Introduction
Metastatic melanoma is the deadliest skin cancer and
has an increasing incidence worldwide (1). It exhibits high metastatic potential
and low response rates to conventional therapies, such as
dacarbazine (2). It also has the
highest mutational load in all cancers (3). Mutations in the BRAF gene which
activate MAPK signaling and subsequently promote unrestricted
melanoma growth, are the most common somatic events with an
incidence of 40–60% in melanoma patients (4). Targeted inhibitors towards BRAF, such
as vemurafenib and dabrafenib, have been developed and approved by
the U.S. Food and Drug Administration for the treatment of
BRAF-mutant melanoma (5). Patients
exhibited a high response rate of ~50% to these BRAF inhibitors
(6–8). In addition, these BRAF inhibitors
improved the median progression-free survival to ~5.1–8.8 months
(6,8–11).
However, resistance develops within 12 months as revealed by the
median progression-free survival. Copy-number alterations,
mutations, or expression changes that result in reactivation of the
MAPK signaling pathway or activation of the PI3K/AKT signaling
pathway are considered to contribute to BRAF inhibitor resistance
in melanoma (5). Therefore, a
combination of BRAF inhibitor and MEK inhibitor was used to treat
BRAF-mutant melanoma. Such a combination was revealed to improve
the objective response rates to >60% and the median
progression-free survival to 9.3–11.4 months (11,8).
However, almost all patients ultimately develop resistance to such
combination therapies. Therefore, it is urgent to develop other new
therapies to overcome BRAF inhibitor resistance in melanoma.
Icariside II (IS) also known as baohuoside I
(Fig. 1A), a glycosyloxy flavone
isolated from Herba Epimedii, has been identified as an
anticancer agent against multiple cancers, such as non-small-cell
lung cancer, multiple myeloma, osteosarcoma, glioblastoma,
epidermoid carcinoma, breast cancer, esophageal carcinoma, prostate
cancer and melanoma. It has been revealed to inhibit cell
proliferation, promote apoptosis, and induce cell cycle arrest by
targeting different signaling pathways including ROS mediated
mitochondrial pathways, EGFR, AKT, MAPK, STAT3, COX-2/PEG2 and
β-catenin, in these cancers (12–14).
In addition, IS has the potential to improve the benefits of
multiple therapies, such as chemotherapies and immunotherapies. Our
previous study indicated that it could further improve
paclitaxel-induced apoptosis by inhibiting TLR4 signaling in human
melanoma cells (15). IS was also
reported to overcome TRAIL resistance by regulating the
ROS/STAT3/cFLIP signaling pathway (16). However, the role of IS in overcoming
BRAF inhibitor resistance remains undefined.
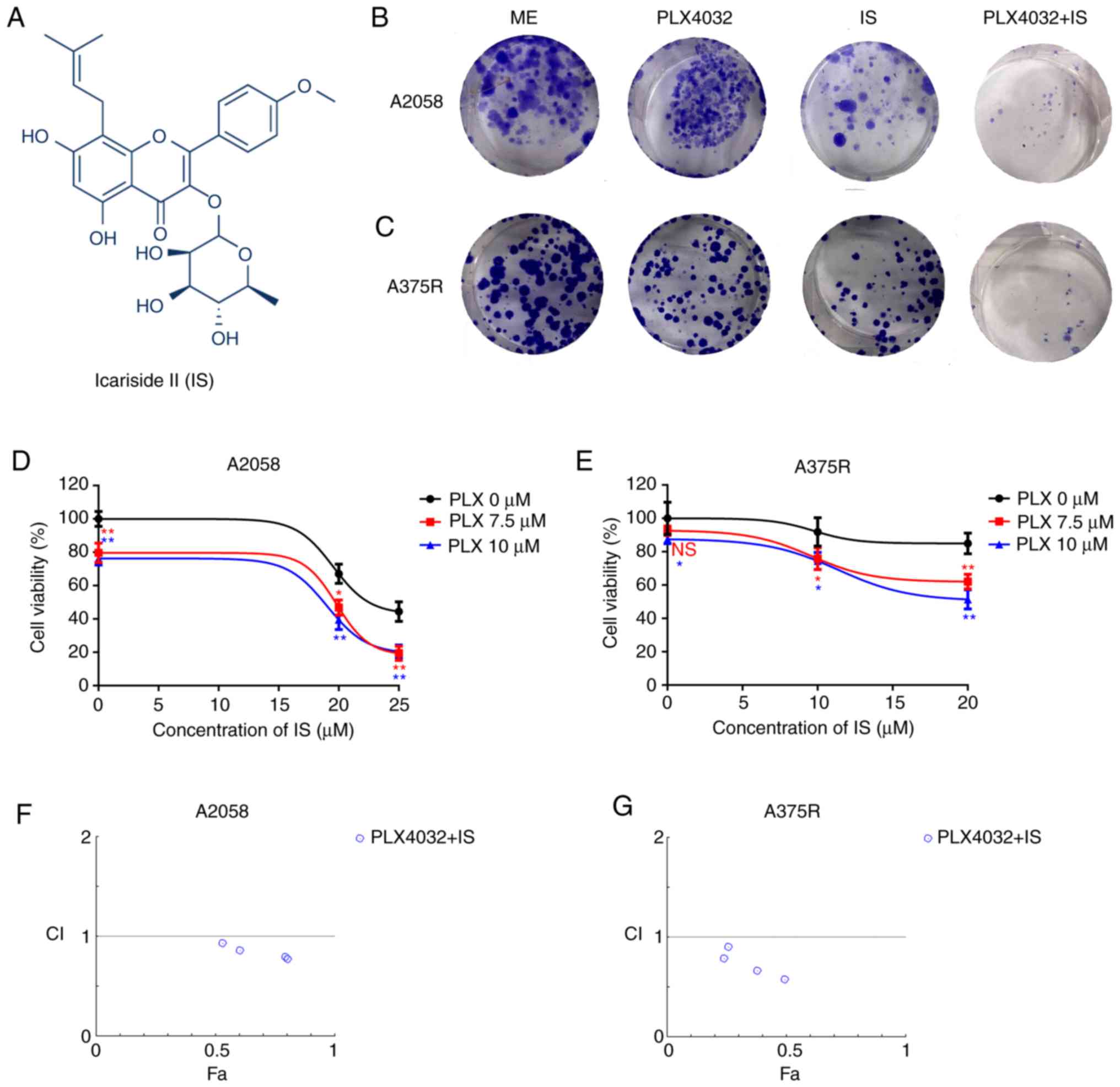 | Figure 1.IS potentiates PLX4032-induced
cytotoxicity in BRAF inhibitor-resistant melanoma cells. (A)
Chemical structure of IS. Both (B) A2058 and (C) A375R cells were
treated with ME, PLX4032 (10 µM), IS (20 µM), and PLX4032 (10
µM)+IS (20 µM), respectively. Nine days later, the long-term
effects of IS and/or PLX4032 were assessed by clonogenic assay.
