Introduction
Measles virus (MV) is a highly contagious enveloped
negative-strand RNA virus, and is the cause of a viral respiratory
infection (1). By contrast, the
attenuated form of MV, which has been used as a vaccine for >60
years (2), inhibits cancer cell
proliferation (3–6). Attenuated MV have been extensively
investigated in numerous clinical trials to treat various types of
cancer, such as ovarian cancer (NCT02364713) (7), glioblastoma multiforme (NCT00390299)
(8) and breast cancer (NCT01503177)
(9); however, despite the potential
of MV in cancer treatment, whether MV can induce efficient toxicity
on chemo- or radioresistant cancer cells is still unknown.
Breast cancer is the most commonly diagnosed type of
cancer among women, and is the second leading cause of female
morbidity and mortality worldwide (10). According to the American Cancer
Society, it was estimated that 271,270 new cases of breast cancer
were diagnosed in the United States in 2019, which accounted for
15% of total cancer diagnoses (11). The current treatment used for breast
cancer includes surgery, chemo- and radiotherapy, which has
increased the survival rate over the past few decades; however,
>30% of patients with breast cancer develop resistance to
treatment and eventually metastasis (12,13).
Therefore, it is crucial to elucidate the mechanisms of treatment
resistance in breast cancer, which have not been fully elucidated.
Various breast cancer chemotherapeutics, including doxorubicin,
5-fluorouracil and carboplatin, as well as radiation therapy induce
cell death by causing DNA damage (14). Thus, abnormal DNA repair is one of
the plausible mechanisms for both chemo- and radioresistance
(15–17).
Unrepaired DNA double strand break (DSB) results in
cell death, while misrepaired DSBs can cause chromosomal
translocations (18). Therefore,
DSB is the most important type of DNA damage that can be caused by
DNA damaging agents and ionizing radiation (IR) (19). Homologous recombination (HR) and
non-homologous end joining (NHEJ) are the major DSB repair pathways
in mammals (20). These factors
require essential factors and lead to different repair outcomes. HR
is typically considered to be the ‘error-free’ pathway as it
incorporates sister chromatids as a template to guide the repair;
thus, HR is restricted to the late S to G2 phase of the cell cycle
(21,22). By contrast, NHEJ is active
throughout the cell cycle (22). We
hypothesized that MV may affect the resistance and DSB repair in
breast cancer. The present study aimed to further evaluate whether
MV may sensitize breast cancer cells to chemo- and radiotherapy by
regulating DSB repair.
Materials and methods
Cell lines and cell culture
MCF7 (cat. no. HTB-22™) and Vero cells (cat. no.
CCL-81™) were cultured at 37°C in a humidified incubator with 5%
CO2 in Eagle's Minimum Essential Medium (cat. no.
30-2003) with 10% FBS for <4 weeks. T47D (cat. no. HTB-133™)
cells were cultured at 37°C in a humidified incubator with 5%
CO2 in RPMI-1640 (cat. no. 30-2001) with 10% FBS (all
from American Tissue Culture Collection) for <4 weeks.
Doxorubicin-resistant (DR) MCF7 cells were generated
by incubating MCF7 cells in medium with 0.25 µM doxorubicin for 2
weeks, followed by 0.5 and 1 µM doxorubicin for 2 weeks at each
concentration. IR-resistant (IRR) MCF7 cells were generated by
treating MCF7 cells with 1-Gy IR twice in one week, followed by
exposure to 2-Gy IR twice a week for two weeks.
Virus propagation and infection
MV-Edm was harvested as previously described
(23). Attenuated measles virus
vaccines were obtained from National Biotec Group Co., Ltd., China.
The virus was propagated in Vero cells (cat. no. CCL-81™; ATCC)
with an MOI 0.01 at 37°C for 2 h. The medium was then replaced, and
the cells were maintained at 32°C for virus propagation. At 3 days
post-infection, the infected cells were scraped into 1 ml Opti-MEM
(cat. no. 51985091; Thermo Fisher Scientific, Inc.). The viral
particles were harvested from Vero cells by snap-freezing in liquid
nitrogen and thawing in a water bath at 37°C for 5 cycles. A 50%
tissue culture infective dose titer was calculated from the 50%
endpoint dilution assay in Vero cells (24). Viral infection was performed as
previously described (25).
Briefly, MCF7 and T47D cells were seeded at a density of
3×105 cells/well in triplicate in 6-well plates with 2
ml complete media. After 24 h, the cells were incubated with 0–0.5
MOI virus for 24 h at 37°C. The cells were trypsinized and
harvested for further experiments.
Analysis of cell viability
MCF7 and T47D cells were seeded at a density of
5×103 cells/well in triplicate in 96-well plates with
100 µl complete media. After 24 h at 37°C, the cells were treated
with doxorubicin for 48 h at 37°C, and the cell viability was
determined using MTT assay. Briefly, 10 µl MTT (Sigma-Aldrich;
Merck KGaA) solution was added to each well, and the plates were
incubated for 3 h at 37°C. DMSO was subsequently added to each well
(100 µl/well) to dissolve the formazan crystals, and the absorbance
was measured at 490 nm using an Epoch™ 2 Microplate
spectrophotometer (BioTek Instruments, Inc.).
NHEJ and HR efficiency assay
A total of 10 µg of NHEJ or HR reporter plasmid was
linearized using NheI and purified using the QIAquick Gel
Extraction kit (cat. no. 28704; Qiagen, Inc.) according to the
manufacturer's instructions. The purified plasmid (1 µg) was
transfected into MCF7 and T47D cells (3×105 cells/ml; 2
ml) using Lipofectamine® 3,000 (cat. no. L3000015;
Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol and incubated at 37°C for 48 h. Stably
transfected cells were selected by incubating in medium with 1
mg/ml geneticin for 2 weeks and subsequently stored in liquid
nitrogen until further use. To measure NHEJ efficiency, stably
transfected cells were seeded at 3×105 cells/ml in a
6-well plate and cultured for 24 h. For the NHEJ or HR assay, 2 µg
I-SceI plasmid (cat. no. 26477; Addgene, Inc.) was transfected into
MCF7 and T47D cells (3×105 cells/ml; 2 ml) using
Lipofectamine® 3,000 to recognize the I-SceI
restriction enzyme site and generate DSB. After incubation at 37°C
for 48 h, positive cells expressing green fluorescent protein
(GFP), which indicated successful DSB repair, were measured using a
FACSCelesta flow cytometer (BD Biosciences) and analyzed using
FlowJo software (V.10; FlowJo, LLC).
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted from MCF7 and T47D cells using the
RNeasy Mini Kit (cat. no. 74104; Qiagen GmbH) and was
reverse-transcribed using the iScript™ RT Supermix (Bio-Rad
Laboratories, Inc.) according to the manufacturer's instructions.
Subsequently the iQ™ SYBR® Green Supermix (Bio-Rad
Laboratories, Inc.) was used for qPCR to detect the mRNA levels of
the NHEJ factors according to the manufacturer's instructions using
a Real-Time PCR system (Eppendorf Thermal Cycler Eco; Eppendorf)
(26). The thermocycling conditions
included initial denaturation at 95°C for 3 min, followed by 35
cycles of denaturation at 95°C for 10 sec and annealing and
extension at 60°C for 50 sec. The relative expression levels were
quantified using the 2−∆∆Cq method (27).
