Introduction
Prostate cancer (PCa) is the second most common type
of cancer in men and the fourth leading cause of cancer-associated
death worldwide, according to GLOBOCAN 2018 statistics (1). In addition, mortality is mainly
associated with the age of diagnosis, degree of histological
differentiation, metastasis and resistance to hormonal therapy,
with the latter being one of the greatest challenges in PCa
management (2,3). Androgen deprivation therapy (ADT),
mainly achieved with gonadotropin-releasing hormone analogs, is the
first-line therapy to treat metastatic PCa. Patients with advanced
PCa initially respond to ADT, showing decreased tumor size and
blood prostate-specific antigen levels (4,5). However,
after two or three years, nearly all patients relapse, progressing
to castration-resistant PCa (CRPC) (3,6).
Increasing evidence has indicated that cancer stem cells (CSCs), a
small subpopulation of malignant cells with stem-like properties,
may serve an important role in the progression of CRPC (7–11).
Functionally, CSCs are defined by their ability of
self-renewal and asymmetric division, giving rise to heterogeneous
cell lines (12). Prostate CSCs are
able to initiate the development of tumors in the metastatic niche
and to differentiate into cancer cells with highly aggressive
phenotypes, contributing to the progression of the disease
(13,14). From a molecular standpoint, CSCs are
characterized by the expression of specific surface markers; in
particular, prostate CSCs express CD133 (also known as prominin-1)
and CD44, and high levels of the multidrug resistance pump ATP
binding cassette subfamily G member 2 and integrin α2β1 (10,15,16).
CSCs contribute to cancer progression due to their
resistance to different therapeutic approaches (17–19).
Prostate CSCs are not sensitive to ADT, due to not expressing the
androgen receptor (AR) (9,10,16).
Furthermore, CSCs exhibit abnormal activation of DNA repair
pathways, low proliferation rates and high expression levels of
multidrug resistance efflux pumps (15–17,19). These
intrinsic characteristics of CSCs may account for the failure of
radio- and chemotherapy in patients with CRPC (16,20,21).
Due to their importance in PCa progression, CSCs
have become a potential target for advanced PCa. However, the
origin and molecular mechanisms of CSCs are not fully understood.
It has been suggested that CSCs may originate from the malignant
transformation of normal prostate stem cells (22); however, several studies have indicated
that CSCs may originate from non-CSCs (23–25).
Notably, numerous molecular networks have been found to induce
reprogramming of non-CSCs into CSCs and have been involved in
epithelial-mesenchymal transition (EMT) (14,26,27).
EMT is a trans-differentiation mechanism, in which
epithelial cells change their phenotype and acquire mesenchymal
features. The main change that occurs during EMT is the loss of
E-cadherin expression, resulting in loss of apicobasal polarity and
cell-cell contact, and increased migration and invasion (28). In cancer cells, several signaling
pathways can initiate and maintain EMT, including the transforming
growth factor-β, Wnt/β-catenin and integrin/integrin-linked kinase
signaling pathways (28–30). These signaling pathways converge on
the activation of EMT transcription factors, which directly inhibit
E-cadherin expression (31,32). Among these transcription factors, zinc
finger E-box-binding homeobox 1 (ZEB1) has been associated with the
activation of several mechanisms leading to resistance to treatment
(33–36). It has been previously demonstrated
that ZEB1 induces EMT in PCa cell lines, promoting the loss of
E-cadherin expression, and increases migration and invasion,
resulting in an aggressive phenotype (37–39).
Accordingly, ZEB1 is expressed at higher levels in the highly
aggressive DU145 cell line compared with in other PCa cell lines,
such as PC3, LNCaP and 22Rv1 cells (38). Knocking down ZEB1 expression in the
DU145 cell line increases E-cadherin expression and decreases
invasion and migration (38).
Furthermore, DU145 cells exhibit some characteristics of CSCs, such
as chemotherapy resistance, and the stable knockdown of ZEB1
sensitizes these cells to docetaxel, a taxane widely used for the
treatment of CRPC (37). Based on the
aforementioned studies, the present study hypothesized that
knocking down ZEB1 in PCa cells could also revert some features
associated with CSCs.
