that L-deprenyl-induced cell death is
mitochondria-dependent will require additional studies evaluating
changes in ROS in AML cells.
Trials of new organelle-targeting drugs designed to
exert an anti-AML effect could pioneer a novel treatment strategy.
However, the amount of time and money required for novel drug
development represents an enormous hurdle. To shorten the time
required to bring a drug to treat AML to the market, the clinically
used drug L-deprenyl was tested in the present study and its novel
targets identified. On the basis of these findings, it may be
concluded that L-deprenyl causes apoptotic cell death in AML
coincident with inhibition of mitochondrial OCR and cytosolic ECAR,
an effect that is not mediated by MAO-B inhibition in vitro
and ex vivo. In the future, regarding the importance of the
development of a drug for controlling intracellular metabolic flux,
L-deprenyl would be a candidate for first-line therapy in AML.
Acknowledgements
Not applicable.
Funding
This study was funded by National Research
Foundation of Korea grants from the Ministry of Science and ICT
(nos. 2016R1A2B4010398, 2016R1A6A3A11935284 and 2017R1A5A2015385)
and by grants from the Ministry of Education (nos. 2014R1A6A1029617
and 2016R1D1A1B03932766) and the research fund of Chungnam National
University.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding authors on reasonable
request.
Authors' contributions
IR, MJR, JYH and GRK made substantial contributions
to the conception and design of the study. IR and MJR were
responsible for the acquisition of data. IR, MJR and JYH assisted
with the analysis and interpretation of data. IR, MJR, JYH and GRK
wrote the manuscript. MJL and XJ performed the whole brain tissue
preparation. BHY and YLL provided the materials for the experiments
and measured the activity of MAO. JH, YJ and SJK conducted the
isolation of mononuclear cells from the bone marrow. ICS, WC and EO
assisted in interpreting the data and provided practical guidance.
GRK and JYH are responsible for the integrity of the study as a
whole. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All animal experiments were conducted in the animal
facility according to institutional guidelines (standard operating
procedure), and approved by the Institutional Animal Care and Use
Committee of Chungnam National University Hospital
(CNUH-015-A0007-2).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests
References
|
1
|
De Kouchkovsky I and Abdul-Hay M: ‘Acute
myeloid leukemia: A comprehensive review and 2016 update’. Blood
Cancer J. 6:e4412016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Löwenberg B and Rowe JM: Introduction to
the review series on advances in acute myeloid leukemia (AML).
Blood. 127:12016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liehr T: Thorough discussion of cancer
research-thoughts against the main stream. Eur J Hum Genet.
25:9022017. View Article : Google Scholar :
|
|
4
|
Wilson CS, Davidson GS, Martin SB, Andries
E, Potter J, Harvey R, Ar K, Xu Y, Kopecky KJ, Ankerst DP, et al:
Gene expression profiling of adult acute myeloid leukemia
identifies novel biologic clusters for risk classification and
outcome prediction. Blood. 108:685–696. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sallmyr A, Fan J, Datta K, Kim KT, Grosu
D, Shapiro P, Small D and Rassool F: Internal tandem duplication of
FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and
misrepair: Implications for poor prognosis in AML. Blood.
111:3173–3182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adam-Vizi V and Chinopoulos C:
Bioenergetics and the formation of mitochondrial reactive oxygen
species. Trends Pharm Sci. 27:639–645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marlein CR, Zaitseva L, Piddock RE,
Robinson SD, Edwards DR, Shafat MS, Zhou Z, Lawes M, Bowles KM and
Rushworth SA: NADPH oxidase-2 derived superoxide drives
mitochondrial transfer from bone marrow stromal cells to leukemic
blasts. Blood. 130:1649–1660. 2017.PubMed/NCBI
|
|
8
|
Watson AS, Riffelmacher T, Stranks A,
Williams O, De Boer J, Cain K, MacFarlane M, McGouran J, Kessler B,
Khandwala S, et al: Autophagy limits proliferation and glycolytic
metabolism in acute myeloid leukemia. Cell Death Discov.
1:150082015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robottom BJ: Efficacy, safety, and patient
preference of monoamine oxidase B inhibitors in the treatment of
Parkinson's disease. Patient Prefer Adherence. 5:57–64. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Youdim MB, Edmondson D and Tipton KF: The
therapeutic potential of monoamine oxidase inhibitors. Nat Rev
Neurosci. 7:295–309. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Magyar K and Szende B: (−)-Deprenyl, A
selective MAO-B inhibitor, with apoptotic and anti-apoptotic
properties. Neurotoxicology. 25:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
ThyagaRajan S, Tran L, Molinaro CA,
Gridley DS, Felten DL and Bellinger DL: Prevention of mammary tumor
development through neuroimmunomodulation in the spleen and lymph
nodes of old female sprague-dawley rats by L-Deprenyl.
