Introduction
Renal cell carcinoma (RCC) is a common malignancy of
the genitourinary system, and is classified into various subtypes
based on histopathological characteristics and each subtype has a
diverse prognosis (1,2). From a clinical viewpoint, three main
RCC subtypes are important in research: Clear cell RCC (ccRCC;
80–90 % cases), papillary RCC (10–15% cases), and chromophobe RCC
(4–5% cases) (1,2). The development of RCC is complex, and
numerous parameters, including anatomical, histological, clinical
and molecular factors contribute to disease outcome. Despite great
improvements in understanding the major molecular mechanisms
involved in RCC, none of the conventional or molecular markers have
demonstrated satisfactory sensitivity and specificity to be
considered as a prognostic indicator of RCC (1,2). Thus,
the majority of studies have focused on the development of suitable
biomarkers to improve diagnosis and prognostication in RCC.
As a result of recent advancements in understanding
of epigenetic modifications, DNA methylation is now regarded as one
of the key findings in the development and progression of cancer.
In RCC, several DNA methylation biomarkers associated with tumor
development, histological subtypes, prognosis and response to
therapy have been reported (3–13);
however, the majority of these markers were identified in cohorts
examined in single-center studies with variable results. Therefore,
their use in routine clinical practice is limited. The development
of high-throughput bioinformatics technology, such as genome-wide
methylation pattern analysis, now enables tumor-specific novel
molecular markers to be identified from numerous samples that can
readily be compared with other cohorts via database sharing.
Establishing the association between biomarkers and
clinical outcomes in RCC is likely to prove useful for developing
effective treatment strategies and improving clinical management.
The aim of the present study was to identify novel biomarkers that
could predict the outcomes of RCC using genome-wide DNA methylation
microarray data derived from patients analyzed in our institute and
from The Cancer Genome Atlas (TCGA), and to evaluate their clinical
application in ccRCC cases with long-term follow-up.
Materials and methods
Subjects and sample collection
A total of 355 human kidney specimens were obtained
from patients (age range: 30–83 years, sample collection: February
1999 to September 2015) with primary, histologically-confirmed
ccRCC who underwent radical or partial nephrectomy from two
independent institutes including: i) 201 human kidney specimens
[164 ccRCC and 37 matched normal control (NC)] from Chungbuk
National University Hospital (CBNUH); and ii) 154 human kidney
specimens (117 ccRCC and 37 matched NC) from Kyungpook National
University Hospital (KPNUH). To enhance the homogeneity of the
study population, patients were enrolled into the study according
to the following criteria: i) clinical and pathological stage T1-T4
without lymph node or distant metastasis; and ii) a minimum
follow-up period of 3 months. Patients whose samples had a positive
surgical margin at final pathology were not included to avoid bias