Materials and methods
Cell culture
The PCa DU145 (ATCC®HTB81™) and LNCaP
clone FGC (ATCC®CRL1740™) cell lines were purchased from
the American Type Culture Collection. DU145 cells were originally
obtained from a brain metastasis of PCa and are insensitive to
androgens, resembling CRPC (40).
LNCaP cells were originally obtained from a lymph node metastasis
of PCa and are responsive to androgens (41). DU145 cells were maintained in DMEM F12
medium and LNCaP cells were maintained in RPMI-1640 medium (both
Gibco; Thermo Fisher Scientific, Inc.). Both culture media were
supplemented with 10% fetal bovine serum (Corning Life Sciences),
streptomycin-penicillin and amphotericin B (Corning, Inc.). Cell
cultures were maintained at 37°C in a humidified atmosphere with 5%
CO2.
Lentiviral transduction
Knockdown of ZEB1 expression in DU145 and LNCaP
cells was achieved using transduction with lentiviral vectors
containing a short hairpin (sh)RNA against ZEB1 [pLenti-U6-shRNA
(hZEB1)-Rsv(RFP-Puro)], or a scrambled shRNA used as a negative
control [pLenti-U6-shRNA (Neg-control)-Rsv(RFP-Puro)]. Pre-packaged
lentiviral particles were purchased ready to use from GenTarget,
Inc., and cells were infected using a standard procedure. Briefly,
106 cells/well were seeded in 6-well plates. After 24 h
at 37°C, cells were incubated with lentiviral particles at a
multiplicity of infection of 3, with 6 µg/ml polybrene
(Sigma-Aldrich; Merck KGaA) in 1 ml culture medium for 24 h at
37°C. Subsequently, cells integrating the vectors were selected
using 2 µg/ml puromycin for 24 h at 37°C.
Western blotting
Whole-cell protein was extracted from cells using
RIPA buffer with cOmplete™ Mini, EDTA-free protease inhibitor
cocktail (Roche Diagnostics), and protein concentration was
determined using a Bradford protein assay. A total of 50 µg
protein/lane was separated by 10% SDS-PAGE and transferred to a
nitrocellulose membrane. Blots were blocked for 1 h at room
temperature with 5% BSA (Winkler Ltda) in 0.2% TBS-Tween and
incubated overnight at 4°C with primary antibodies diluted in
blocking buffer. After washing three times in 0.2% TBS-Tween, bound
primary antibodies were detected with HRP-conjugated secondary
antibodies incubated for 1.5 h at room temperature, and revealed
with an enhanced chemiluminescence detection kit for HRP (EZ-ECL;
Biological Industries). Chemiluminescence was detected using the
Fusion FX image system (VilberLourmat) and the optical density of
the bands was analyzed using the software ImageJ v1.51 (National
Institutes of Health).
The antibodies used were as follows: SOX2 (1:1,000;
cat. no. ab92494; Abcam), Krüppel-like factor 4 (KLF4; 1:1,000;
cat. no. ab215036; Abcam), CD44 (1:5,000; cat. no. ab51037; Abcam),
CD133 (1:500; cat. no. Pas-38014; Thermo Fisher Scientific, Inc.),
ZEB1 (1:1,000; cat. no. Pa5-28221; Thermo Fisher Scientific, Inc.),
E-cadherin (1:1,000; cat. no. 610181; BD Biosciences), β-actin
(1:5,000; cat. no. 691002; MP Biomedicals, LLC), anti-mouse HRP
(1:10,000; cat. no. 115-035-003; Jackson ImmunoResearch
Laboratories, Inc.) and anti-rabbit HRP (1:10,000; cat. no.
111-035-003; Jackson ImmunoResearch Laboratories, Inc.).