Neuroimmunomodulation. 20:141–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lajkó E, Polgár L, Láng O, Lengyel J,
Kőhidai L and Magyar K: Basic cell physiological activities (cell
adhesion, chemotaxis and proliferation) induced by selegiline and
its derivatives in Mono Mac 6 human monocytes. J Neural Transm
(Vienna). 119:545–556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chi Y, Lindgren V, Quigley S and Gaitonde
S: Acute myelogenous leukemia with t(6;9)(p23;q34) and marrow
basophilia: An overview. Arch Pathol Lab Med. 132:1835–1837.
2008.PubMed/NCBI
|
|
16
|
Lee BH, Tothova Z, Levine RL, Anderson K,
Buza-Vidas N, Cullen DE, McDowell EP, Adelsperger J, Fröhling S,
Huntly BJ, et al: FLT3 mutations confer enhanced proliferation and
survival properties to multipotent progenitors in a murine model of
chronic myelomonocytic leukemia. Cancer Cell. 12:367–380. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sharpe MA and Baskin DS: Monoamine oxidase
B levels are highly expressed in human gliomas and are correlated
with the expression of HiF-1α and with transcription factors Sp1
and Sp3. Oncotarget. 7:3379–3393. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jayanthi S, Deng X, Noailles PA, Ladenheim
B and Cadet JL: Methamphetamine induces neuronal apoptosis via
cross-talks between endoplasmic reticulum and
mitochondria-dependent death cascades. FASEB J. 18:238–251. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pfeiffer T, Schuster S and Bonhoeffer S:
Cooperation and competition in the evolution of ATP-producing
pathways. Science. 292:504–507. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oliver D: The structure and activity of
selegiline, its functional groups, and congeners. Bioorg Chem.
2002.
|
|
22
|
Sriskanthadevan S, Jeyaraju DV, Chung TE,
Prabha S, Xu W, Skrtic M, Jhas B, Hurren R, Gronda M, Wang X, et
al: AML cells have low spare reserve capacity in their respiratory
chain that renders them susceptible to oxidative metabolic stress.
Blood. 125:2120–2130. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Samudio I, Harmancey R, Fiegl M,
Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W,
Duvvuri S, Taegtmeyer H and Andreeff M: Pharmacologic inhibition of
fatty acid oxidation sensitizes human leukemia cells to apoptosis
induction. J Clin Invest. 120:142–156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jacque N, Ronchetti AM, Larrue C, Meunier
G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M,
et al: Targeting glutaminolysis has antileukemic activity in acute
myeloid leukemia and synergizes with BCL-2 inhibition. Blood.
126:1346–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yen K, Travins J, Wang F, David MD, Artin
E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E, et al:
AG-221, a first-in-class therapy targeting acute myeloid leukemia
harboring oncogenic IDH2 mutations. Cancer Discov. 7:478–493. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang F, Travins J, DeLaBarre B,
Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A,
Liu W, Gliser C, et al: Targeted inhibition of mutant IDH2 in
leukemia cells induces cellular differentiation. Science.
340:622–626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
ThyagaRajan S, Felten SY and Felten DL:
Antitumor effect of L-deprenyl in rats with carcinogen-induced
mammary tumors. Cancer Lett. 123:177–183. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thyagarajan S, Meites J and Quadri SK:
Deprenyl reinitiates estrous cycles, reduces serum prolactin, and
decreases the incidence of mammary and pituitary tumors in old
acyclic rats. Endocrinology. 136:1103–1110. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
ThyagaRajan S and Quadri SK: L-Deprenyl
inhibits tumor growth, reduces serum prolactin, and suppresses
brain monoamine metabolism in rats with carcinogen-induced mammary
tumors. Endocrine. 10:225–232. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tatton WG and Chalmers-Redman RM:
Modulation of gene expression rather than monoamine oxidase
inhibition: (−)-deprenyl-related compounds in controlling
neurodegeneration. Neurology. 47:S171–S183. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Simon L, Szilágyi G, Bori Z, Telek G,
Magyar K and Nagy Z: Low dose (−)deprenyl is cytoprotective: It
maintains mitochondrial membrane potential and eliminates oxygen
radicals. Life Sci. 78:225–231. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yi H, Maruyama W, Akao Y, Takahashi T,
Iwasa K, Youdim MB and Naoi M: N-Propargylamine protects SH-SY5Y
cells from apoptosis induced by an endogenous neurotoxin,
N-methyl(R)salsolinol, through stabilization of mitochondrial
membrane and induction of anti-apoptotic Bcl-2. J Neural Trans.
113:21–32. 2006. View Article : Google Scholar
|
|
33
|
Liberti MV and Locasale JW: The Warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|