The following primers were used: β-actin forward,
5′-ACCAACTGGGACGACATGGAG-3′ and reverse,
5′-GTGAGGATCTTCATGAGGTAGTC-3′; 70 kDa subunit of Ku antigen (Ku70)
forward, 5′-ATGGCAACTCCAGAGCAGGTG-3′ and reverse,
5′-AGTGCTTGGTGAGGGCTTCCA-3′; 86 kDa subunit of Ku antigen (Ku80)
forward, 5′-TGACTTCCTGGATGCACTAATCGT-3′ and reverse,
5′-TTGGAGCCAATGGTCAGTCG-3′; catalytic subunit of a nuclear
DNA-dependent serine/threonine protein kinase (DNA-PKcs) forward,
5′-CCAAGTCCAACACCAAGTAGCCACCCA-3′ and reverse,
5′-CCGCCATGCCGCCGAGTCCC-3′; X-ray repair cross-complementing 4
(XRCC4) forward, 5′-CCCTCACAGAAACACAACTCA-3′ and reverse,
5′-CAAGGAGGTGGCCACTAGTT-3; XRCC4-like factor (XLF) forward,
5′-ACAAGGTCTAATGCACCCCA-3′ and reverse,
5′-GGGTTGCAGCCTTAGAAAAGT-3′; DNA ligase IV forward,
5′-CACCTTGCGTTTTCCACGAA-3′ and reverse,
5′-CAGATGCCTTCCCCCTAAGTTG-3′; and p53-binding protein 1 (53BP1)
forward, 5′-CCAGCACCAACAAGAGC-3′ and reverse,
5′-GGATGCCTGGTACTGTTTGG-3′.
Western blot analysis
MCF7 and T47D cells were pelleted and resuspend in
hypotonic buffer (20 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM
MgCl2). The cells were lysed by adding 10% NP40 and
vortexing. The cell lysate was centrifuged for 15 min at 1,200 × g
at 4°C, and the supernatant (cytoplasmic fraction) was removed. The
nuclear pellet was lysed in lysis buffer (10 mM Tris, pH 7.4, 150
mM NaCl, 2 mM Na3VO4, 1% Triton X-100, 1 mM EDTA, 0.1% SDS, 0.5%
deoxycholate, 1 mM NaF, 10% glycerol) with 1X protease cocktail
(Roche Diagnostics) for 30 min on ice with vortexing and
centrifuged at 14,000 × g at 4°C, following which the protein
concentration of the supernatant (nuclear fraction) was determined
using Bradford assay. The samples (20 µg/lane) were separated using
SDS-PAGE and transferred to PVDF membranes (Roche Diagnostics).
After blocking with 3% BSA in 1X PBS with 0.1% Tween-20, the
membrane was incubated with primary antibodies, followed by
incubation with horseradish peroxidase-conjugated secondary
antibodies. The signals were detected using an enhanced
chemiluminescence reagent (Thermo Fisher Scientific, Inc.), and the
proteins were visualized using the ChemiDoc MP imaging system
(Bio-Rad Laboratories, Inc.) and analyzed using ImageJ version 1.51
software (National Institutes of Health). The following antibodies
(1:1,000 dilution) were used: Anti-Ku70 (cat. no. 4588; Cell
Signaling Technology, Inc.), anti-Ku80 (Cell Signaling Technology,
Inc.), anti-DNA-PKcs (cat. no. 38168; Cell Signaling Technology,
Inc.), anti-XLF (cat. no. 2854; Cell Signaling Technology, Inc.),
anti-XRCC4 (cat. no. sc-136124; Santa Cruz Biotechnology, Inc.),
anti-DNA ligase IV (cat. no. 14649; Cell Signaling Technology,
Inc.), anti-53BP1 (cat. no. 4937; Cell Signaling Technology, Inc.),
anti-lamin B1 (cat. no. 65986; Abcam) and anti-origin recognition
complex subunit 2 (ORC2; cat. no. 4736; Cell Signaling Technology,
Inc.).
Chromatin fractionation assay
DNA damage was induced using 2-Gy of IR or 0.5 µM
doxorubicin, and the cells were allowed to recover for 2 h to allow
the DNA repair mechanism to assemble. Subsequently, MCF7 cells were
lysed in buffer A (50 mM HEPES-KOH, pH 8.0, 150 mM NaCl, 10%
glycerol, 3 mM MgCl2, 1 mM EGTA, 1% NP-40, 1 mM DTT)
with 1X protease inhibitor cocktail (Roche Diagnostics) on ice for
20 min and centrifuged at 1,200 × g at 4°C for 3 min. The cell
pellets were collected and lysed in buffer B (10 mM Tris-HCl, 200
mM NaCl, 1 mM EDTA, 1 mM EGTA) with 1X protease inhibitor cocktail
and centrifuged at 1,200 × g at 4°C for 3 min. The sediment was
resuspended in buffer C (50 mM Tris-HCl, pH 8, 20 mM NaCl, 1 mM
MgCl2, 0.1% SDS, 1% NP-40) supplemented with 1X protease
inhibitor cocktail and denatured with SDS loading buffer.
DNA pull-down assay
A 50% DNA cellulose (Invitrogen; Thermo Fisher
Scientific, Inc.) mixture was washed in washing buffer [50 mM
HEPES, pH 8, 100 mM potassium acetate, 0.5 mM magnesium acetate, 1
mM ATP, 1 mM DTT, 0.1 mg/ml bovine serum albumin (Sigma-Aldrich;
Merck KGaA)] three times. MCF7 and T47D cells (3×105
cells/ml; 2 ml) were cultured in the presence or absence of 0.5
multiplicity of infection (MOI) MV-Edm for 24 h and lysed using
RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1%
SDS, 25 mM Tris, pH 7.4) supplemented with 1X protease inhibitor
cocktail. The 53BP1 protein was immunoprecipitated from the cell
extract using an anti-53BP1 antibody (cat. no. 4937; Cell Signaling
Technology, Inc.) followed by A/G protein binding beads (Thermo
Fisher Scientific, Inc.). A total of 0.1 µg 53BP1 was added to 20
µl of the DNA cellulose mixture. The reactions were rotated at room
temperature for 3 h, and the DNA cellulose was centrifuged at 1,000
× g for 1 min and was washed twice with washing buffer. The pellet
was resuspended in 20 µl 2X SDS buffer, and DNA-bound 53BP1 was
analyzed using western blot analysis. The non-biotinylated
DNA-bound 53BP1 was used as negative control.
CRISPR-Cas9-mediated DNA ligase IV
deletion
A pool of 3 plasmids, each encoding the Cas9 coding
gene and ligase IV-specific 20 nt guide RNA (cat. no. sc-401372;
Santa Cruz Biotechnology, Inc.), targeting the 5′ constitutive exon
within the ligase IV gene, was transfected into MCF7-DR and
MCF7-IRR cells using Lipofectamine® 3,000 and selected
as previously described (28).
Cells were trypsinized and seeded in 96-well plate at densities of
100, 300 and 500 cells/ml and incubated for 14 days at 37°C. Single
clones were selected, expanded and screened for DNA ligase IV
expression using western blot analysis (29). The CRISPR/Cas9-Ctr plasmid (cat. no.
sc-418922; Santa Crus Biotechnology, Inc.), which encoded a single
scrambled gRNA sequence, was used as negative control. For each
knockout, two clones (KO1 and KO2) were selected for further
experiments.