RNA extraction and reverse
transcription-quantitative (q)PCR
Total RNA was extracted from cells using TRIzol
(Ambion; Thermo Fisher Scientific, Inc.). A total of 3,000 ng cDNA
was synthesized using the cDNA Affinity Script QPCR kit (Agilent
Technologies, Inc.), according to the manufacturer's protocol, and
100 ng cDNA was amplified by qPCR using the Brilliant II SYBR Green
qPCR Master Mix kit (Agilent Technologies, Inc.) according to the
manufacturer's protocol. For qPCR, the thermocycling conditions
were as follows: Initial denaturation at 95°C for 10 min, followed
by 40 cycles of denaturation at 95°C for 15 sec, annealing at 60°C
for 15 sec and extension at 72°C for 15 sec. The housekeeping gene
pumilio RNA binding family member 1 was used as a normalizer
(42) and the results were analyzed
using the 2−ΔΔCq method (43). The primer sequences used for qPCR are
presented in Table I.
 | Table I.Primer sequences used for
quantitative PCR. |
Table I.
Primer sequences used for
quantitative PCR.
| Gene | Forward primer | Reverse primer |
|---|
| ZEB1 |
5′-TTCACAGTGGAGAGAAGCCA-3′ |
5′-GCCTGGTGATGCTGAAAGAG-3′ |
| CD44 |
5′-CACGTGGAATACACCTGCCA-3′ |
5′-GACAAGTTTTGGTGGCAGGT-3′ |
| CD133 |
5′-TCAATTTTGGATTCATATTT-3′ |
5′-ACTCCCATAAAGCTGGACCC-3′ |
| SOX2 |
5′-GTCTAGCCTCGTCGATGAAC-3′ |
5′-AACCCCAAGATGCACAACTC-3′ |
| KLF4 |
5′-CCCCGTGTGTTTACGGTAGT-3′ |
5′-AGAGTTCCCATCTCAAGGCA-3′ |
| PUM1 |
5′-CGTACGTGAGGCGTAAGTAA-3′ |
5′-CGGTCGTCCTGAGGATAAAA-3′ |
Colony formation assay
Cells cultured under adherent conditions were
detached using 0.25% trypsin at 37°C for 10 min, seeded at
2×104 cells/plate in 6-well plates and incubated at 37°C
in a humidified atmosphere with 5% CO2. After 15 days,
cells were fixed for 10 min at room temperature with cold 100%
methanol, stained with crystal violet (0.5% crystal violet in 25%
methanol) for 10 min at room temperature, washed and air-dried at
room temperature. The resulting colonies were photographed using an
Olympus SZ60 stereoscopic light microscope (Olympus Corporation),
and the images were analyzed using ImageJ v1.51 (National
Institutes of Health). Groups of ≥50 cells were considered as
colonies, and the types of colonies formed were classified
according to their morphology in holoclones, meroclones and
paraclones, as previously described by Barrandon and Green
(44).
Prostatosphere formation assay
Cells from cultures maintained under adherent
conditions were washed with PBS and detached using acutase
(eBioscience; Thermo Fisher Scientific, Inc.) for 7 min at 37°C.
The collected cells were centrifuged at 300 × g for 5 min at room
temperature, and the pellet was mechanically disaggregated using a
micropipette and collected through a 40-µm cell strainer (BD
Falcon; Becton, Dickinson and Company). Cells (1×105)
were seeded in 6-cm dishes coated with 1% agarose in a culture
medium suitable for inducing cell growth under non-adherent
conditions, as previously described (15). Prostatospheres were photographed every
other day for a total of 7 days using an Olympus SIG60 stereoscopic
light microscope (Olympus Corporation; magnification, ×100) and
analyzed using the software AxioVision v4.8.1 (Carl Zeiss AG), as
described by Acikgoz et al (45). Prostatosphere 3D volume was calculated
from 2D images using the following formula: (length × width ×
width) × (3.1416/6).
Statistical analysis
Data analysis was performed using GraphPad Prism 6.0
software (GraphPad Software, Inc.). Data are expressed as the mean
± standard deviation of at least three independent experiments, and
the Mann-Whitney U test was used to analyze differences between
groups. P≤0.05 was considered to indicate a statistically
significant difference.