in survival analysis. The pathology of samples was independently
reviewed by a pathologist unaware of the intended use of the
clinical data. The specimens were obtained from the CBNUH and
KPNUH, members of the National Biobank of Korea, which is supported
by the Ministry of Health, Welfare and Family Affairs. The
collection and analysis of all samples were approved by the
Institutional Review Board of the CBNUH (approval no. 2010-01-001)
and KPNUH (approval no. KNUMC 2016-05-021). Informed consent was
obtained from each patient. The study design and validation
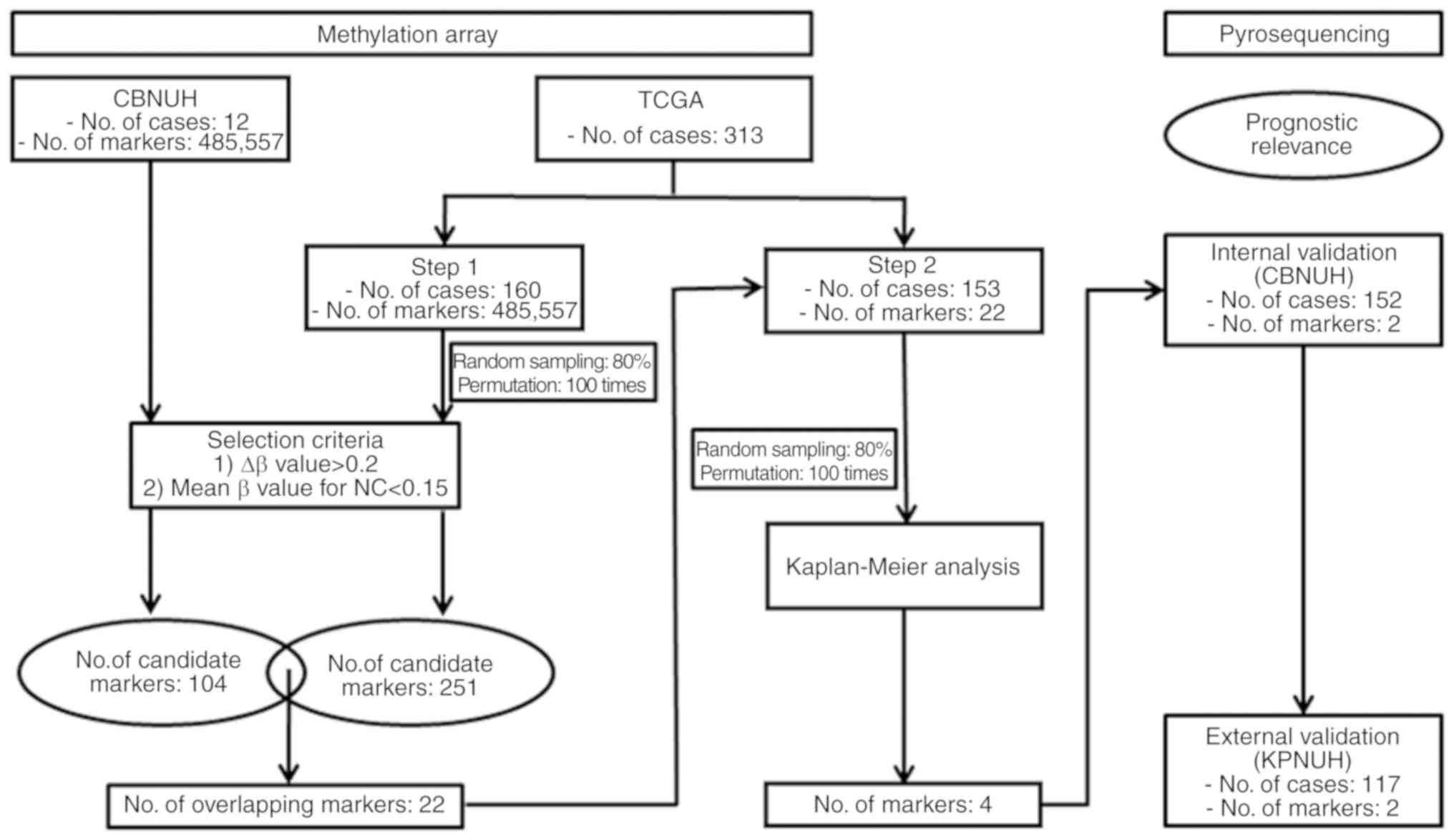
strategies were presented in Fig.
1. Tumors were categorized based on pathological stage and
histological subtype, as previously described (14,15).
Nuclear differentiation was graded according to the Fuhrman Nuclear
Grade system (16). All patients
were followed-up and managed according to standard recommendations
(1). Progression was defined as
lymph node involvement and/or metastatic disease as determined by a
computed tomography scan or bone scan.
DNA methylation profiling
Genomic DNA (gDNA) from patient's samples was
extracted according to the manufacture's protocol by using the
Wizard Genomic DNA Purification System (Promega Corporation,
Madison, WI, USA). Bisulfite-modified gDNA was prepared using the
EZ DNA Methylation-Lightning kit (Zymo Research, Irvine, CA, USA)
according to the manufacturer's instructions. The methylation
status was assayed using the Infinium HumanMethylation450 BeadChip
array (Infinium Methylation 450K; Illumina, Inc., San Diego, CA,
USA), which assesses the methylation status of >480,000 CpG
sites distributed across the whole genome. A total of 24 matched
DNA samples (pairs of NC and ccRCC from 12 patients) were used for
DNA methylation profiling. Fluorescence signals corresponding to C-
or T-nucleotides were measured (GenomeStudio Methylation Module,
V1.8, Illumina, Inc., San Diego, CA, USA), and the data were used
to assign a quantitative measure of methylation of specific CpG
islands (β value): Ranging from 0 (complete unmethylation) to 1
(complete methylation). The complete sets of microarray methylation
data are available online (http://www.ncbi.nlm.nih.gov/geo/; accession no.
GSE92482.
Array profiling data from TCGA
For comparisons with the array data from the
selected CBNUH patient samples and to investigate the prognostic
relevance of the identified methylated CpG islands, the TCGA kidney
renal clear cell carcinoma (KIRC) level 3 data set, also obtained
using the Infinium HumanMethylation450 BeadChip, was investigated
(https://cancergenome.nih.gov/).
Methylation profiles of 313 ccRCC patient samples were available
within the KIRC dataset. The dataset was divided into two groups:
i) Training set, 160 pairs of ccRCC and NC matched samples; and ii)
test set, an addition 153 ccRCC samples without matched NC samples.
The training set data were used for comparisons with the candidate
CpG sites identified with the CBNUH samples, and the test set data
were used for assessing the prognostic relevance of those
candidates. Baseline characteristics of the KIRC patients are
provided in Table SI.