Caspase-3/7 activity assay
Caspase-3/7 activity was measured using the
Caspase-Glo® 3/7 Assay System according to the
manufacturer's protocol (Promega Corporation). MCF7-IRR, MCF7-DR,
T47D-IRR and T47D-DR cells (1×104 cells/well in 96-well
plate) were treated with MV-Edm for 24 h, followed by exposure to
2-Gy IR or 0.1 µM doxorubicin. Subsequently, 100 µl
Caspase-Glo® 3/7 reagent was added in each well and
gently mixed with the cells, and incubated at room temperature for
1.5 h before caspase-3/7 activity was measured using a luminometer
(490 nm excitation, 570 nm emission) (FLUOstar Optima).
Statistical analysis
Data are presented as the mean ± standard error of
the mean from three experimental repeats. GraphPad Prism v7.0
software (GraphPad Software, Inc.) was used to plot the graphs and
analyze the data. Student's t-test was used for comparisons between
two groups. One-way or two-way ANOVA with the Bonferroni post hoc
test was used to compare multiple groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
MV impairs NHEJ efficiency in breast
cancer cells
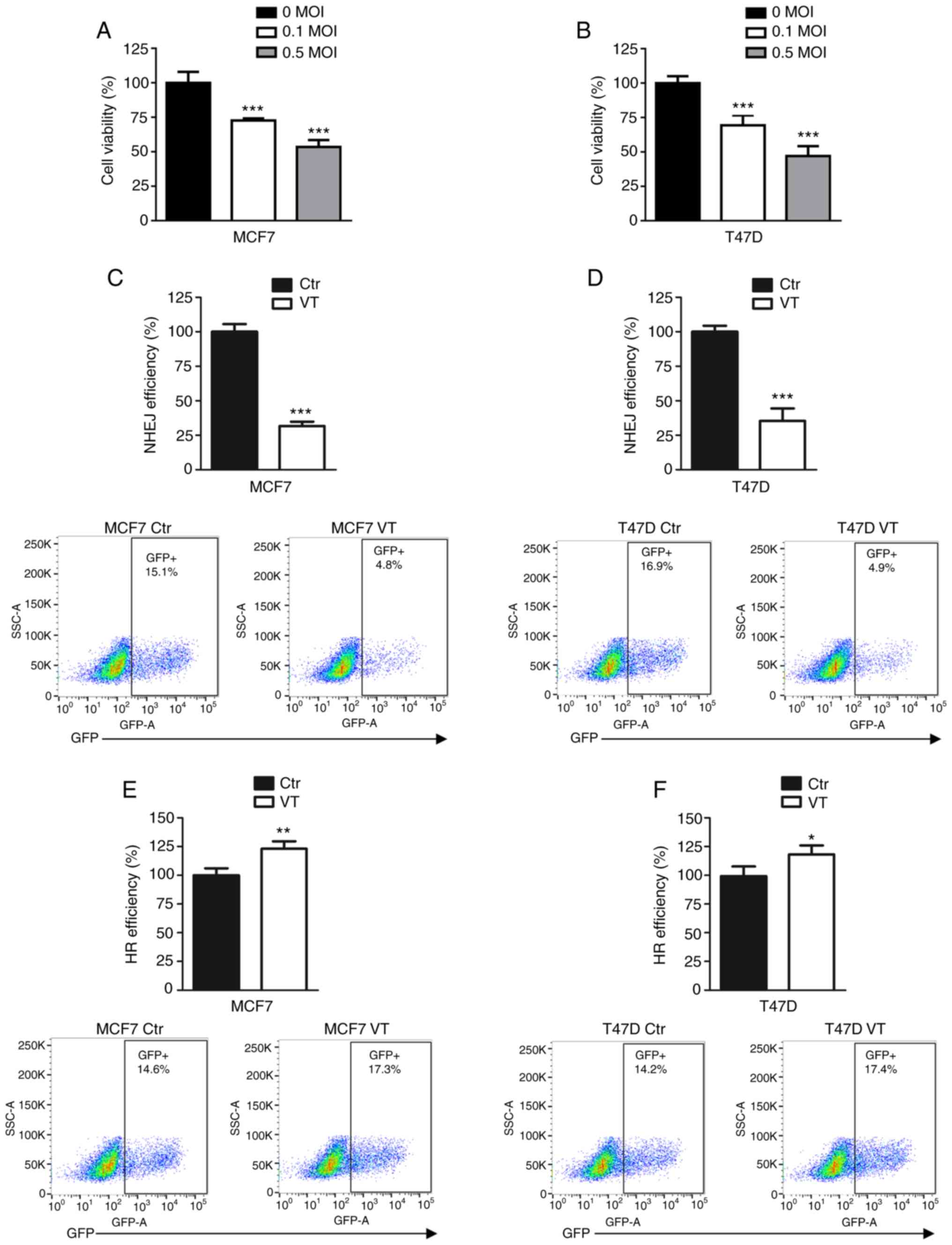
To determine the cell viability inhibition induced
by MV-Edm, the MCF7 and T47D cell lines were infected with 0, 0.1
and 0.5 MOI MV-Edm. As presented in Fig. 1A and B, the viral infection
significantly inhibited the viability of the two breast cancer cell
lines compared with that of uninfected cells.
To investigate which DSB repair pathway was affected
by MV, MCF7 and T47D cells were treated with MV-Edm and analyzed
using NHEJ and HR efficiency assays. These reporter assays
integrate inducible DSB in chromosomal DNA, and the successful
repair of DSB results in GFP expression. NHEJ and HR efficiency was
measured using flow cytometry, and the results revealed that 0.5
MOI MV-Edm decreased NHEJ efficiency to ~30% of that observed in
control groups in the MCF7 and T47D cell lines (Fig. 1C and D). The viral infection
increased HR efficiency by 10–20% compared with the respective
control groups (Fig. 1E and F).
These results suggested that impaired NHEJ may be a mechanism of
inhibition of cell viability induced by MV-Edm in breast cancer
cells.
MV decreases 53BP1 mRNA expression
levels in breast cancer cells
To elucidate how MV-Edm affected NHEJ efficiency,
the mRNA expression levels of NHEJ-associated factors were
investigated, including Ku70, Ku80, DNA-PKcs, XRCC4 and XLF, as
well as the pathway factor 53BP1, which favors NHEJ (30). In both breast cancer cell lines, 0.5
MOI MV-Edm reduced the mRNA expression levels of 53BP1, but not
those of other NHEJ factors (Fig. 2A
and B). Western blot analysis demonstrated that the protein
expression level of 53BP1 was decreased in MV-Edm-infected T47D and
MCF7 cells (Fig. 2C), indicating
that MV-Edm may impair NHEJ efficiency by downregulating 53BP1.
 | Figure 2.MV decreases the mRNA expression
levels of 53BP1 in breast cancer cells. (A and B) The mRNA
expression levels of Ku70, Ku80, XRCC4, ligase IV, DNA-PKcs, XLF
and 53BP1 in (A) MCF7 and (B) T47D cells infected with 0.5 MOI
MV-Edm for 24 h were determined using quantitative PCR.