Both (D) A2058 and (E) A375R cells were treated with various
concentrations of PLX4032 (0, 7.5, 10 µM) and/or IS (0, 20 and 25
µM in A2058 cells; 0, 10 and 20 µM in A375R cells) for 48 h and
cell viabilities were detected by MTT assay. The combination index
of PLX4032 and IS in (F) A2058 and (G) A375R cells was calculated
by CompuSyn. CI<1 indicates a synergistic effect; CI=1 indicates
additivity; CI >1 indicates an antagonistic effect. Data were
presented as the mean ± standard error of the mean. A two-way
ANOVA, with post hoc Tukey's test was applied for comparisons.
*P<0.05 and **P<0.01. IS, icariside II; ME, medium
control. |
In the present study, it was demonstrated that IS
potentiated PLX4032-induced cytotoxicity, apoptosis, and autophagy
in melanoma cells with either intrinsic or acquired resistance to
BRAF inhibitors. Furthermore, it was revealed that IS significantly
increased ROS production, and subsequently inhibited the expression
of microphthalmia-associated transcription factor (MITF) and
tyrosine-protein kinase Met (c-Met), well-known factors that
contribute to BRAF inhibitor resistance. Therefore, IS serves as a
potential agent that overcomes BRAF inhibitor resistance in
melanoma.
Materials and methods
Cell culture and BRAF
inhibitor-resistant melanoma cell line establishment
The human melanoma cell lines, A2058 and A375, were
purchased from the American Type Culture Collection and cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS) and 1% penicillin/streptomycin (all from
Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C in a 5%
CO2 humidified incubator. Vemurafenib (PLX4032)
(MedChemExpress, Inc.) resistant A375 melanoma cells (A375R) were
generated by adding increasing concentrations of PLX4032 into A375
cells for more than half a year (17). The final concentration of PLX4032
used to maintain A375R was 2 µM. To verify whether A375R cells were
resistant to PLX4032, both parental A375 and resistant A375R cells
were treated with various concentrations (0.1, 0.5, 1, 5 and 10 µM)
of PLX4032 for 48 h, and the cell viabilities were detected by MTT
assay. Furthermore, both parental A375 and resistant A375R cells
were treated with different concentrations (0, 2 and 5 µM) of
PLX4032 for 24 h, and cell apoptosis was assessed by Annexin V and
7-AAD staining.
Cell viability assay
Cell viability was determined by MTT assay. Cells
were seeded into 96-well plates at a density of 5,000 cells/well
and cultured in an incubator overnight at 37°C in 5%
CO2. Then various concentrations of PLX4032 (0, 7.5 and
10 µM) and/or IS (0, 20 and 25 µM in A2058 cells; 0, 10 and 20 µM
in A375R cells) were added into corresponding wells, and incubated
for 48 h at 37°C. For NAC/GSH treatment experiment, cells were
pre-treated with 5 mM NAC/1 mM GSH for 1 h, and then treated with
PLX4032 (10 µM) and IS (20 µM) for another 18 h. Next, 20 µl MTT
solution (Sigma-Aldrich; Merck KGaA) with a concentration of 5
mg/ml was added to every well and incubated at 37°C for 2–4 h.
Subsequently, 100 µl DMSO was added to each well to dissolve the
formed formazan crystal. Finally, the optical density (OD) was
detected at 492 nm using Infinite F200 Pro microplate reader (Tecan
Group, Ltd.). The cell viabilities were calculated via the
following formula: Cell viability (%)=(mean OD treatment/mean OD
control) × 100.
Cell apoptosis assay
The cell apoptosis rates were determined by PE
Annexin V Apoptosis Detection kit I (BD Biosciences) via flow
cytometry according to the manufacturer's instructions. Briefly,
cells were seeded in 6-well plates at a density of 2×105
cells/well, and cultured in an incubator overnight at 37°C in 5%
CO2. Then PLX4032 (0 and 10 µM) and/or IS (0 and 20 µM)
were added into corresponding wells, and incubated for 24 or 72 h
at 37°C. Next, the cells were harvested and washed three times with
PBS (Gibco; Thermo Fisher Scientific, Inc.), and resuspended with
100 µl 1X binding buffer, and then 5 µl PE Annexin V and 20 µl
7-Amino-Actinomycin (7-AAD) were added into each tube and incubated
at room temperature in the dark for 20 min. Finally, the labeled
cells were washed with 1X binding buffer, and the data were
obtained by a flow cytometer (Attune NxT; Thermo Fisher Scientific,
Inc.) and analyzed by FlowJo software v10.0 (Tree Star, Inc.).
Cellular ROS detection
A total of 20×104 cells were seeded into
each well of a 6-well plate and incubated overnight. Then the cells
were treated with vehicle control, IS, PLX4032, and IS+PLX4032 for
24 h. The treated cells were loaded with DCFH-DA probes for 20 min
at 37°C, followed by a three-time wash with DMEM without FBS.
Finally, the signals were detected by a flow cytometer and analyzed
by FlowJo software v10.0.