Pyrosequencing (PSQ) analysis
The DNA methylation status of the candidate CpG
island loci identified using the Infinium HumanMethylation450 array
were specifically assessed by PSQ analysis of 331 human kidney
samples (CBNUH, 152 ccRCC and 25 matched NCs; KPNUH, 117 ccRCC and
37 matched NCs) using a PyroMark Q96 ID instrument (Qiagen, Inc.,
Valencia, CA, USA) according to the manufacturer's instructions.
Each primer was designed using Pyrosequencing Assay Design Software
v2.0 (Qiagen, Inc.). PSQ primers were designed to encompass the CpG
island loci determined from the Illumina Infinium array. The primer
sequences and amplification conditions are described in Table SII.
Statistical analysis
The levels of DNA methylation were normalized using
quantile normalization in R (v2.10.0, http://www.r-project.org/). The criteria applied for
the selection of ccRCC-specific methylated CpG island loci were as
follows: i) a difference in the levels of DNA methylation between
ccRCC and NC (∆β value) >0.2; and ii) a mean β-value for
NC<0.15. Using the same selection criteria, the training set
from the TCGA data was used for comparisons with genes identified
from CBNUH ccRCC samples. With the same selection criteria of CBNUH
samples, a permutation test (80% random sampling; 100 times) was
conducted to reduce unduly identified CpG island loci. Only CpG
island loci that possessed >50-times difference were selected
for further analysis. Among the candidate CpG island loci
identified, those common to the CBNUH ccRCC samples and the TCGA
training set were selected as candidate prognostic indicators of
ccRCC. The prognostic relevance of candidate CpG island loci was
explored with the TCGA test dataset using Kaplan-Meier analysis
(100 times permutation, 80% sampling) and those loci that had
statistical significance >50 times were regarded as targets for
further validation with ccRCC samples by PSQ.
The differences in continuous variables between
groups were assessed using a two-sample t-test or ANOVA trend
analyses using polynomial contrasts and data were presented as the
mean ± standard deviation. Receiver operating characteristic (ROC)
curves were used to estimate the capability of candidate markers in
predicting the of progression of disease, and to determine the
optimal cut-off point for dividing patients into subgroups
(hypomethylation or hypermethylation) with the highest combined
sensitivity and specificity. A multivariate Cox proportional
hazards regression model was used to evaluate the prognostic value
of candidate markers adjusted for well-known clinicopathological
factors (sex, age, body mass index, grade and stage). Statistical
analysis was performed using SPSS software v21.0 (IBM Corp. Armonk,
NY, USA) and R v2.12.1 software. P<0.05 was considered to
indicate a statistically significant difference.
Results
Baseline characteristics
The baseline characteristics of the 164 CBNUH
patients with ccRCC were presented in Table I. The mean age and median
progression-free survival period of the patients for PSQ were
64.3±13.8 years and 27.2 months (ranging 3.0-188.9 months),
respectively; 43 patients (28.2%) experienced disease
progression.
 | Table I.Baseline characteristics of the
patients enrolled in the present study. |
Table I.
Baseline characteristics of the
patients enrolled in the present study.
| Variables | Microarray
(n=12) | Pyrosequencing
(n=152) |
|---|
| Age (years, mean ±
SD) | 57.3±11.3 |
64.3±13.8 |
| Gender [n (%)] |
|
|
|
Male | 8 (66.7) | 110 (72.4) |
|
Female | 4 (33.3) | 42
(27.6) |
| BMI
kg/m2 (mean ± SD) | 25.9±3.3 | 24.6±3.6 |
| Fuhrman grade, [n
(%)] |
|
|
| G1 | 2 (16.7) | 34
(22.4) |
| G2 | 7 (58.3) | 68
(44.7) |
| G3 | 2 (16.7) | 45
(29.6) |
| G4 | 1 (8.3) | 5
(3.3) |
| T stage, [n
(%)] |
|
|
| T1 | 4 (33.3) | 112 (73.7) |
| T2 | 4 (33.3) | 14 (9.2) |
| T3 | 4 (33.3) | 26
(15.1) |
| T4 | – | 3
(2.0) |
| Progression-free
survival (months) Median (range) |
| 27.2
(3.0-188.9) |
| Progression [n
(%)] |
|
|
| No |
| 109 (71.8) |
|
Yes |
| 43
(28.2) |
Identification of differentially
methylated genes in ccRCC and NC
Genome-wide microarray methylation profiles of ccRCC
samples and corresponding NC samples from 12 CBNUH cases were
compared to identify ccRCC-specific methylated genes. After
applying selection criteria (∆β value >0.2 and mean β-value in
NC<0.15), 104 unique CpG island loci that were hypermethylated
in ccRCC compared with the NCs were identified (Table SIII). The 104 selected candidate
markers were then validated by comparing with the TCGA training
data set from 160 pairs of ccRCC and matched NC samples subjected
to the aforementioned selection criteria. In the TCGA training set,
251 CpG island loci were hypermethylated in ccRCC compared with the
NCs (Table SIV). Of these, 22 CpG
island loci overlapped with those identified as ccRCC-specific in
the 12 CBNUH cases (Table SV). The
prognostic importance of the 22 candidate CpG island loci were then
examined with the TCGA test dataset. In the 100-times permutation
test of the Kaplan-Meier analysis, 4/22 CpG island loci were
selected as targets for validation by PSQ using 152 CBNUH ccRCC
samples. The candidate genes at the four loci were as follows: Zinc
finger protein 492 (ZNF492; at two CpG island loci), twist
family bHLH transcription factor 1 (TWIST1), and G
protein-coupled receptor 149 (GPR149).