***P<0.001. (C) Western blot analysis of 53BP1 expression
following infection with 0.5 MOI MV-Edm for 24 h. β-actin was used
as the internal control. MV, measles virus; VT, virus transfection;
Edm, Edmonston-B; MOI, multiplicity of infection; DNA-PKcs,
DNA-dependent protein kinase catalytic subunit; XRCC4, X-ray repair
cross-complementing 4; XLF, XRCC4-like factor; 53BP1, p53-binding
protein 1. |
MV-Edm inhibits the assembly of NHEJ
factors
Impaired 53BP1 mRNA and protein expression by MV-Edm
infection results in decreased NHEJ efficiency, which may be due to
the activation of an unfavored pathway. To determine whether MV-Edm
impedes the NHEJ pathway in cells, in which NHEJ was previously
incorporated for DSB repair, DNA damage was induced using 2-Gy IR,
and the cells were allowed to recover for 2 h to allow the DNA
repair mechanism to assemble. The chromatin binding activity of the
NHEJ-associated factors was determined using a chromatin
fractionation assay, and the results demonstrated that 0.5 MOI
MV-Edm treated cells exhibited a notable decrease in chromatin
binding of DNA-PKcs, XRCC4, XLF and ligase IV in MCF7 cells
compared with that in the control cells (Fig. 3A). A similar result was also
observed in doxorubicin-treated MCF7 cells (Fig. 3B).
 | Figure 3.MV inhibits the assembly of NHEJ
factors. (A and B) Chromatin assembly of NHEJ key factors in MCF7
cells. Chromatin-bound proteins were detected using western
blotting analysis in cells infected with 0.5 MOI MV-Edm for 24 h
followed by (A) 2-Gy IR or (B) 0.5 µM doxorubicin treatment. ORC2
was used as an internal control. (C and D) DNA pull-down assay for
53BP1 in (C) IR- or doxorubicin-treated MCF7 cells infected with
0.5 MOI MV-Edm for 24 h. MV, measles virus; NHEJ, non-homologous
end joining; MOI, multiplicity of infection; VT, virus
transfection; Edm, Edmonston-B; IR, ionizing radiation; DNA-PKcs,
DNA-dependent protein kinase catalytic subunit; XRCC4, X-ray repair
cross-complementing 4; XLF, XRCC4-like factor; 53BP1, p53-binding
protein 1. |
To determine whether MV-Edm affected the DNA binding
activity of 53BP1, biotinylated DNA was used to pull down the 53BP1
immunoprecipitate from MV-Edm-infected and control cells. As
presented in Fig. 3C and D, MV-Edm
did not affect the 53BP1-DNA interaction in MCF7 cells after IR and
doxorubicin treatment. These observations suggested that MV-Edm may
induce NHEJ defects in breast cancer cells by reducing NHEJ
incorporation during DSB repair by downregulating 53BP1, as well as
disrupting the NHEJ protein complex during NHEJ.
Combination of MV and IR/doxorubicin
overcomes radio- and chemoresistance in breast cancer cells
As abnormal DNA repair contributes to radio- and
chemoresistance in various types of cancer, we hypothesized that
MV-Edm, which significantly impairs NHEJ in breast cancer cells,
may inhibit cell viability in treatment-resistant cells. First,
MCF7-IRR, T47D-IRR, MCF7-DR and T47D-DR cell lines were established
(Fig. S1). Cells resistant to IR
or doxorubicin exhibited a ~40–60% increase in NHEJ efficiency
compared with that in the respective parental cells (P<0.001;
Fig. S2).
To determine whether MV-Edm affected NHEJ efficiency
in resistant cell lines, the IRR and DR cell lines were infected
with 0.5 MOI MV-Edm, and the NHEJ efficiency compared with that in
the control cells was investigated. As presented in Fig. 4A-D, MV-Edm significantly decreased
NHEJ efficiency in the IRR and DR cell lines. Subsequently, cell
viability was measured in the resistant cell lines infected with
MV-Edm following IR or doxorubicin treatment. The results
demonstrated that MV-Edm significantly inhibited cell viability in
MCF7-IRR cells; in particular, 0.5 MOI MV-Edm decreased cell
survival by 4.36-fold at 2 Gy (Fig.
4E) compared with 0 MOI MV-Edm treatment. In addition, MCF7-DR
cells were re-sensitized to doxorubicin by MV-Edm infection, and
cell survival decreased by 4.56-fold at 0.2 µM compared with 0 MOI
MV-Edm treatment (Fig. 4F). Similar
re-sensitization to IR and doxorubicin by MV-Edm was observed in
T47D-IRR cells by 3.70-fold at 2 Gy and in T47D-DR cells 3.61-fold
at 0.2 µM compared with 0 MOI MV-Edm treatment (Fig. 4G and H). To further elucidate the
mechanism underlying IR- and doxorubicin-induced cell death, the
caspase-3/7 activity assay was used to determine apoptosis
following IR or doxorubicin treatment in the absence or presence of
MV-Edm. As presented in Fig. S3,
combination treatment with MV-Edm and IR or doxorubicin induced a
significant dose-dependent increase of caspase-3/7 activation in
resistant cells. These results suggested that MV-Edm may serve a
promising role as a re-sensitizing agent for radio- or
chemotherapy.
 | Figure 4.Combination of MV and IR/doxorubicin
overcomes radio- and chemoresistance in breast cancer cells. (A-D)
Quantification of GFP generated by NHEJ in (A) MCF7-IRR, (B)
T47D-IRR, (C) MCF7-DR and (D) T47D-DR cells treated with vehicle or
0.5 MOI virus transfection. The GFP level was normalized to that of
cells treated with the vehicle. **P<0.01 and ***P<0.001 vs.
Ctr. (E and F) MV-Edm re-sensitized (E) MCF7-IRR and (F) T47D-IRR
cells to IR. Cell viability was evaluated using MTT assay following
infection with MV-Edm at a MOI of 0, 0.1 or 0.5 and IR treatment.
***P<0.001. (G and H) MV-Edm re-sensitized (G) MCF7-DR and (H)
T47D-DR cells to doxorubicin. Cell viability was evaluated using
MTT assay. Cells were infected with MV-Edm at a MOI of 0, 0.1 or
0.5, followed by doxorubicin treatment. ***P<0.001. GFP, green
fluorescent protein; MOI, multiplicity of infection; NHEJ,
non-homologous end joining; IR, ionizing radiation; DR, doxorubicin
resistant; IRR, ionizing radiation resistant; MV, measles virus;
Edm, Edmonston-B. |
MV overcomes radio- and
chemoresistance by inhibiting NHEJ
To determine whether MV-Edm may overcome IR or
doxorubicin resistance by impeding the NHEJ pathway, NHEJ-deficient
cells were constructed by knocking down its key factor, ligase IV.
Using a commercially available CRISPR/cas9 plasmid with a ligase IV
guiding sequence, ligase IV-deficient MCF7-IRR and MCF7-DR cells
[MCF7-IRR-knockout (KO)1, MCF7-IRR-KO2, MCF7-DR-KO1 and
MCF7-DR-KO2] were generated. Western blot analysis of DNA ligase IV
demonstrated that there was no detectable ligase IV protein
expression in the KO cells compared with that in the parental cell
lines transfected with CRISPR/cas9-Ctr (Fig. 5A and B).