Western-blotting
The treated cells were lysed in RIPA lysis buffer
(Beyotime Institute of Biotechnology) containing 10% PMSF (Beyotime
Institute of Biotechnology) at 4°C for 15 min with intermittent
vortex, and then centrifuged at 4°C at 12,000 × g for 10 min. The
supernatants were immediately collected. The protein concentrations
were measured by a BCA Protein Assay kit (Life Technology; Thermo
Fisher Scientific, inc.), according to the manufacturer's
instructions. In addition, 5X loading buffer (Beyotime Institute of
Biotechnology) was added into the collected supernatants at a ratio
of 1:4, and the mixture was heated at 100°C for 10 min. Total
amounts of 30 µg cellular proteins were electrophoresed on 10%
SDS-PAGE (Beyotime Institute of Biotechnology), and subsequently
transferred onto polyvinylidene difluoride (PVDF) membranes (EMD
Millipore), which was followed by blocking with 5% nonfat milk at
room temperature for 1 h. Then the membranes were incubated with
the primary antibody at 4°C overnight, and followed by an
incubating step with a secondary goat anti-rabbit antibody
conjugated with horseradish peroxidase (HRP; product code ab6721;
dilution 1:4,000; Abcam) for 1 h at room temperature. Finally, the
bands were measured via ECL technology (Fujifilm LAS-3000; Fujifilm
Corporation). The band intensities were quantified by ImageJ
software (v1.37; National Institutes of Health). β-Actin was used
as a loading control, and the relative band intensities were
normalized to the medium control (ME) group. Primary antibodies
against p53 (product code ab32389; dilution 1:1,000),
phosphorylated (p)-p53 (product code ab1431; dilution 1:1,000),
LC3B (product code ab192890; dilution 1:1,000), Beclin1 (product
code ab207612; dilution 1:1,000), Bax (product code ab32503;
dilution 1:1,000), Bcl-2 (product code ab32124; dilution 1:1,000),
MITF (product code ab232415; dilution 1:1,000), c-Met (product code
ab51067; dilution 1:1,000), and β-actin (product code ab8227;
dilution 1:1,000) were supplied by Abcam.
Mitochondrial membrane potential
detection
The treated cells were stained with the JC-1
fluorescent probe (Beyotime Institute of Biotechnology) following
the manufacturer's instructions. The mitochondrial membrane
potential was detected by a flow cytometer and analyzed by FlowJo
software v10.0. We referred to the study of Chen et al to
draw the cut-off line (18).
Mitochondrial ROS detection
A2058 or A375R cells were seeded into a 6-well plate
at a concentration of 2×105 cells/well, and treated with
vehicle, PLX4032, IS, or PLX4032+IS for 24 h. The treated cells
were incubated with MitoROS™ 580 (AAT Bioquest, Inc.) working
solution for 30 min and washed 3 times with Hanks' solution with 20
mM HEPES buffer (AAT Bioquest, Inc.). The mean fluorescence
intensity was detected by a cytometer and analyzed by FlowJo
software v10.0.
Clonogenic assay
A total of 1,000 A2058 or A375R cells were seeded
into each well of a 6-well plate and treated with vehicle, PLX4032,
IS, or PLX40332+IS for 9 days. The medium was changed every 3 days.
Finally, the treated cells were fixed with 4% paraformaldehyde for
1 h at 4°C and stained with 0.05% crystal violet solution for 15
min at room temperature.
Statistical analysis
All data were analyzed by GraphPad Prism 6 (GraphPad
Software, Inc.) and presented as the mean ± standard deviation of
three independent experiments. For comparison of two independent
groups, Student's t-test was used. For comparison of more than two
independent groups, one-way ANOVA (one factor) or two-way ANOVA
(two factors) and subsequent Tukey's post hoc analysis were
applied. P<0.05 was considered to indicate a statistically
significant difference.
Results
IS potentiates PLX4032-induced
cytotoxicity in BRAF inhibitor-resistant melanoma cells
To verify the effects of IS on BRAF
inhibitor-resistant melanoma, the effects of PLX4032 and/or IS on
the cell proliferation of two BRAF inhibitor-resistant melanoma
cell lines, A2058 (intrinsic resistance) (19) and A375R (acquired resistance,
Fig. S1A and B) were first
evaluated. In the clonogenic assay, it was revealed that either
PLX4032 or IS alone could inhibit the proliferation of both A2058
and A375R cells. Moreover, the combination of PLX4032 and IS
revealed increased inhibition of cell proliferation compared to
PLX4032 or IS alone (Fig. 1B and
C). It was also demonstrated that in the MTT assay, various
concentrations of PLX4032 or IS alone could decrease the cell
viability of both A2058 and A375R cells, and combined IS and
PLX4032 further reduced the cell viability of both A2058 and A375R
cells (P<0.05, Fig. 1D and E).
Moreover, IS and PLX4032 exhibited synergetic effects in inhibiting
the proliferation of both A2058 and A375R cells (Fig. 1F and G).
IS enhances PLX4032-induced apoptosis
and autophagy in BRAF inhibitor-resistant melanoma cells
The effects of PLX4032 and/or IS on the apoptosis of
BRAF inhibitor-resistant cells were further examined by staining
with Annexin V and 7-AAD. As revealed in Fig. 2A and B, cells treated with either
PLX4032 or IS alone exhibited a higher apoptosis rate than
ME-treated cells (P<0.05). In addition, the highest apoptosis
rate was observed in cells treated with both PLX4032 and IS
(P<0.05, Fig. 2A and B). In
addition, the highest expression of autophagy-related proteins,
LC3B and Beclin1, was revealed in the PLX4032+IS group in both
A2058 and A375R cells (Fig. 2C and
D).
 | Figure 2.IS enhances PLX4032-induced apoptosis
and autophagy in BRAF inhibitor-resistant melanoma cells. (A) A2058
or (B) A375R cells were treated with ME, PLX4032 (10 µM), IS (20
µM), and PLX4032 (10 µM)+IS (20 µM), respectively, for 24 or 72 h,
and cell apoptosis was detected by Annexin V and 7-AAD staining.