PSQ analysis and internal validation
of prognostic relevance in the clinical samples
To verify the prognostic relevance of candidate
genes using a different methodology in a larger, different set of
samples, we performed PSQ analyses on bisulfite-modified gDNA
obtained from 177 CBNUH human kidney specimens (152 ccRCC and 25
matched NC). PSQ analysis was performed on two of the four
candidate CpG island regions in the present study, including one of
the two ZNF492 loci (Infinium HumanMethylation450 target ID,
cg01485075) and GPR149 (cg00046499). Analysis of the
candidate gene TWIST1 (cg26818735) was not possible due to
technical variability across multiple PSQ assays of complex
sequences of the homopolymer for PSQ.
To examine the association between methylation
patterns and clinicopathological factors, methylation values for
each gene were compared between samples. The methylation levels of
ZNF492 and GPR149 were significantly higher in the
ccRCC specimens than in the NCs (P<0.05). Increased methylation
levels of ZNF492 and GPR149 were significantly
associated with advanced pathological T stage (P<0.05).
Decreased methylation levels of ZNF492 were associated with
higher body mass index; however, no significant association was
observed with GPR149. The methylation levels of
ZNF492 and GPR149 exhibited significantly increased
methylation compared with control samples and had a tendency to
increase with higher grade; however, differences between low and
high grade tumors in GPR149 methylation were not
statistically significant (Fig. 2,
Table SVI).
The value of candidate methylation markers for the
prediction of disease progression was determined using the area
under the ROC curve (AUC). The AUC values for ZNF492 and
GPR149 were 0.716 [95% confidence interval (CI),
0.636-0.787; P<0.001] and 0.781 (95% CI, 0.707-0.842;
P<0.001), respectively (Fig.
S1). To validate the prognostic relevance of candidate
methylation markers, the methylation values of ZNF492 and
GPR149 were dichotomized (hypomethylation or
hypermethylation) using optimal cut-off points (ZNF492:
18.11 and GPR149: 29.31), and survival analysis was
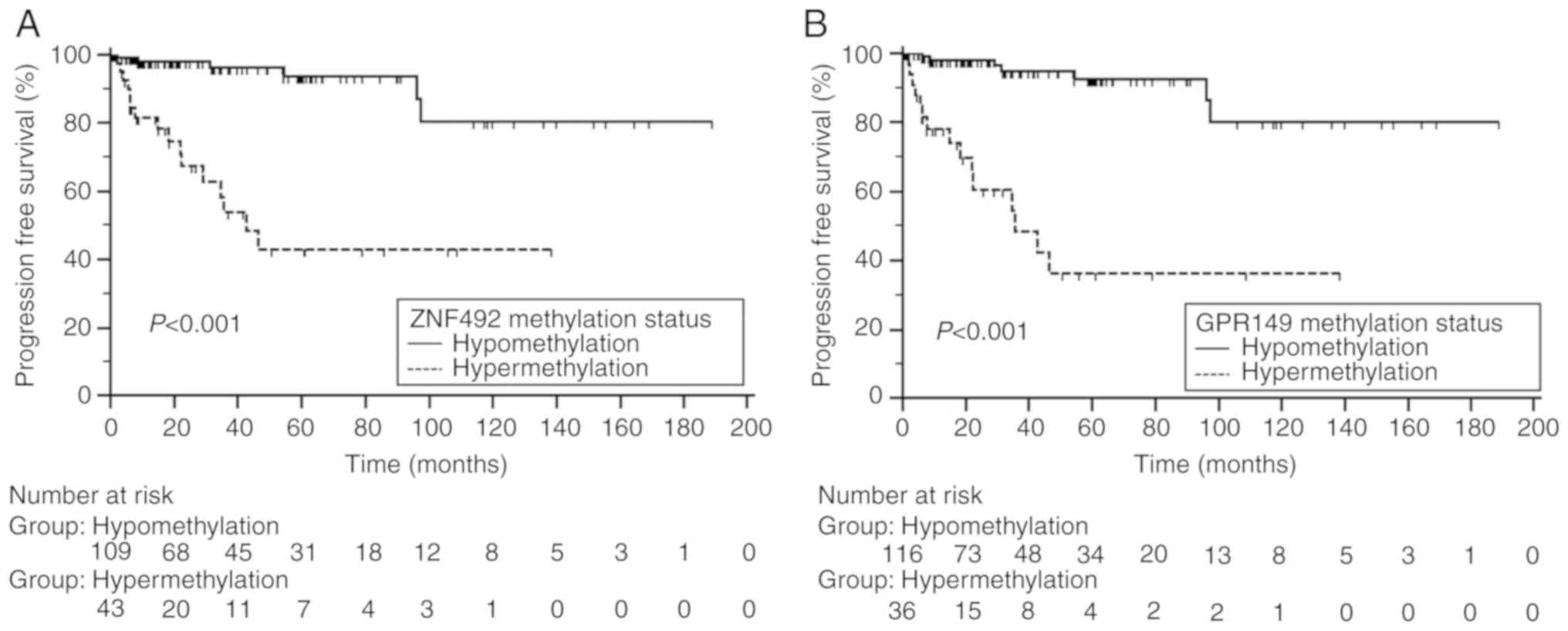
performed. Kaplan-Meier estimates identified significant
differences in time-to-progression according to the methylation
status of ZNF492 and GPR149 (log-rank test,
P<0.05; Fig. 3). In the
multivariate Cox regression analyses, the methylation status of
ZNF492 [hazard ratio (HR), 5.44; 95% CI, 2.04-14.53;
P=0.001] and of GPR149 (HR, 7.07; 95% CI, 2.65-18.89;
P<0.001) was suggested as independent predictors of disease
progression (Table II).