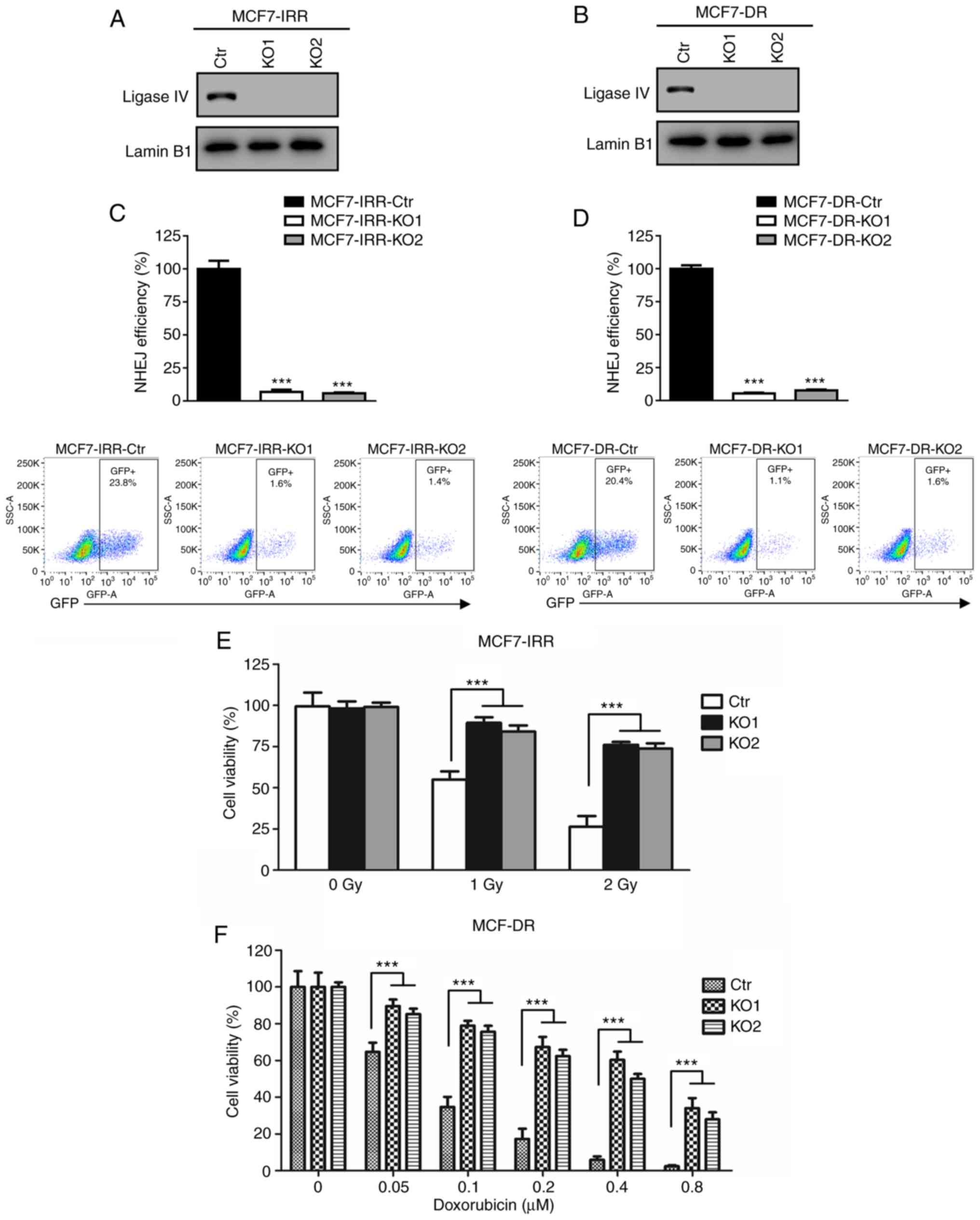 | Figure 5.MV overcomes radio- and
chemoresistance via inhibition of NHEJ. (A and B) Western blot
analysis of DNA ligase IV expression in (A) MCF7-IRR-Ctr,
MCF7-IRR-KO1 and MCF7-IRR-KO2 cells and in (B) MCF7-DR-Ctr,
MCF7-DR-KO1 and MCF7-DR-KO2 cells. (C) Quantification of GFP
generated by NHEJ in MCF7-IRR-Ctr, MCF7-IRR-KO1 and MCF7-IRR-KO2
cells. The GFP level was normalized to that of MCF7-IRR-Ctr cells.
***P<0.001 vs. Ctr. (D) Quantification of GFP generated by NHEJ
in MCF7-DR-Ctr, MCF7-DR-KO1 and MCF7-DR-KO2 cells. The GFP level
was normalized to that of MCF7-DR cells. ***P<0.001 vs. Ctr. (E
and F) MV-Edm failed to re-sensitize ligase IV-deficient (E)
MCF7-IRR cells to IR and (F) MCF7-DR cells to doxorubicin.
MCF7-IRR-Ctr, MCF7-IRR-KO1, MCF7-IRR-KO2, and MCF7-DR-Ctr,
MCF7-DR-KO1 and MCF7-DR-KO2 cells were infected with MV-Edm at 0.5
MOI for 24 h followed by IR and doxorubicin treatment,
respectively. Cell viability was evaluated using MTT assay and
cells were either normalized to MV-Edm-treated MCF7-IRR-Ctr without
IR exposure or MV-Edm-treated MCF7-DR-Ctr with 0 µM doxorubicin
treatment. ***P<0.001. MV, measles virus; NHEJ, non-homologous
end joining; IR, ionizing radiation; IRR, ionizing radiation
resistant; Ctr, control; KO, knockout; DR, doxorubicin resistant;
Edm, Edmonston-B; GFP, green fluorescent protein. |
To verify the ligase IV deficient cell lines, NHEJ
efficiency was compared between the ligase IV-KO and the control
cell lines. MCF7-IRR-KO1 and MCF7-IRR-KO2 exhibited near complete
loss of NHEJ compared with that in the MCF7-IRR-Ctr cells (Fig. 5C). As demonstrated in Fig. 5D, MCF7-DR-KO1 and MCF7-DR-KO2 were
also NHEJ-deficient. Therefore, NHEJ-deficient radio- and
chemoresistant cells were successfully constructed to elucidate the
re-sensitizing mechanism of MV-Edm.
To verify the role of the NHEJ pathway in the
re-sensitization to IR or doxorubicin induced by MV-Edm, the cell
viability in MCF7-IRR-Ctr and MCF7-IRR-KO cells was investigated.
As presented in Fig. 5E, following
infection with 0.5 MOI MV-Edm, both MCF7-IRR-KO cell lines
exhibited a significant increase in IR resistance compared with
that in the MCF7-IRR-Ctr cells. These results suggested that the
effects of MV-Edm on IR resistance in breast cancer cells required
efficient NHEJ.
Subsequently, MCF7-DR-Ctr, MCF7-DR-KO1 and
MCF7-DR-KO2 cell lines were infected with 0.5 MOI MV-Edm, and cell
viability was measured following doxorubicin treatment. Consistent
with the results presented in Fig.
5E, MCF7-DR-KO cells were more resistant to doxorubicin
compared with MCF7-DR-Ctr cells (Fig.