The statistical analysis of the apoptosis rates of (A) A2058 and
(B) A375R were performed by GraphPad Prism 6. (C) A2058 and (D)
A375R cells were treated with ME, PLX4032 (10 µM), IS (20 µM), and
PLX4032 (10 µM)+IS (20 µM), respectively, for 8 h, and the protein
levels of LC3B and Beclin1 were assessed by western blotting. Data
were presented as the mean ± standard error of the mean. For
comparison of two independent groups, Student's t-test was used.
For comparison of more than two independent groups, one-way ANOVA
and subsequent Tukey's post hoc analysis were applied. *P<0.05.
**,##,&&P<0.01. The ‘*’ refers to comparisons
with the ME group. The ‘#’ refers to comparisons with the PLX4032
group. The ‘&’ refers to comparisons with the IS group. IS,
icariside II; ME, medium control. |
IS induces p53-dependent Bax
accumulation and mitochondrial depolarization in BRAF
inhibitor-resistant melanoma cells
Having revealed the effect of IS on cell
proliferation, apoptosis and autophagy in BRAF inhibitor-resistant
melanoma cells, the underlying mechanisms were next determined. As
indicated in Fig. 3A and B, IS
significantly decreased the percentages of JC-1 aggregates and
increased the proportion of JC-1 monomers in A2058 cells,
indicating that IS could induce mitochondrial depolarization
(P<0.05). However, PLX4032 had little effect on mitochondrial
membrane potential (P>0.05, Fig. 3A
and B). In addition, it was revealed that the combination of
PLX4032 and IS promoted the depolarization of mitochondria to a
greater extent than IS or PLX4032 alone (P<0.05, Fig. 3A and B).
 | Figure 3.IS induces p53-dependent Bax
accumulation and mitochondrial depolarization in BRAF
inhibitor-resistant melanoma cells. (A) A2058 cells were treated
with ME, PLX4032 (10 µM), IS (20 µM), and PLX4032 (10 µM)+IS (20
µM), respectively, for 18 h, and then stained with the JC-1
fluorescent probe. The mitochondrial membrane potential was
detected by flow cytometry. (B) The percentages of JC-1 monomers in
A2058 cells were analyzed by GraphPad Prism 6. A2058 cells were
treated with ME, PLX4032 (10 µM), IS (20 µM), and PLX4032 (10
µM)+IS (20 µM), respectively. (C) Twenty-four hours later, the
protein levels of Bcl-2 and Bax were assessed by western blotting.
(D) Eight hours later, the protein levels of p53 and phosphorylated
p53 were assessed by western blotting. Data were presented as mean
± standard error of the mean. For comparison of two independent
groups, Student's t-test was used. For comparison of more than two
independent groups, one-way ANOVA and subsequent Tukey's post hoc
analysis were applied. **,##,&&P<0.01. The
‘*’ refers to comparisons with the ME group. The ‘#’ refers to
comparisons with the PLX4032 group. The ‘&’ refers to
comparisons with the IS group. IS, icariside II; p-p53,
phosphorylated p53; ME, medium control. |
The expression of two apoptotic regulators in the
Bcl-2 family, Bax and Bcl-2, which are co-localized on the outer
membrane of mitochondria and regulate the opening of mitochondrial
voltage-dependent anion channels, thus moderating the mitochondrial
membrane potential and release of cytochrome c, was then
examined (20). As revealed in
Fig. 3C, IS with or without PLX4032
significantly downregulated Bcl-2 expression and upregulated Bax
levels in A2058 cells.
Given that p53 participates in apoptosis by directly
interacting with and neutralizing Bcl-2 and Bcl-xL in mitochondria,
and directly activating Bax in the cytosol (21), it was hypothesized that IS led to
apoptosis by p53-dependent inhibition of Bcl-2, activation of Bax
and subsequent mitochondrial membrane permeabilization. As
anticipated, PLX4032 did not activate p53, while IS or IS+PLX4032
significantly upregulated the expression of p-p53 in A2058 cells
(Fig. 3D).
IS promotes mitochondria-dependent ROS
production, while ROS scavengers partially reverse IS-induced
enhancement of the response to PLX4032 in BRAF inhibitor-resistant
melanoma cells
Since higher ROS generation has been reported to
sensitize resistant cells to BRAF inhibitors, the ROS levels in all
four groups, i.e. ME, PLX4032, IS, PLX4032+IS, were then assessed.
As anticipated, higher ROS levels were detected in the IS and
PLX4032+IS groups than in the ME or PLX4032 alone group (P<0.05,
Fig. 4A). Given the role of IS on
the mitochondrial membrane, it was hypothesized that IS induced-ROS
was generated from mitochondria. To verify this hypothesis,
mitochondrial ROS levels were assessed in all groups in both A2058
and A375R cells. In accordance with total ROS generation, IS with
or without PLX4032 significantly increased mitochondrial ROS levels
(P<0.05; Fig. 4B).