| Table II.Univariate and Multivariate Cox
regression analyses for determining disease outcome based on the
methylation status of each gene. |
Table II.
Univariate and Multivariate Cox
regression analyses for determining disease outcome based on the
methylation status of each gene.
|
| Model
1b | Model
2c |
|---|
|
|
|
|
|---|
|
| Univariate | Multivariate | Univariate | Multivariate |
|---|
|
|
|
|
|
|
|---|
|
Variablesa | HR (95%
CI)a | P-value | HR (95%
CI)b | P-value | HR (95%
CI)a | P-value | HR (95%
CI)b | P-value |
|---|
| Age | 1.02
(0.98-1.05) |
0.373 | – | – | 1.02
(0.98-1.05) |
0.373 | – | – |
| Sex | 0.779
(0.30-2.00) |
0.603 | – | – | 0.779
(0.30-2.00) |
0.603 |
|
|
| BMI | 0.37
(0.16-0.86) |
0.021 | 0.35
(0.12-1.04) |
0.058 | 0.37
(0.16-0.86) |
0.021 | 0.22
(0.07-0.65) |
0.007 |
| Grade | 5.39
(2.19-13.23) | <0.001 | 5.39
(2.19-13.23) | <0.001 | 5.39
(2.19-13.23) | <0.001 | 1.85
(0.55-6.29) |
0.323 |
| Stage |
| <0.001 |
|
0.001 |
| <0.001 |
|
0.003 |
| T1 | Reference | – | Reference |
| Reference | – | Reference |
|
| T2 | 5.65
(1.71-18.67) | – | 6.37
(1.56-25.71) |
| 5.65
(1.71-18.67) | – | 7.43
(1.89-29.28) |
|
| T3/4 | 10.33
(3.81-28.01) | – | 7.75
(2.56-23.47) |
| 10.33
(3.81-28.01) | – | 5.78
(1.77-18.94) |
|
| ZNF492 | 9.32
(3.62-23.91) | <0.001 | 5.44
(2.04-14.53) |
0.001 | – | – | – | – |
| GPR149 | – | – | – | – | 10.51
(4.26-25.93) | <0.001 | 7.07
(2.65-18.89) | <0.001 |
External validation of prognostic
relevance in the clinical samples
The validity of two genes as methylation markers in
ccRCC was also evaluated using an independent set of PQS data from
KPNUH samples, which included ccRCC (n=117) and matched NC (n=37)
tissues (Table SVII). The
association between the two methylation markers and
clinicopathological characteristics revealed similar results to
those of the CBNUH dataset (Table
SVIII). The AUC values for ZNF492 and GPR149 were
0.717 [95% CI, 0.542-0.891; P=0.014] and 0.866 (95% CI,
0.746-0.986; P<0.001), respectively (Fig. S1). Patients with hypermethylated
genes had shorter durations of progression than those of
hypomethylated status (log-rank test, P<0.05, respectively;
Fig. 4). According to the Cox
multivariate regression analysis, the methylation status of
ZNF492 (HR), 3.89; 95% CI, 1.04-14.61; P=0.044] and of
GPR149 (HR, 33.67; 95% CI, 5.87-192.88; P<0.001) were
significantly associated with disease progression (Table SIX).
Discussion
DNA methylation is a crucial mechanism for the
development and progression of cancer. The methylation status of
various genes can be used as a biomarker for risk assessment, early
detection, prediction of prognosis, and therapy response in
numerous types of cancer (17,18).