5F). Thus, these results demonstrated that the MV-Edm-induced
re-sensitization to IR and doxorubicin was achieved by inhibition
of NHEJ.
Discussion
Despite the advances in surgery, chemo- and
radiotherapy, treatment of breast cancer remains a clinical and
scientific challenge due to chemo- and radioresistance (31). However, alternative treatments or
methods to overcome resistance are limited. Therefore,
identification of novel approaches and understanding the mechanism
of resistance are essential to improve breast cancer therapy.
MV has been recognized as a promising system to
develop potent and safe anticancer therapies (32). Infecting patients with a replicating
virus may raise a number of safety issues, such as immune
suppression (33); however,
attenuated MV virotherapy is less active compared with a
replicating virus and tumor-selective by interacting with the MV
CD46 receptor, which is upregulated in tumor cells (34,35).
By proteolytically creating inactive C3b and C4b complement
proteins, upregulated CD46 prevents the tumor cells from complement
lysis (36). Consistently, CD46
expression is negatively associated with prognosis in breast
cancer, as well as in other types of cancer, such as ovarian and
prostate cancer, multiple myeloma and colorectal cancer (37–41).
In particular, patients with CD46-positive breast tumors present
with a significantly shorter progression-free and overall survival
time compared with those with CD46-negative tumors (37). A genetic study of nectin-4, which is
another MV receptor upregulated in breast cancer and glioblastoma,
demonstrated that downregulation of nectin-4 by microRNA(miR)-31
and miR-128 markedly impaired the infection rate of MV-Edm in
vitro and in vivo (42).
Attenuated MV is non-persistent and
non-transmissible, which further supports the application and
refinement of MV in the treatment of various types of cancer, such
as ovarian cancer and recurrent or refractory multiple myeloma
(43–47). The safety of MV-Edm derivatives has
been demonstrated in clinical trials with minimal toxicity
(48). Ovarian cancer and
glioblastoma multiforme were first selected to investigate the
preclinical toxicity of MV-Edm in 2002 and 2003, respectively
(44,49). Subsequent studies have used suitable
animal models, such as the interferon type I receptor-deficient
CD46 Ge mouse, Rhesus macaques and squirrel monkeys, which are
permissive to MV-Edm, to elucidate dosing strategies and routes of
administration, and no virus-associated toxicity was observed
(50–52). The first phase I clinical trial of
the MV vaccine was conducted in Switzerland in cutaneous patients
with T-cell lymphoma (53). Even
with a low dose of MV, 4/5 patients exhibited partial regression
with no dose-limiting toxicity (53). This promising study resulted in
subsequent multiple phase I/II clinical trials in ovarian cancer
(NCT02364713 and NCT02068794) (8,54),
glioblastoma multiforme (NCT00390299) (8), multiple myeloma (NCT00450814)
(55), mesothelioma (NCT01846091)
(56), squamous cell head and neck
cancer (NCT02192775) (57), breast
cancer (NCT01503177) (58) and
malignant peripheral nerve sheath tumors, (NCT02700230) (59). Recombinant MV-Edm derivatives,
including MV-carcinoembryonic antigen and MV-thyroidal sodium
iodide symporter, induced a 2-fold increase of median overall
survival rate with no dose-limiting toxicity in a total of 37
patients with ovarian cancer (60).
Similar low toxicity was observed in trials involving glioblastoma
multiforme and multiple myeloma (60). Despite the verified safety profile
of the MV strains, there are several difficulties for measles-based
therapeutics, including immunity to MV and MV-triggered immune
responses (61,62). Various strategies have been proposed
and validated to protect virus infusion and increase safety, such
as cell-based delivery vehicles and combination therapy with
immunosuppressant drugs (60).
The results of the present study confirmed that
MV-Edm inhibited breast cancer cell viability. Notably, the results
demonstrated that the viral infection affected the efficiency of
the NHEJ pathway in breast cancer cells. Association between high
NHEJ efficiency and low responsiveness to cancer treatments has
been observed in various types of cancer, including ovarian cancer,
cervical carcinoma, head and neck squamous cell carcinoma and lung
cancer (63). In breast cancer
cells, NHEJ efficiency is not significantly higher compared with
that in normal mammary epithelial cells (64). However, increases in the interaction
between Ku and DNA-PKcs proteins have been demonstrated to occur
via non-coding RNA LINP1, which results in efficient NHEJ, thus
enhancing radio- and chemoresistance in triple-negative breast
cancer cells (65). The results of
the present study demonstrated that, mechanistically, MV-Edm
attenuated NHEJ by downregulating 53BP1 expression and assembly of
the NHEJ complex in resistant cells. Since abnormal DNA repair is
associated with chemo- and radioresistance, DR and IRR breast
cancer cell lines were constructed to evaluate the potential of
attenuated MV in overcoming treatment resistance. The results
demonstrated that inhibition of NHEJ by MV-Edm significantly
improved the sensitivity of resistant breast cancer cells to
doxorubicin and IR therapy.
The present study provides a novel approach for
re-sensitizing breast cancer cells to chemo- and radiotherapy, and
provides other researchers with the knowledge to investigate
whether MV overcomes chemoresistance in various subtypes of breast
cancer, as well as in other types of cancer. In addition, it is
also important to elucidate whether MV acts in synergy with other
DNA-damaging targeting agents.
In conclusion, the results of the present study
demonstrated that MV-Edm inhibited the NHEJ pathway in breast
cancer cells. To the best of our knowledge, this is the first
report that associates the DNA DSB repair pathway with MV-Edm
infection. Mechanistically, MV-Edm impeded 53BP1 expression and
NHEJ factor assembly on DNA. Notably, MV-Edm may re-sensitize IRR
or DR breast cancer cells by impairing NHEJ efficiency. Therefore,
MV-Edm is a potential treatment option in radio- and chemotherapy
in breast cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81601756), the China
Postdoctoral Science Foundation (grant no. 2017M611328), the Jilin
Provincial Department of Education ‘Thirteenth Five-Year Plan’
Science and Technology Research Planning Project (grant no.
JJKH20170830KJ) and Jilin University Bethune Project B (grant no.
2015323).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. Additional data are available
from the corresponding author on reasonable request.
Authors' contributions
BY, JS and XX conceived and designed the
experiments. BY, DZ and ZS performed the experiments and analyzed
the data. JS wrote the manuscript and provided the funding. XX
reviewed, revised and approved the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MV-Edm
|
Edmonston-B vaccine strain of measles
virus
|
|
NHEJ
|
non-homologous end joining
|
|
HR
|
homologous recombination
|
|
DSB
|
DNA double strand break
|
|
IR
|
ionizing radiation
|
References
|
1
|
Enders G: Paramyxoviruses. Medical
Microbiology. Baron S: 4th. University of Texas Medical Branch at
Galveston; Galveston, TX: 1996
|
|
2
|
Enders JF and Peebles TC: Propagation in
tissue cultures of cytopathogenic agents from patients with
measles. Proc Soc Exp Biol Med. 86:277–286. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grote D, Russell SJ, Cornu TI, Cattaneo R,
Vile R, Poland GA and Fielding AK: Live attenuated measles virus
induces regression of human lymphoma xenografts in immunodeficient
mice. Blood. 97:3746–3754. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen A, Zhang Y, Meng G, Jiang D, Zhang H,
Zheng M, Xia M, Jiang A, Wu J, Beltinger C and Wei J: Oncolytic
measles virus enhances antitumour responses of adoptive CD8+NKG2D+
cells in hepatocellular carcinoma treatment. Sci Rep. 7:51702017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Msaouel P, Opyrchal M, Domingo Musibay E
and Galanis E: Oncolytic measles virus strains as novel anticancer
agents. Expert Opin Biol Ther. 13:483–502. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Allen C, Opyrchal M, Aderca I, Schroeder
MA, Sarkaria JN, Domingo E, Federspiel MJ and Galanis E: Oncolytic
measles virus strains have significant antitumor activity against
glioma stem cells. Gene Ther. 20:444–449. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galanis E, Hartmann LC, Cliby WA, Long HJ,
Peethambaram PP, Barrette BA, Kaur JS, Haluska PJ Jr, Aderca I,
Zollman PJ, et al: Phase I trial of intraperitoneal administration
of an oncolytic measles virus strain engineered to express
carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res.