Results
Knockdown of ZEB1 in the PCa DU145
cell line
To determine whether ZEB1 could revert some of the
CSC features in PCa cells, knockdown of ZEB1 expression in the PCa
DU145 cell line was performed using transduction with lentiviral
vectors expressing a shRNA targeting ZEB1 (DU145 sh-ZEB1). Scramble
shRNA was used as a control (DU145 sh-Scr). The cells transduced
with ZEB1 shRNA formed cohesive groups in adherent conditions
(Fig. 1A) and sh-ZEB1 significantly
decreased ZEB1 mRNA (Fig. 1B) and
protein expression (Fig. 1C and D)
compared with sh-Scr. ZEB1 is a known transcriptional repressor of
E-cadherin. Therefore, to evaluate whether knocking down ZEB1
expression in these cells modified E-cadherin expression,
E-cadherin protein expression was determined using western blot
analysis. As shown in Fig. 1C and D,
ZEB1-knockdown significantly increased E-cadherin expression.
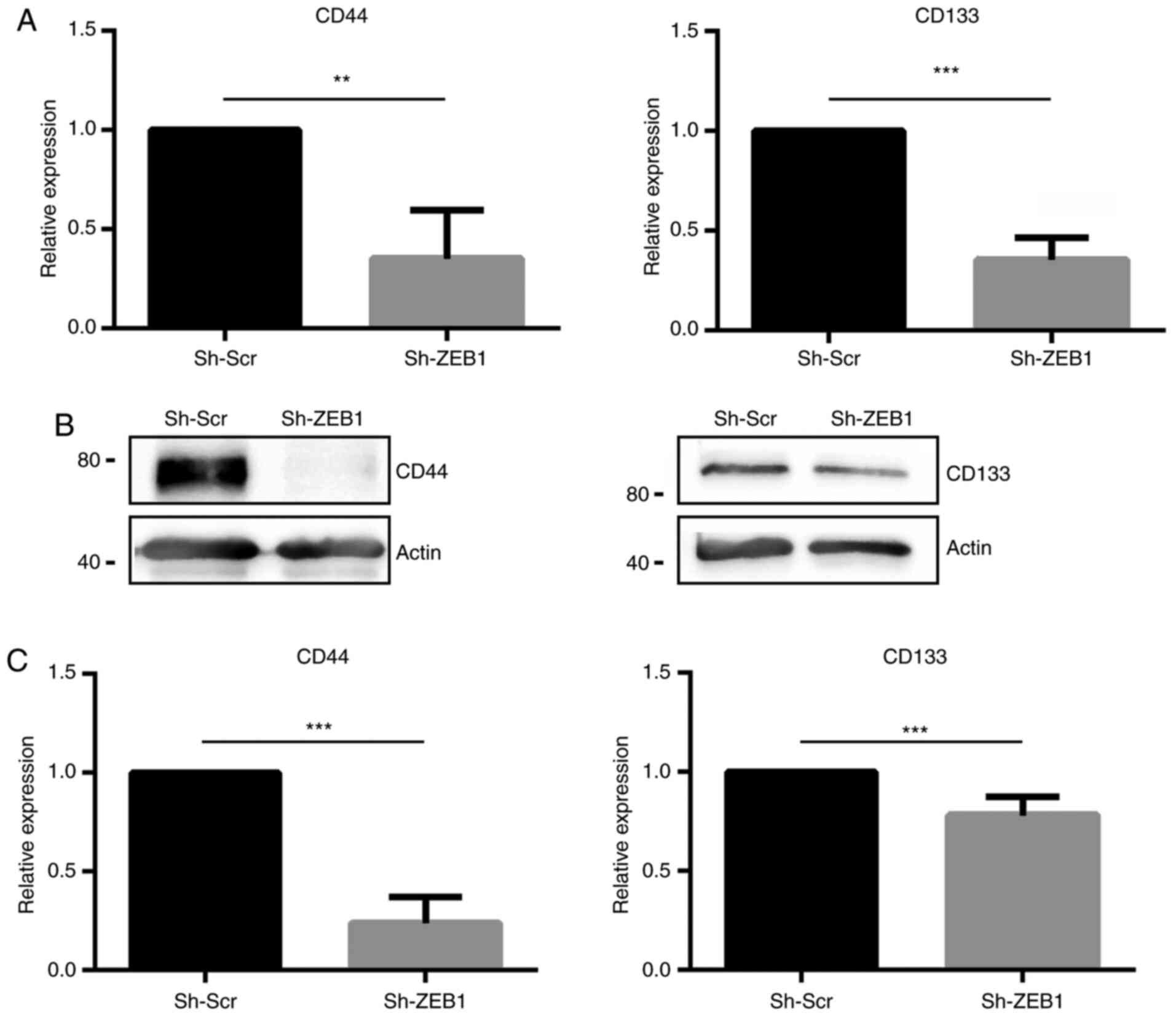
Knockdown of ZEB1 in DU145 cells
decreases the expression levels of the CSC markers CD44 and
CD133
In the established ZEB1-knockdown cell line, the
expression levels of the CSC markers CD44 and CD133 were evaluated.