In the present study, novel methylation markers that were able to
predict the likelihood of disease progression were identified based
on genome-wide DNA methylation array datasets of clinical samples
from our own institute and from TCGA. Close associations with
unfavorable characteristics of disease, including advanced stage
were noted. Furthermore, the methylation status of each of these
markers was revealed to be an independent indicator of progression
in two cohorts of patients with ccRCC.
Increasing evidence has indicated that the
methylation status of individual or combinations of genes are
associated with different outcomes in patients with RCC and may
serve as independent indicators of prognosis (3–6). In a
study of 179 patients who underwent radical or partial nephrectomy
for ccRCC, Ras association domain-containing protein 1 methylation
status was significantly associated with higher grade and advanced
stage. Additionally, this gene served as an independent variable
for the prediction of cancer-specific survival in patients with
ccRCC (6). Similarly, another study
revealed that the combined methylation status of collagen α-1 (XIV)
and basonuclin was independently associated with a poorer
prognosis, and served as a better prognostic indicator than tumor
stage or grade (4).
Previously, microarray-based clinical studies have
sought novel molecules that could enhance diagnostic and prognostic
ability alone or with conventional biomarkers (7–11). In
a genome-wide study with kidney tissues from 96 patients, a set of
DNA methylation markers was demonstrated to reliably discriminate
between malignant and benign lesions, and classical histological
subtypes (7). In addition, these
findings were validated using a microarray data set from TCGA
comprising >1,000 kidney samples. In addition, other studies
have suggested the possible roles of methylation signatures for the
detection and prognostic prediction of RCC (8–11).
Microarray-based DNA methylation profiling may therefore provide
insight into the disease and, most importantly, reveal biomarkers
for the detection and prognostication of RCC. In the present study,
using methylation profiles of our clinical samples and those in
TCGA, we selected CpG island loci commonly hypermethylated in ccRCC
compared with NCs. The potential of these candidate loci as
indicators of prognosis was first explored in a test set of TCGA
data and independently validated with a different technique (PSQ)
in two independent ccRCC cohorts with long-term follow-up. Our
methodological approach for gene selection and prognostic
validation using multiple steps with different independent data
sets was selected to enhance the reliability of our results as much
as possible. Furthermore, limiting patient enrollment to those with
ccRCC increased the homogeneity of the samples, which may enhance
the likelihood of identifying differences. Our candidate markers
are likely to be applicable across various ethnicities as they were
determined from data derived from Western and Eastern populations.
The results of the present study are therefore promising; however,
further investigation is required to determine the false prediction
rate of these markers in particular.
Limitations of the present study must be addressed.
Only 12 samples were used for genome-wide microarray methylation
profiling to identify candidate markers. To overcome the limited
sample number, a large TCGA dataset was utilized simultaneously.
Additionally, validation was performed using PSQ in a larger set of
different samples from our institute and others. These types of
multiple validation strategies with different datasets may improve
the reliability of our results. Additionally, the effects of
methylation status on mRNA and protein expression should have been
evaluated. Gene expression and immunohistochemical analysis could
not be performed due to the lack of available tissue samples.
Although the association between alterations in methylation and
gene expression of selected markers was not determined in the
present study, preliminary data analysis with TCGA data revealed
that the expression of ZNF492 was significantly lower in
cancer tissues than in NC samples (Fig. S2). Furthermore, our study
investigated hypermethylation genes instead those hypomethylated.
Global hypomethylation or locus-specific hypermethylation in CpG
island-rich promoters is a common phenomenon of cancer. Numerous
sites of hypomethylation that vary between normal and RCC tissues
could be identified with a microarray dataset. In the present
study, we aimed to analyze 2–5 genes at most with validation in
clinical samples. Providing that too many candidate genes are
selected, it would be difficult to validate their clinical
relevance in clinical samples. In this regard, we focused on the
hypermethylation rather than the hypomethylation of genes.
To the best of our knowledge, this is the first
report to demonstrate that ZNF492 and GPR149 act as
methylation-induced prognostic indicators in RCC. Currently,
limited information regarding the specific function of these genes
is available in the literature. ZNF492, also known as
ZNF115, is a member of the zinc finger protein family and is
located at 19p12. There are eight different classes of zinc finger
motifs and each class has a distinct biological role (19). Evidence accumulated over several
decades has demonstrated crucial roles for zinc finger proteins in
the initiation and progression of cancer in numerous type of
cancers (20–24). As an example of the role of
methylation, one zinc finger family member ZNF545 was
reported to serve as a tumor suppressor by inducing cell apoptosis,
inhibiting ribosome biogenesis, and suppressing nuclear factor-κB
and activator protein-1 signaling in nasopharyngeal, esophageal,
lung, gastric, colon, and breast cancer upon methylation (21). In addition, a combination of
methylated CpG island sites corresponding to those of zinc finger
genes was able to predict survival outcome in patients with gastric
cancer (22). GPR149 is a
member of the G protein-coupled receptor (GPCR) family, which is
located at 3q25.2. GPCRs comprise a large family of cell-surface
receptors and control various features of tumorigenesis, as well as
many cancer-associated signaling pathways (25,26).