70:875–882. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hutzen B, Raffel C and Studebaker AW:
Advances in the design and development of oncolytic measles
viruses. Oncolytic Virother. 4:109–118. 2015.PubMed/NCBI
|
|
9
|
Iankov ID, Kurokawa CB, D'Assoro AB, Ingle
JN, Domingo-Musibay E, Allen C, Crosby CM, Nair AA, Liu MC, Aderca
I, et al: Inhibition of the Aurora A kinase augments the anti-tumor
efficacy of oncolytic measles virotherapy. Cancer Gene Ther.
22:438–444. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Patel S: Breast cancer: Lesser-known
facets and hypotheses. Biomed Pharmacother. 98:499–506. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
DeSantis CE, Ma J, Gaudet MM, Newman LA,
Miller KD, Goding Sauer A, Jemal A and Siegel RL: Breast cancer
statistics, 2019. CA Cancer J Clin. 69:438–451. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rivera E and Gomez H: Chemotherapy
resistance in metastatic breast cancer: The evolving role of
ixabepilone. Breast Cancer Res. 12 (Suppl 2):S22010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun J, Guo Y, Fu X, Wang Y, Liu Y, Huo B,
Sheng J and Hu X: Dendrobium candidum inhibits MCF-7 cells
proliferation by inducing cell cycle arrest at G2/M phase and
regulating key biomarkers. Onco Targets Ther. 9:21–30.
2015.PubMed/NCBI
|
|
14
|
Cheung-Ong K, Giaever G and Nislow C:
DNA-damaging agents in cancer chemotherapy: Serendipity and
chemical biology. Chem Biol. 20:648–659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khanna A: DNA damage in cancer
therapeutics: A boon or a curse? Cancer Res. 75:2133–2138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao D, Herman JG and Guo M: The clinical
value of aberrant epigenetic changes of DNA damage repair genes in
human cancer. Oncotarget. 7:37331–37346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
O'Connor MJ: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cannan WJ and Pederson DS: Mechanisms and
consequences of double-strand DNA break formation in chromatin. J
Cell Physiol. 231:3–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jeggo PA and Löbrich M: DNA double-strand
breaks: Their cellular and clinical impact? Oncogene. 26:7717–7719.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haber JE: Partners and pathwaysrepairing a
double-strand break. Trends Genet. 16:259–264. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rothkamm K, Krüger I, Thompson LH and
Löbrich M: Pathways of DNA double-strand break repair during the
mammalian cell cycle. Mol Cell Biol. 23:5706–5715. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hinz JM, Yamada NA, Salazar EP, Tebbs RS
and Thompson LH: Influence of double-strand-break repair pathways
on radiosensitivity throughout the cell cycle in CHO cells. DNA
Repair (Amst). 4:782–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duprex WP, McQuaid S, Hangartner L,
Billeter MA and Rima BK: Observation of measles virus cell-to-cell
spread in astrocytoma cells by using a green fluorescent
protein-expressing recombinant virus. J Virol. 73:9568–9575. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramakrishnan MA: Determination of 50%
endpoint titer using a simple formula. World J Virol. 5:85–86.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McDonald CJ, Erlichman C, Ingle JN,
Rosales GA, Allen C, Greiner SM, Harvey ME, Zollman PJ, Russell SJ
and Galanis E: A measles virus vaccine strain derivative as a novel
oncolytic agent against breast cancer. Breast Cancer Res Treat.
99:177–184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo Y, Fu X, Huo B, Wang Y, Sun J, Meng L,
Hao T, Zhao ZJ and Hu X: GATA2 regulates GATA1 expression through
LSD1-mediated histone modification. Am J Transl Res. 8:2265–2274.
2016.PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo Y, Fu X, Jin Y, Sun J, Liu Y, Huo B,
Li X and Hu X: Histone demethylase LSD1-mediated repression of
GATA-2 is critical for erythroid differentiation. Drug Des Devel
Ther. 9:3153–3162. 2015.PubMed/NCBI
|
|
29
|
Meng Y, Chen CW, Yung MMH, Sun W, Sun J,
Li Z, Li J, Li Z, Zhou W, Liu SS, et al: DUOXA1-mediated ROS
production promotes cisplatin resistance by activating ATR-Chk1
pathway in ovarian cancer. Cancer Lett. 428:104–116. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Daley JM and Sung P: 53BP1, BRCA1, and the
choice between recombination and end joining at DNA double-strand
breaks. Mol Cell Biol. 34:1380–1388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sledge GW, Mamounas EP, Hortobagyi GN,
Burstein HJ, Goodwin PJ and Wolff AC: Past, present, and future
challenges in breast cancer treatment. J Clin Oncol. 32:1979–1986.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Russell SJ and Peng KW: Measles virus for
cancer therapy. Curr Top Microbiol Immunol. 330:213–241.
2009.PubMed/NCBI
|
|
33
|
Laksono BM, de Vries RD, McQuaid S, Duprex
WP and de Swart RL: Measles virus host invasion and pathogenesis.
Viruses. 8:2102016. View Article : Google Scholar
|
|
34
|
Msaouel P, Iankov ID, Dispenzieri A and
Galanis E: Attenuated oncolytic measles virus strains as cancer
therapeutics. Curr Pharm Biotechnol. 13:1732–1741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sugiyama T, Yoneda M, Kuraishi T, Hattori
S, Inoue Y, Sato H and Kai C: Measles virus selectively blind to
signaling lymphocyte activation molecule as a novel oncolytic virus
for breast cancer treatment. Gene Ther. 20:338–347. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Noris M and Remuzzi G: Overview of
complement activation and regulation. Semin Nephrol. 33:479–492.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maciejczyk A, Szelachowska J,
Szynglarewicz B, Szulc R, Szulc A, Wysocka T, Jagoda E, Lage H and
Surowiak P: CD46 expression is an unfavorable prognostic factor in
breast cancer cases. Appl Immunohistochem Mol Morphol. 19:540–546.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Surowiak P, Materna V, Maciejczyk A,
Kaplenko I, Spaczynski M, Dietel M, Lage H and Zabel M: CD46
expression is indicative of shorter revival-free survival for
ovarian cancer patients. Anticancer Res. 26:4943–4948.