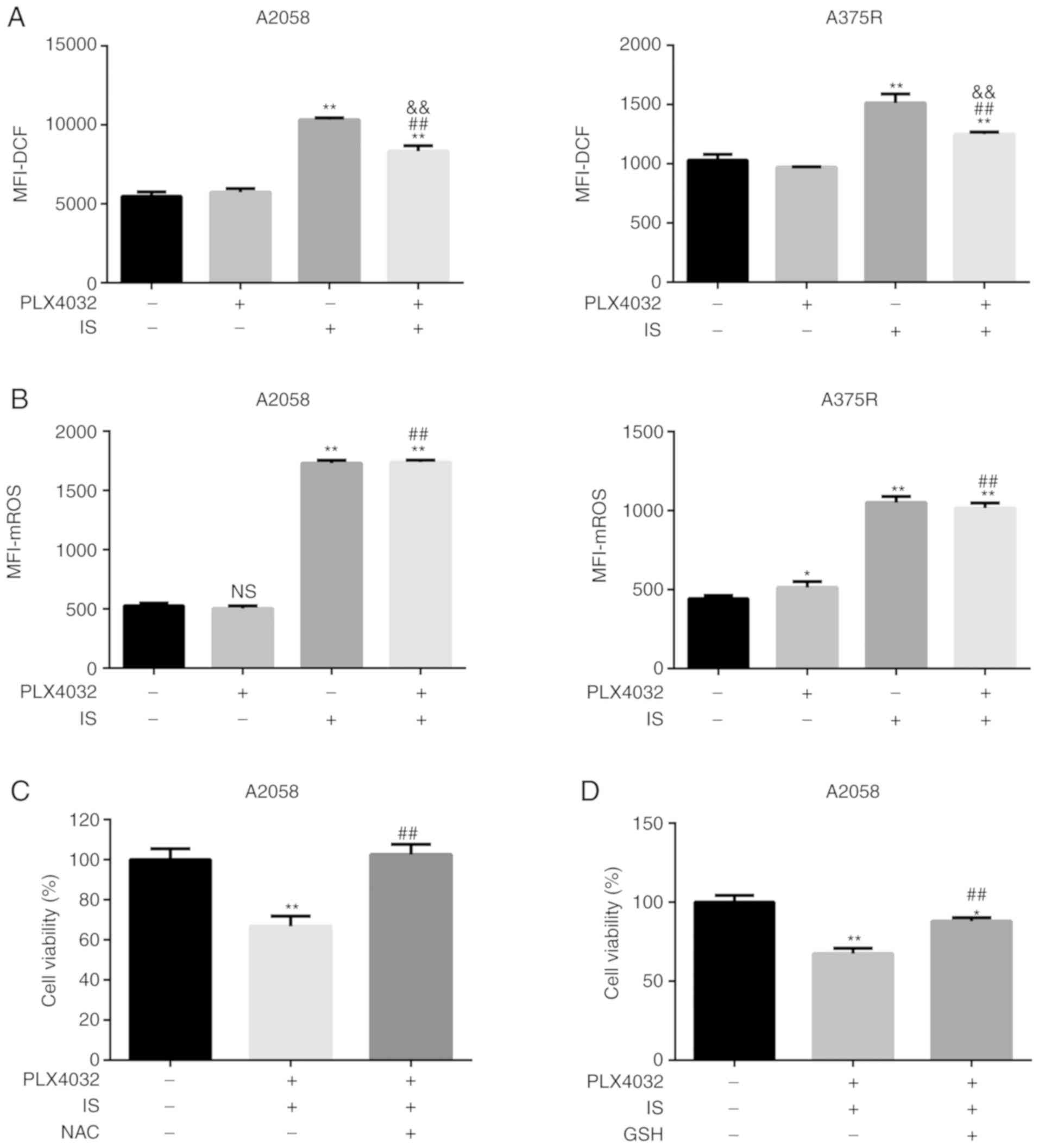 | Figure 4.IS promotes mitochondria-dependent
ROS production in BRAF inhibitor-resistant melanoma cells. (A) Both
A2058 and A375R cells were treated with ME, PLX4032 (10 µM), IS (20
µM), and PLX4032 (10 µM)+IS (20 µM), respectively, for 24 h. ROS
levels were examined using a ROS assay kit. (B) Mitochondrial ROS
levels were examined using a MitoROS™ 580 kit. A2058 cells were
pre-treated with (C) NAC (5 mM) or (D) GSH (1 mM) for 1 h, and then
treated with ME or PLX4032 (10 µM)+IS (20 µM) for 18 h. The cell
viabilities were assessed by MTT assay. Data were presented as the
mean ± standard error of the mean. For comparison of two
independent groups, Student's t-test was used. For comparison of
more than two independent groups, one-way ANOVA and subsequent
Tukey's post hoc analysis were applied. *P<0.05.
**,##,&&P<0.01. The ‘*’ refers to comparisons
with the ME group. The ‘#’ refers to comparisons with the PLX4032
group. The ‘&’ refers to comparisons with the IS group. IS,
icariside II; ROS, reactive oxygen species; NAC, N-acetyl cysteine;
GSH, glutathione; ME, medium control. |
To further confirm the role of ROS in the
IS-improved response to PLX4032, NAC and GSH, ROS scavengers, were
then used to eliminate ROS secretion, and it was detected whether
combination of PLX4032 and IS inhibition of cell viability could be
reversed. As indicated in Fig. 4C and
D, the cell viability of NAC/GSH+PLX4032+IS-treated cells was
significantly higher than that of PLX4032+IS-treated cells
(P<0.05).
IS reduces the expression of MITF and
c-Met by promoting ROS production
MITF and c-Met are well-known factors that
contribute to BRAF inhibitor resistance (22–24).
Furthermore, c-Met is a direct transcriptional target of MITF in
melanocytes and melanoma cells (25). Therefore, the effects of IS on the
expression of both MITF and c-Met, were then verified. As
demonstrated in Fig. 5A-D,
decreased mRNA and protein levels of MITF and c-Met were observed
in the IS and IS+PLX4032 groups than in the ME group in both A2058
and A375R cells (P<0.05). However, PLX4032 alone could not
markedly downregulate MITF expression (Fig. 5A-D). PLX4032 significantly increased
c-Met expression in A2058 cells compared to the ME group, while it
significantly decreased c-Met expression in the A375R cells
(Fig. 5A-D). To verify that IS
induced downregulation of MITF and c-Met by increasing ROS
production, H2O2, exogenous ROS, was used to
promote ROS generation and it was revealed that
H2O2 could significantly downregulate MITF
and c-Met expression in both A2058 and A375R cells (Fig. 5E). These results indicated that IS
may improve the response to PLX4032 partially by increasing ROS
production and subsequently inhibiting MITF and c-Met expression in
resistant melanoma cells.
 | Figure 5.IS reduces the expression of MITF and
c-Met by promoting ROS production. Both (A) A2058 and (B) A375R
cells were treated with ME, PLX4032 (10 µM), IS (20 µM), and
PLX4032 (10 µM)+IS (20 µM), respectively, for 8 h, and the mRNA
levels of MITF and c-Met were detected by qRT-PCR. Both (C) A2058
and (D) A375R cells were treated with ME, PLX4032 (10 µM), IS (20
µM), and PLX4032 (10 µM)+IS (20 µM), respectively, for 24 h, and
the protein levels of MITF and c-Met were detected by western
blotting. (E) A2058 cells were treated with ME or
H2O2 (250 µM) for 6 h, and the protein levels
of MITF and c-Met were examined by western blotting. Data were
presented as the mean ± standard error of the mean. For comparison
of two independent groups, Student's t-test was used. For
comparison of more than two independent groups, one-way ANOVA and
subsequent Tukey's post hoc analysis were applied.