Reverse transcription-qPCR revealed that DU145 cells transduced
with sh-ZEB1 exhibited significantly decreased mRNA expression
levels of CD44 and CD133 compared with DU145 cells transduced with
sh-Scr (Fig. 2A). These results were
verified using western blot analysis (Fig. 2B), revealing that ZEB1-knockdown
induced a significant decrease of ~75 and 20% of CD44 and CD133
protein expression, respectively (Fig.
2C).
Knockdown of ZEB1 in DU145 cells
decreases SOX2 expression
Since ZEB1-knockdown decreased the expression levels
of the CSC markers CD44 and CD133, which in turn are controlled by
CSC transcription factors, such as KLF4 and SOX2, the present study
further investigated whether ZEB1-knockdown affected the expression
levels of these transcription factors. ZEB1-knockdown in the DU145
cell line significantly decreased the expression levels of SOX2, at
both the mRNA and protein level; however, the expression levels of
KLF4 were not significantly changed at the mRNA level, but were
significantly decreased at the protein level (Fig. 3).
Effect of ZEB1-knockdown on the colony
forming ability of DU145 cells
One of the main characteristics of CSCs is the
ability of self-renewal. To determine if ZEB1-knockdown in PCa
cells affected this ability, a colony formation assay was
performed, in which single cells that self-renew will form colonies
of the clones. ZEB1-knockdown significantly decreased the number of
colonies formed by DU145 cells (Fig. 4A
and B). Furthermore, the types of colonies formed by DU145
cells transduced with sh-ZEB1 were different compared with those in
cells transduced with sh-Scr, with respect to size and morphology.
The colonies were classified into holoclones, meroclones and
paraclones (44): Holoclones are
colonies of small and densely compact cells, with regular edges,
paraclones consist of larger and elongated cells that grow in a
scattered way, with irregular edges, and meroclones have an
intermediate morphology, between a paraclone and a holoclone. DU145
cells transduced with either sh-Scr or sh-ZEB1 formed all three
types of the colonies (Fig. 4C);
however, cells transduced with sh-Scr formed mainly holoclones,
whereas cells transduced with sh-ZEB1 formed a high percentage of
paraclones (Fig. 4D). In all cases,
the mean number of cells per colony was significantly higher in the
colonies formed by cells transduced with sh-Scr compared with that
in colonies with cells transduced with sh-ZEB1 (Fig. 4E).
Effect of ZEB1-knockdown on the
anchorage-independent growth ability of DU145 cells
Prostate CSCs can grow in an anchorage-independent
manner forming prostatospheres when cultured in soft agar (46). Prostatospheres formed by cells
transduced with sh-Scr and sh-ZEB1 were obtained following 7 days
of anchorage-independent growth. There was a higher number of
prostatospheres formed by cells transduced with sh-Scr compared
with cells transduced with sh-ZEB1 after 1 and 3 days (Fig. 5A and B). Furthermore, the
prostatospheres formed by cells transduced with sh-Scr were bigger
compared with those formed by cells transduced with sh-ZEB1 after 5
and 7 days (Fig. 5C and D).