2006.PubMed/NCBI
|
|
39
|
Su Y, Liu Y, Behrens CR, Bidlingmaier S,
Lee NK, Aggarwal R, Sherbenou DW, Burlingame AL, Hann BC, Simko JP,
et al: Targeting CD46 for both adenocarcinoma and neuroendocrine
prostate cancer. JCI Insight. 3:e1214972018. View Article : Google Scholar
|
|
40
|
Sherbenou DW, Aftab BT, Su Y, Behrens CR,
Wiita A, Logan AC, Acosta-Alvear D, Hann BC, Walter P, Shuman MA,
et al: Antibody-drug conjugate targeting CD46 eliminates multiple
myeloma cells. J Clin Invest. 126:4640–4653. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cho YS, Do MH, Kwon SY, Moon C, Kim K, Lee
K, Lee SJ, Hemmi S, Joo YE, Kim MS and Jung C: Efficacy of
CD46-targeting chimeric Ad5/35 adenoviral gene therapy for
colorectal cancers. Oncotarget. 7:38210–38223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Geekiyanage H and Galanis E: MiR-31 and
miR-128 regulates poliovirus receptor-related 4 mediated measles
virus infectivity in tumors. Mol Oncol. 10:1387–1403. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Carlson SK, Classic KL, Hadac EM, Dingli
D, Bender CE, Kemp BJ and Russell SJ: Quantitative molecular
imaging of viral therapy for pancreatic cancer using an engineered
measles virus expressing the sodium-iodide symporter reporter gene.
AJR Am J Roentgenol. 192:279–287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Peng KW, TenEyck CJ, Galanis E, Kalli KR,
Hartmann LC and Russell SJ: Intraperitoneal therapy of ovarian
cancer using an engineered measles virus. Cancer Res. 62:4656–4662.
2002.PubMed/NCBI
|
|
45
|
Shoji K, Yoneda M, Fujiyuki T, Amagai Y,
Tanaka A, Matsuda A, Ogihara K, Naya Y, Ikeda F, Matsuda H, Sato H
and Kai C: Development of new therapy for canine mammary cancer
with recombinant measles virus. Mol Ther Oncolytics. 3:150222016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Russell SJ: Replicating vectors for cancer
therapy: A question of strategy. Semin Cancer Biol. 5:437–443.
1994.PubMed/NCBI
|
|
47
|
Delpeut S, Sisson G, Black KM and
Richardson CD: Measles virus enters breast and colon cancer cell
lines through a PVRL4-mediated macropinocytosis pathway. J Virol.
91:e02191–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Baldo A, Galanis E, Tangy F and Herman P:
Biosafety considerations for attenuated measles virus vectors used
in virotherapy and vaccination. Hum Vaccin Immunother.
12:1102–1116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Phuong LK, Allen C, Peng KW, Giannini C,
Greiner S, TenEyck CJ, Mishra PK, Macura SI, Russell SJ and Galanis
EC: Use of a vaccine strain of measles virus genetically engineered
to produce carcinoembryonic antigen as a novel therapeutic agent
against glioblastoma multiforme. Cancer Res. 63:2462–2469.
2003.PubMed/NCBI
|
|
50
|
Peng KW, Frenzke M, Myers R, Soeffker D,
Harvey M, Greiner S, Galanis E, Cattaneo R, Federspiel MJ and
Russell SJ: Biodistribution of oncolytic measles virus after
intraperitoneal administration into Ifnar-CD46Ge transgenic mice.
Hum Gene Ther. 14:1565–1577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Myers R, Harvey M, Kaufmann TJ, Greiner
SM, Krempski JW, Raffel C, Shelton SE, Soeffker D, Zollman P,
Federspiel MJ, et al: Toxicology study of repeat intracerebral
administration of a measles virus derivative producing
carcinoembryonic antigen in rhesus macaques in support of a phase
I/II clinical trial for patients with recurrent gliomas. Hum Gene
Ther. 19:690–698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Myers RM, Greiner SM, Harvey ME, Griesmann
G, Kuffel MJ, Buhrow SA, Reid JM, Federspiel M, Ames MM, Dingli D,
et al: Preclinical pharmacology and toxicology of intravenous
MV-NIS, an oncolytic measles virus administered with or without
cyclophosphamide. Clin Pharmacol Ther. 82:700–710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Heinzerling L, Künzi V, Oberholzer PA,
Kündig T, Naim H and Dummer R: Oncolytic measles virus in cutaneous
T-cell lymphomas mounts antitumor immune responses in vivo and
targets interferon-resistant tumor cells. Blood. 106:2287–2294.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ruf B and Lauer UM: Assessment of current
virotherapeutic application schemes: ‘hit hard and early’ versus
‘killing softly’? Mol Ther Oncolytics. 2:150182015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Calton CM, Kelly KR, Anwer F, Carew JS and
Nawrocki ST: Oncolytic viruses for multiple myeloma therapy.
Cancers (Basel). 10:1982018. View Article : Google Scholar
|
|
56
|
Robinson S and Galanis E: Potential and
clinical translation of oncolytic measles viruses. Expert Opin Biol
Ther. 17:353–363. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Achard C, Surendran A, Wedge ME,
Ungerechts G, Bell J and Ilkow CS: Lighting a fire in the tumor
microenvironment using oncolytic immunotherapy. EBioMedicine.
31:17–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Galanis E, Atherton PJ, Maurer MJ, Knutson
KL, Dowdy SC, Cliby WA, Haluska P Jr, Long HJ, Oberg A, Aderca I,
et al: Oncolytic measles virus expressing the sodium iodide
symporter to treat drug-resistant ovarian cancer. Cancer Res.
75:22–30. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Robertson KA, Nalepa G, Yang FC, Bowers
DC, Ho CY, Hutchins GD, Croop JM, Vik TA, Denne SC, Parada LF, et
al: Imatinib mesylate for plexiform neurofibromas in patients with
neurofibromatosis type 1: A phase 2 trial. Lancet Oncol.
13:1218–1224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Msaouel P, Opyrchal M, Dispenzieri A, Peng
KW, Federspiel MJ, Russell SJ and Galanis E: Clinical trials with
oncolytic measles virus: Current status and future prospects. Curr
Cancer Drug Targets. 18:177–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Russell SJ, Federspiel MJ, Peng KW, Tong
C, Dingli D, Morice WG, Lowe V, O'Connor MK, Kyle RA, Leung N, et
al: Remission of disseminated cancer after systemic oncolytic
virotherapy. Mayo Clin Proc. 89:926–933. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gauvrit A, Brandler S, Sapede-Peroz C,
Boisgerault N, Tangy F and Gregoire M: Measles virus induces
oncolysis of mesothelioma cells and allows dendritic cells to
cross-prime tumor-specific CD8 response. Cancer Res. 68:4882–4892.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sishc BJ and Davis AJ: The role of the
core non-homologous end joining factors in carcinogenesis and
cancer. Cancers (Basel). 9:812017. View Article : Google Scholar
|
|
64
|
Mao Z, Jiang Y, Liu X, Seluanov A and
Gorbunova V: DNA repair by homologous recombination, but not by
nonhomologous end joining, is elevated in breast cancer cells.
Neoplasia. 11:683–691. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lees-Miller SP, Beattie TL and Tainer JA:
Noncoding RNA joins Ku and DNA-PKcs for DNA-break resistance in
breast cancer. Nat Struct Mol Biol. 23:509–510. 2016. View Article : Google Scholar : PubMed/NCBI
|