*,#,&P<0.05.
**,##,&&P<0.01. The ‘*’ refers to comparisons
with the ME group. The ‘#’ refers to comparisons with the PLX4032
group. The ‘&’ refers to comparisons with the IS group. IS,
icariside II; ME, medium control; MITF, microphthalmia-associated
transcription factor; c-Met, tyrosine-protein kinase Met. |
Discussion
IS, a natural compound extracted from Herba
Epimedii, exhibits multiple biological and pharmacological
activities, including anti-inflammatory, anti-osteoporosis,
anti-aging, and anticancer properties (12). As an anticancer agent, it is
reported to inhibit cell proliferation and induce cell apoptosis in
diverse cancer types (12,26). Furthermore, it has been demonstrated
to overcome resistance to traditional therapies, such as
chemotherapies (15,16). In the present study, it was revealed
that IS improved the therapeutic benefits of PLX4032 in melanoma
cells with intrinsic or acquired resistance to BRAF inhibitors.
Autophagy, a highly conserved catabolic process
mediated by lysosomes, plays an important role in maintaining the
internal environment homeostasis and cell survival (27,28).
It was also reported to promote cell death (28). In the present study, it was revealed
that IS with or without PLX4032 significantly promoted the
expression of autophagy-related proteins. It was surmised that IS
induces ROS generation and p53 activation to promote both apoptosis
and autophagy, thus inducing cell death.
ROS generation plays an important role in inducing
mitochondrial apoptosis by regulating apoptotic proteins, such as
Bcl-2 and Bax (29,30). ROS levels were reported to be
significantly upregulated in BRAF inhibitor-treated BRAF V600E
melanoma cells and BRAF inhibitor-resistant melanoma cells
irrespective of the presence of BRAF inhibitors (30). In the present study, it was revealed
that PLX4032 alone exerted little effect on ROS generation and
mitochondrial depolarization in melanoma cells with either
intrinsic or acquired resistance to BRAF inhibitor resistance. The
possible reason is that the ROS levels in resistant melanoma cells
are already high, and these cells exhibited no response to BRAF
inhibitors. In addition, higher ROS generation has been widely
reported to sensitize resistant cells to BRAF inhibitors.
Elesclomol, a pro-oxidative drug, was revealed to increase
intracellular ROS production and melanoma cell death in a
dose-dependent manner. Elesclomol was further indicated to
significantly inhibit tumor growth in SCID mice xenografted with
vemurafenib-resistant A375 cells and tumor fragments from melanoma
patients by decreasing cell proliferation and inducing cell
apoptosis (31). A100, a
ROS-activated prodrug, was demonstrated to sensitize several
dabrafenib-resistant melanoma cell lines to dabrafenib by inducing
DNA damage and inhibiting cell proliferation (31). TRAM-34, a potassium channel
inhibitor, was also revealed to improve the therapeutic benefits of
vemurafenib by increasing intracellular ROS levels, decreasing
mitochondrial membrane potential, and enhancing pro-apoptotic
pathways in melanoma (32). In
accordance with these findings, it was revealed that IS combined
with PLX4032 could significantly increase ROS production, and
subsequently lead to p53-dependent activation of apoptotic proteins
and mitochondrial depolarization. In addition, ROS scavengers, NAC
and GSH, reversed IS-induced enhancement of the response to PLX4032
in BRAF inhibitor-resistant melanoma cells.
Notably, it was revealed that both PLX4032 and IS
could inhibit cell proliferation and induce apoptosis in
PLX4032-resistant melanoma. However, IS could increase ROS
generation and subsequently increase Bax and decrease Bcl-2, while
PLX4032 had little effect on ROS generation and subsequent Bax and
Bcl-2 expression. The mechanisms of IS and PLX4032 in melanoma
treatment may be different. It is well-known that MEK-ERK signaling
is one of the major targets of PLX4032. It was revealed that high
concentration of PLX4032 could still inhibit MEK-ERK signaling in
PLX4032-resistant melanoma cells, though this effect was not as
significant as that in parental melanoma cells (data not
shown).
MITF is an indispensable factor in pigmentation and
in the development of the melanocytic lineage (33). High MITF levels inhibit
proliferation by cell cycle arrest, and cells lacking MITF display
invasive properties (34,35). However, MITF has been revealed to
play an oncogenic role in the tumorigenesis of melanoma, since it
is amplified in 15% of metastatic melanomas (36). MITF was further reported to be
upregulated by BRAF/MEK inhibitors in a MAPK-dependent manner at an
early stage of patient treatment (37). In addition, high MITF levels were
demonstrated to allow melanoma cells to avoid BRAF/MEK
inhibitor-induced cell death even when the MAPK signaling pathway
was completely blocked, thus contributing to MAPK inhibitor
resistance in melanoma (38,39).
The present study revealed that PLX4032 could not decrease MITF
expression in BRAF inhibitor-resistant melanoma cells, while IS
with or without PLX4032 significantly decreased MITF expression.
Therefore, it is highly possible that IS overcomes BRAF inhibitor
resistance by downregulating MITF expression in melanoma.