Therefore, ZEB1-knockdown in DU145 cells affected their ability to
generate prostatospheres in anchorage-independent cultures.
Effect of ZEB1-knockdown on the
expression levels of CSC markers, clonogenicity and prostatosphere
forming ability in LNCaP cells
The DU145 cell line is characterized by a lack of
the AR and by their high aggressiveness (47), which are intrinsic characteristics of
CRPC. To evaluate if knocking down ZEB1 exerted the same effects in
androgen-sensitive cells, ZEB1 expression was knocked down in
another PCa cell line, LNCaP. In these cells, the expression levels
of E-cadherin, CD44, CD133, SOX2 and KLF4 were investigated, and
the results revealed that ZEB1-knockdown significantly increased
E-cadherin protein expression and significantly decreased SOX2
protein expression; however, no significant changes were observed
in the expression levels of CD44, CD133 and KLF4 (Fig. 6A and B). To assess the ability of
self-renewal of the LNCaP ZEB1-knockdown cells, a colony formation
assay was performed. ZEB1-knockdown in LNCaP cells did not affect
the number or size of the colonies formed (Fig. 6C-E). However, it modified the
morphology of the colonies, forming significantly fewer holoclones
and more meroclones compared with the control cells (Fig. 6F). Finally, to evaluate the ability of
these cells for anchorage-independent growth, a prostatosphere
formation assay was performed. As shown in Fig. 6G-I, the LNCaP control cells cultured
in soft agar formed bigger prostatospherescompared with the LNCaP
cells transduced with sh-ZEB1 after 5 and 7 days, although by day 7
the prostatospheres were smaller compared with day 5 as the LNCaP
cells disaggregated.
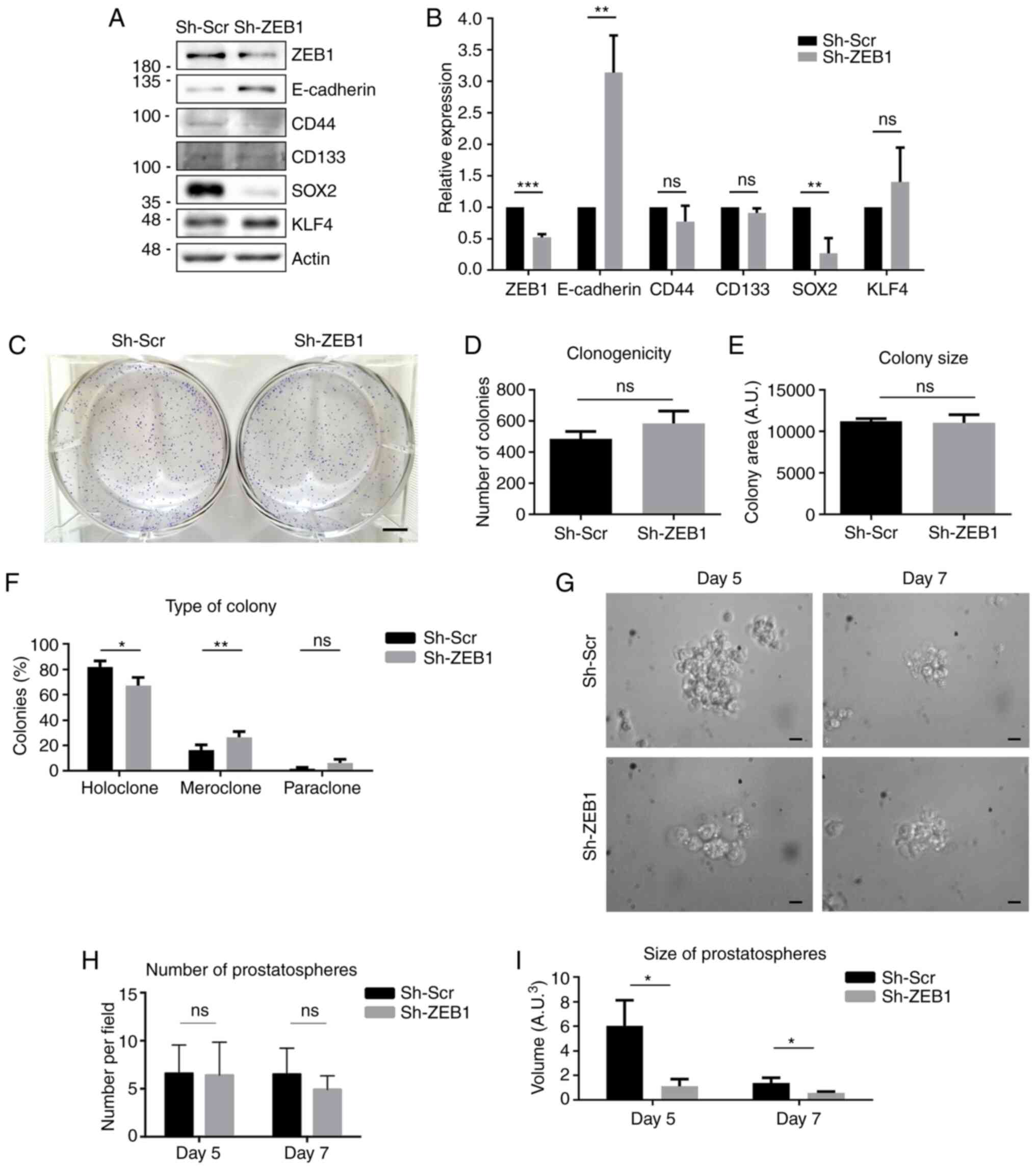 | Figure 6.Effect of ZEB1-knockdown in the
prostate cancer LNCaP cell line. (A) Representative western blots
for ZEB1, E-cadherin, CD44, CD133, SOX2 and KLF4 protein expression
in LNCaP cells transduced with a lentiviral vector carrying Sh-ZEB1