With aid of genome-wide approaches, numerous biomolecules have
recently emerged as novel biomarkers in several human tumors;
however, in the majority of cases, the association between a
specific disease and a putative biomarker has not been yet
determined. Similarly, we have not investigated the functional
roles of ZNF492 and GPR149 in the tumorigenesis and
progression of RCC, which poses as a limitation of the present
study. However, establishing novel biomarkers, even of unknown
function, and the potential application in the detection,
prognostic prediction and use as therapeutic targets is likely to
prove valuable in various types of human disease. Therefore,
functional studies are required to determine the full potential of
the putative biomarkers identified in the present study.
Supplementary Material
Supporting Data
Acknowledgements
The abstract was presented at the 66th Annual
Meeting of the Korean Urological Association Oct 12-Oct 14 2016 in
Seoul, published as abstract no. O-047 in Korean Urological
Association Abstracts: 2016.
Funding
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education (grant nos. 2015R1D1A1A01057786
and 2018R1D1A1B07043906).
Availability of data and materials
The datasets generated and analyzed during the
current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=azovkgyunvcdrij&acc=GSE92482.
Authors' contributions
YJK, SCL, TGK and WJK made substantial contributions
to the concept of the present study. YJK, WJ, TGK and WJK conducted
the experiments. Validation of the data was conducted by YJB, JSK
and SPS. YJK, WJ, HYY, SMK, SKL, SPS, HWK, WTK, SJY, HSS, KHR, SWK,
YSH and GSY performed data analysis. YJK, WJ, YJB and JSK collected
data. YJK drafted the manuscript. YJK, WJ, XMP, HYH, SMK, SKL, HWK,
WTK, SJY, HSS, KHR, SWK, YSH, GSY, SCL, TGK and WJK reviewed the
manuscript. SCL, TGK and WJK provided guidance and supervision. YJK
acquired funding for the study.
Ethics approval and consent to
participate
The collection and analysis of all samples were
approved by the CBNUH (approval no. 2010-01-001) and KPNUH
(approval no. KNUMC 2016-05-021). Institutional Review Board and
informed consent was obtained from each subject. The study
methodologies conformed with the standards set by the Declaration
of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CBNUH
|
Chungbuk National University
Hospital
|
|
ccRCC
|
clear cell renal cell carcinoma
|
|
CI
|
confidence interval
|
|
gDNA
|
genomic DNA
|
|
GPCRs
|
G protein-coupled receptors
|
|
GPR149
|
G protein-coupled receptor 149
|
|
HR
|
hazard ratio
|
|
KIRC
|
kidney renal clear cell carcinoma
|
|
KPNUH
|
Kyungpook National University
Hospital
|
|
NC
|
normal control
|
|
PSQ
|
pyrosequencing
|
|
ROC
|
receiver operating characteristic
|
|
AUC
|
area under the ROC curve
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TWIST1
|
twist family bHLH transcription factor
1
|
|
ZNF492
|
zinc finger protein 492
|
References
|
1
|
Ljungberg B, Bensalah K, Canfield S,
Dabestani S, Hofmann F, Hora M, Kuczyk MA, Lam T, Marconi L,
Merseburger AS, et al: EAU guidelines on renal cell carcinoma: 2014
update. Eur Urol. 67:913–924. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Srinivasan R, Ricketts CJ, Sourbier C and
Linehan WM: New strategies in renal cell carcinoma: Targeting the
genetic and metabolic basis of disease. Clin Cancer Res. 21:10–17.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eggers H, Steffens S, Grosshennig A,
Becker JU, Hennenlotter J, Stenzl A, Merseburger AS, Kuczyk MA and
Serth J: Prognostic and diagnostic relevance of hypermethylated in
cancer 1 (HIC1) CpG island methylation in renal cell carcinoma. Int
J Oncol. 40:1650–1658. 2012.PubMed/NCBI
|
|
4
|
Morris MR, Ricketts C, Gentle D,
Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F and
Maher ER: Identification of candidate tumour suppressor genes
frequently methylated in renal cell carcinoma. Oncogene.