In addition, MITF may be regulated by ROS
generation. Ko and Cho demonstrated that Phytol suppressed
melanogenesis by promoting ROS production, and subsequently
activating ERK, followed by proteasomal degradation of MITF
(40). Furthermore, MITF-negative
melanoma cells were revealed to have lower survival rates than
MITF-positive melanoma cells under ROS stress (41). In the present study, the regulatory
role of ROS on MITF was confirmed, since it was revealed that
exogenous ROS, H2O2, could significantly
decrease MITF expression.
C-Met, an important member of the receptor tyrosine
kinase (RTK) family, is the ligand of HGF. Activated c-Met was
reported to enhance cell proliferation in BRAF inhibitor-resistant
melanoma cells (42). The
combination of BRAF inhibitor and c-Met inhibitor revealed
significant therapeutic benefit in BRAF-mutant melanoma cells
(43). In addition, c-Met is
directly transcriptionally regulated by MITF in melanocytes and
melanoma cells (25). In the
present study, it was revealed that IS with or without PLX4032
could decrease c-Met expression in BRAF inhibitor-resistant
melanoma cells However, IS could inhibit c-Met expression to a
larger degree in A2058 cells when compared to A375R cells. A2058
cells have intrinsic resistance to PLX4032, while A375R cells have
acquired resistance to PLX4032. This may be, because they are
different cell lines. In addition, H2O2 could
significantly decrease c-Met. Therefore, it is highly possible that
IS improves the response to BRAF inhibitors by downregulating MITF
and c-Met expression in a ROS-dependent manner.
Notably, IS may sensitize BRAF inhibitor resistance
by other mechanisms besides inducing ROS generation and regulating
MITF expression. Our previous studies demonstrated that IS could
inhibit EGFR signaling (14) and
STAT3 signaling (16) in cancer
cells. Since EGFR and STAT3 were involved in BRAF inhibitor
resistance in melanoma, it was hypothesized that IS may reverse
BRAF inhibitor resistance through EGFR and STAT3 signaling. Further
studies are necessary to verify this hypothesis.
In conclusion, the present study demonstrated that
IS overcomes BRAF inhibitor resistance by improving ROS generation
and subsequently inducing ROS-dependent apoptosis and autophagy and
downregulating MITF expression in BRAF inhibitor-resistant melanoma
cells.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Dr Jianhua Huang from the
Department of Integrative Medicine, Huashan Hospital, Fudan
University, Shanghai, China, for his valuable suggestions on the
design of the project.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81673917 and
81973582) and the Shanghai Science and Technology Committee
(13JC1401401).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JX and JW designed the research and interpreted the
data. XL and ZL performed the experiments and wrote the manuscript.
ML, JC and SH analyzed the data. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
IS
|
icariside II
|
|
ROS
|
reactive oxygen species
|
|
NAC
|
N-acetyl cysteine
|
|
GSH
|
glutathione
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
A375R cells
|
A375 BRAF inhibitor-resistant
cells
|
|
ME
|
medium control
|
|
RTK
|
receptor tyrosine kinase
|
|
MITF
|
microphthalmia-associated
transcription factor
|
|
c-Met
|
tyrosine-protein kinase Met
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wilson MA and Schuchter LM: Chemotherapy
for Melanoma. Cancer Treat Res. 167:209–229. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alexandrov LB, Nik-Zainal S, Wedge DC,
Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A,
Borresen-Dale AL, et al: Signatures of mutational processes in
human cancer. Nature. 500:415–421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Colombino M, Capone M, Lissia A, Cossu A,
Rubino C, De Giorgi V, Massi D, Fonsatti E, Staibano S, Nappi O, et
al: BRAF/NRAS mutation frequencies among primary tumors and
metastases in patients with melanoma. J Clin Oncol. 30:2522–2529.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luebker SA and Koepsell SA: Diverse
mechanisms of BRAF inhibitor resistance in melanoma identified in
clinical and preclinical studies. Front Oncol. 9:2682019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sosman JA, Kim KB, Schuchter L, Gonzalez
R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ,
Flaherty KT, et al: Survival in BRAF V600-mutant advanced melanoma
treated with vemurafenib. N Engl J Med. 366:707–714. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Combined BRAF and MEK inhibition versus BRAF
inhibition alone in melanoma. N Engl J Med. 371:1877–1888. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Flaherty KT, Infante JR, Daud A, Gonzalez
R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N,
et al: Combined BRAF and MEK inhibition in melanoma with BRAF V600
mutations. N Engl J Med. 367:1694–1703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WJ Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robert C, Karaszewska B, Schachter J,
Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R,
Grange F, Mortier L, et al: Improved overall survival in melanoma
with combined dabrafenib and trametinib. N Engl J Med. 372:30–39.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khan M, Maryam A, Qazi JI and Ma T:
Targeting apoptosis and multiple signaling pathways with Icariside
II in cancer cells. Int J Biol Sci. 11:1100–1112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song J, Shu L, Zhang Z, Tan X, Sun E, Jin
X, Chen Y and Jia X: Reactive oxygen species-mediated mitochondrial
pathway is involved in Baohuoside I-induced apoptosis in human
non-small cell lung cancer. Chem Biol Interact. 199:9–17. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu J, Zuo F, Du J, Wong PF, Qin H and Xu
J: Icariside II induces apoptosis via inhibition of the EGFR
pathways in A431 human epidermoid carcinoma cells. Mol Med Rep.
8:597–602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu J, Guan M, Wong PF, Yu H, Dong J and Xu
J: Icariside II potentiates paclitaxel-induced apoptosis in human
melanoma A375 cells by inhibiting TLR4 signaling pathway. Food Chem
Toxicol. 50:3019–3024. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du J, Wu J, Fu X, Tse AK, Li T, Su T and
Yu ZL: Icariside II overcomes TRAIL resistance of melanoma cells
through ROS-mediated downregulation of STAT3/cFLIP signaling.