or Sh-Scr. (B) Quantification of optical density of western blots.
(C) Representative image of colony formation assay using LNCaP
Sh-ZEB1 and Sh-Scr cells. Scale bar, 5 mm. (D) Number of colonies
formed after 2 weeks of growth under limiting dilution conditions.
(E) Size of the colonies formed by each cell condition. (F)
Quantification of the different types of colonies formed. (G)
Representative images of the prostatospheres formed in each
condition at days 5 and 7 after seeding. Scale bar, 20 µm. (H)
Number of prostatospheresformed after 5 and 7 days of growth under
non-adherent conditions. (I) Quantification of the size of the
prostatospheres formed by each cell line at 5 and 7 days after
seeding. Data are expressed as the mean ± SD (n=3). *P≤0.05,
**P≤0.01 and ***P≤0.001 (Mann-Whitney U test). ns, not significant;
ZEB1, ZEB1, zinc finger E-box-binding homeobox 1; Sh-ZEB1, short
hairpin RNA against ZEB1; Sh-Scr, scrambled short hairpin RNA;
KLF4, Krüppel-like factor 4; A.U., arbitrary units. |
Discussion
The amount of CSCs within a tumor varies and may be
important for the prognosis of the disease (16,48–50).
Studies in several types of cancer, such as melanoma, breast, colon
and prostate cancer, have described CSCs as tumor-initiating cells,
as they can generate a new tumor in distant organs in an
appropriate cellular environment and contribute to cancer
aggressiveness due to their radio- and chemo-resistance, driving
recurrence following conventional therapy (17,18,51).
EMT is accompanied by a reactivation of signaling
pathways involved in self-renewal, such as the Wnt and Notch
signaling pathway, which facilitate changes in the phenotypic
profile of cells, with some of them acquiring a more aggressive
and/or mesenchymal phenotype, favoring metastasis and invasiveness
(52,53). ZEB1 is a transcription factor that
modulates EMT, repressing E-cadherin expression and favoring the
expression of mesenchymal markers, in coordination with other
transcription factors from the SNAIL and TWIST family (38,54,55).