29:2104–2117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Vlodrop IJ, Baldewijns MM, Smits KM,
Schouten LJ, van Neste L, van Criekinge W, van Poppel H, Lerut E,
Schuebel KE, Ahuja N, et al: Prognostic significance of Gremlin1
(GREM1) promoter CpG island hypermethylation in clear cell renal
cell carcinoma. Am J Pathol. 176:575–584. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawai Y, Sakano S, Suehiro Y, Okada T,
Korenaga Y, Hara T, Naito K, Matsuyama H and Hinoda Y: Methylation
level of the RASSF1A promoter is an independent prognostic factor
for clear-cell renal cell carcinoma. Ann Oncol. 21:1612–1617. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lasseigne BN, Burwell TC, Patil MA, Absher
DM, Brooks JD and Myers RM: DNA methylation profiling reveals novel
diagnostic biomarkers in renal cell carcinoma. BMC Med. 12:2352014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arai E, Ushijima S, Tsuda H, Fujimoto H,
Hosoda F, Shibata T, Kondo T, Imoto I, Inazawa J, Hirohashi S and
Kanai Y: Genetic clustering of clear cell renal cell carcinoma
based on array-comparative genomic hybridization: Its association
with DNA methylation alteration and patient outcome. Clin Cancer
Res. 14:5531–5539. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ricketts CJ, Morris MR, Gentle D, Shuib S,
Brown M, Clarke N, Wei W, Nathan P, Latif F and Maher ER:
Methylation profiling and evaluation of demethylating therapy in
renal cell carcinoma. Clin Epigenetics. 5:162013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Slater AA, Alokail M, Gentle D, Yao M,
Kovacs G, Maher ER and Latif F: DNA methylation profiling
distinguishes histological subtypes of renal cell carcinoma.
Epigenetics. 8:252–267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei JH, Haddad A, Wu KJ, Zhao HW, Kapur P,
Zhang ZL, Zhao LY, Chen ZH, Zhou YY, Zhou JC, et al: A
CpG-methylation-based assay to predict survival in clear cell renal
cell carcinoma. Nat Commun. 6:86992015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arai E, Kanai Y, Ushijima S, Fujimoto H,
Mukai K and Hirohashi S: Regional DNA hypermethylation and DNA
methyltransferase (DNMT) 1 protein overexpression in both renal
tumors and corresponding nontumorous renal tissues. Int J Cancer.
119:288–296. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ellinger J, Kahl P, Mertens C, Rogenhofer
S, Hauser S, Hartmann W, Bastian PJ, Büttner R, Müller SC and von
Ruecker A: Prognostic relevance of global histone H3 lysine 4
(H3K4) methylation in renal cell carcinoma. Int J Cancer.
127:2360–2366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Greene FL: The American Joint Committee on
Cancer: Updating the strategies in cancer staging. Bull Am Coll
Surg. 87:13–15. 2002.PubMed/NCBI
|
|
15
|
Lopez-Beltran A, Scarpelli M, Montironi R
and Kirkali Z: 2004 WHO classification of the renal tumors of the
adults. Eur Urol. 49:798–805. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fuhrman SA, Lasky LC and Limas C:
Prognostic significance of morphologic parameters in renal cell
carcinoma. Am J Surg Pathol. 6:655–663. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome-biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ushijima T: Detection and interpretation
of altered methylation patterns in cancer cells. Nat Rev Cancer.
5:223–231. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krishna SS, Majumdar I and Grishin NV:
Structural classification of zinc fingers: Survey and summary.
Nucleic Acids Res. 31:532–550. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jen J and Wang YC: Zinc finger proteins in
cancer progression. J Biomed Sci. 23:532016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng Y, Liang P, Geng H, Wang Z, Li L,
Cheng SH, Ying J, Su X, Ng KM, Ng MH, et al: A novel 19q13
nucleolar zinc finger protein suppresses tumor cell growth through
inhibiting ribosome biogenesis and inducing apoptosis but is
frequently silenced in multiple carcinomas. Mol Cancer Res.
10:925–936. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deng J, Liang H, Ying G, Dong Q, Zhang R,
Yu J, Fan D and Hao X: Poor survival is associated with the
methylated degree of zinc-finger protein 545 (ZNF545) DNA promoter
in gastric cancer. Oncotarget. 6:4482–4495. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jen J, Lin LL, Chen HT, Liao SY, Lo FY,
Tang YA, Su WC, Salgia R, Hsu CL, Huang HC, et al: Oncoprotein
ZNF322A transcriptionally deregulates alpha-adducin, cyclin D1 and
p53 to promote tumor growth and metastasis in lung cancer.
Oncogene. 35:2357–2369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang L, Hamilton SR, Sood A, Kuwai T,
Ellis L, Sanguino A, Lopez-Berestein G and Boyd DD: The previously
undescribed ZKSCAN3 (ZNF306) is a novel ‘driver’ of colorectal
cancer progression. Cancer Res. 68:4321–4330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bar-Shavit R, Maoz M, Kancharla A, Nag JK,
Agranovich D, Grisaru-Granovsky S and Uziely B: G protein-coupled
receptors in cancer. Int J Mol Sci. 17:E13202016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lappano R and Maggiolini M: G
protein-coupled receptors: Novel targets for drug discovery in
cancer. Nat Rev Drug Discov. 10:47–60. 2011. View Article : Google Scholar : PubMed/NCBI
|