Oncotarget. 7:52218–52229. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen G, Yang Y, Xu C and Gao S: A Flow
Cytometry-based Assay for measuring mitochondrial membrane
potential in cardiac myocytes after hypoxia/reoxygenation. J Vis
Exp. 2018.
|
|
19
|
Feng JH, Nakagawa-Goto K, Lee KH and Shyur
LF: A Novel plant sesquiterpene lactone derivative, DETD-35,
suppresses BRAFV600E mutant melanoma growth and overcomes acquired
vemurafenib resistance in mice. Mol Cancer Ther. 15:1163–1176.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Westphal D, Kluck RM and Dewson G:
Building blocks of the apoptotic pore: How Bax and Bak are
activated and oligomerize during apoptosis. Cell Death Differ.
21:196–205. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vaseva AV and Moll UM: The mitochondrial
p53 pathway. Biochim Biophys Acta. 1787:414–420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vergani E, Vallacchi V, Frigerio S, Deho
P, Mondellini P, Perego P, Cassinelli G, Lanzi C, Testi MA,
Rivoltini L, et al: Identification of MET and SRC activation in
melanoma cell lines showing primary resistance to PLX4032.
Neoplasia. 13:1132–1142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aida S, Sonobe Y, Tanimura H, Oikawa N,
Yuhki M, Sakamoto H and Mizuno T: MITF suppression improves the
sensitivity of melanoma cells to a BRAF inhibitor. Cancer Lett.
409:116–124. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Filitis DC, Rauh J and Mahalingam M: The
HGF-cMET signaling pathway in conferring stromal-induced
BRAF-inhibitor resistance in melanoma. Melanoma Res. 25:470–478.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McGill GG, Haq R, Nishimura EK and Fisher
DE: c-Met expression is regulated by Mitf in the melanocyte
lineage. J Biol Chem. 281:10365–10373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen M, Wu J, Luo Q, Mo S, Lyu Y, Wei Y
and Dong J: The anticancer properties of herba epimedii and its
main bioactive componentsicariin and icariside II. Nutrients.
8(pii): E5632016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizushima N and Klionsky DJ: Protein
turnover via autophagy: Implications for metabolism. Annu Rev Nutr.
27:19–40. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Das G, Shravage BV and Baehrecke EH:
Regulation and function of autophagy during cell survival and cell
death. Cold Spring Harb Perspect Biol. 4(pii):
a0088132012.PubMed/NCBI
|
|
29
|
Franke JC, Plotz M, Prokop A, Geilen CC,
Schmalz HG and Eberle J: New caspase-independent but ROS-dependent
apoptosis pathways are targeted in melanoma cells by an
iron-containing cytosine analogue. Biochem Pharmacol. 79:575–586.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Quast SA, Berger A and Eberle J:
ROS-dependent phosphorylation of Bax by wortmannin sensitizes
melanoma cells for TRAIL-induced apoptosis. Cell Death Dis.
4:e8392013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Corazao-Rozas P, Guerreschi P, Jendoubi M,
Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M,
Balayssac S, Rocchi S, et al: Mitochondrial oxidative stress is the
Achille's heel of melanoma cells resistant to Braf-mutant
inhibitor. Oncotarget. 4:1986–1998. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bauer D, Werth F, Nguyen HA, Kiecker F and
Eberle J: Critical role of reactive oxygen species (ROS) for
synergistic enhancement of apoptosis by vemurafenib and the
potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death
Dis. 8:e25942017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hodgkinson CA, Moore KJ, Nakayama A,
Steingrimsson E, Copeland NG, Jenkins NA and Arnheiter H: Mutations
at the mouse microphthalmia locus are associated with defects in a
gene encoding a novel basic-helix-loop-helix-zipper protein. Cell.
74:395–404. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Carreira S, Goodall J, Aksan I, La Rocca
SA, Galibert MD, Denat L, Larue L and Goding CR: Mitf cooperates
with Rb1 and activates p21Cip1 expression to regulate cell cycle
progression. Nature. 433:764–769. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carreira S, Goodall J, Denat L, Rodriguez
M, Nuciforo P, Hoek KS, Testori A, Larue L and Goding CR: Mitf
regulation of Dia1 controls melanoma proliferation and
invasiveness. Genes Dev. 20:3426–3439. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Garraway LA, Widlund HR, Rubin MA, Getz G,
Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J,
et al: Integrative genomic analyses identify MITF as a lineage
survival oncogene amplified in malignant melanoma. Nature.
436:117–122. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Smith MP, Brunton H, Rowling EJ, Ferguson
J, Arozarena I, Miskolczi Z, Lee JL, Girotti MR, Marais R, Levesque
MP, et al: Inhibiting Drivers of Non-mutational drug tolerance is a
salvage strategy for targeted melanoma therapy. Cancer Cell.
29:270–284. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Muller J, Krijgsman O, Tsoi J, Robert L,
Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes
Foppen MH, et al: Low MITF/AXL ratio predicts early resistance to
multiple targeted drugs in melanoma. Nat Commun. 5:57122014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Smith MP, Ferguson J, Arozarena I, Hayward
R, Marais R, Chapman A, Hurlstone A and Wellbrock C: Effect of
SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J
Natl Cancer Inst. 105:33–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ko GA and Cho SK: Phytol suppresses
melanogenesis through proteasomal degradation of MITF via the
ROS-ERK signaling pathway. Chem Biol Interact. 286:132–140. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu F, Fu Y and Meyskens FJ Jr: MiTF
regulates cellular response to reactive oxygen species through
transcriptional regulation of APE-1/Ref-1. J Invest Dermatol.
129:422–431. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Straussman R, Morikawa T, Shee K,
Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J,
Frederick DT, et al: Tumour micro-environment elicits innate
resistance to RAF inhibitors through HGF secretion. Nature.
487:500–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Etnyre D, Stone AL, Fong JT, Jacobs RJ,
Uppada SB, Botting GM, Rajanna S, Moravec DN, Shambannagari MR,
Crees Z, et al: Targeting c-Met in melanoma: Mechanism of
resistance and efficacy of novel combinatorial inhibitor therapy.
Cancer Biol Ther. 15:1129–1141. 2014. View Article : Google Scholar : PubMed/NCBI
|