In the present study, the effect of ZEB1-knockdown
on the expression levels of E-cadherin and pluripotency genes
commonly expressed in embryonic cells, SOX2 (56,57) and
KLF4 (58,59), was investigated. It was found that
silencing ZEB1 in DU145 cells induced an increase in E-cadherin and
a decrease in SOX2 expression. However, ZEB1 silencing did not
regulate the expression levels of KLF4. Stoichiometric SOX2 and
KLF4 expression is sufficient for pluripotency in the absence of
OCT4 (60). However, knocking down
SOX2 by itself results in a decrease of stemness and tumor growth,
and induces tumor regression in several types of cancer, such a
colorectal, breast and lung cancer (61). Considering that SOX2 has been
described as one of the transcription factors that is overexpressed
in more aggressive cancer cells (62,63),
targeting ZEB1 and the consequent decrease of SOX2 expression may
impair CSC self-renewal and maintenance in a variety of tumors,
including PCa. However, it is not clear whether this effect may be
a result of the EMT process or be EMT-independent. Previous studies
have reported that SOX2 increases cell proliferation and survival
by inducing EMT (64,65). Forced SOX2 expression increases the
expression levels of the EMT transcription factors TWIST, SNAI1 and
SNAI2 in pancreatic cancer cell lines (66). In PCa, SOX2-knockdown decreases the
expression levels of SNAI1 and SNAI2, and inhibits migration and
prostatosphere formation (67).
Furthermore, knocking down SNAI1 in pancreatic cancer cells
increases E-cadherin expression and downregulates SOX2 expression,
as well as decreases tumor size in vivo (68). In the PCa PC3 cells, silencing
E-cadherin increases the formation of prostatospheres and the
expression levels of CD44 and SNAI1 (69). Notably, knocking down SNAI1 in these
cells results in a decrease in prostatosphere formation and
clonogenicity (69), which is similar
to the observed phenotype of the DU145 cells with ZEB1-knockdown in
the present study. This suggested that transcriptional factors
involved in EMT may be key for the induction of CSC features. On
the other hand, a previous study has reported temporary SOX2 and
KLF4 expression, mainly during colonization in the metastatic niche
by CSCs, which was absent or low during EMT (70).
Consistent with the results of the present study in
DU145 cells, knockdown of ZEB1 in LNCaP cells increased E-cadherin
and decreased SOX2 expression. However, in LNCaP cells, no changes
were observed in the expression levels of the CSC markers, CD44 and
CD133. Prostate CSCs are characterized by a molecular signature
that includes positive expression of CD44 and CD133 (15). In the present study, it was found that
LNCaP cells expressed very low levels of CD44 and CD133. By
contrast, DU145 cells were positive for CD44 and CD133 expression,
and their expression levels were downregulated by ZEB1-knockdown. A
possible explanation for the different results observed may be due
to the intrinsic characteristics of both cell lines. LNCaP cells
are androgen-sensitive cells derived from a lymph node metastasis
(41), representing an earlier stage
of PCa, whereas DU145 are androgen-insensitive cells, derived from
a brain metastasis (40); therefore,
they are more representative of CRPC. Androgen sensitivity of PCa
cells serves a role in CSC phenotype and ZEB1 expression. In
androgen-sensitive cells, androgens promote the expression levels
of ZEB1 via the binding of the AR to androgen response elements
present in the ZEB1 promoter (71).
Prostate CSCs do not express AR and other prostate epithelial
differentiation markers (9,16). However, it has been demonstrated that
ZEB1 expression may be induced in AR-null cells (72). Furthermore, in our previous study,
ZEB1 expression in PCa cell lines was characterized, revealing that
DU145 cells expressed higher levels of ZEB1 compared with LNCaP
cells (38). The aforementioned
studies, together with the results of the present study, indicated
that DU145 cells may be enriched in cell populations that display
CSC properties, such as chemotherapy and anoikis resistance,
whereas LNCaP cells did not display these characteristics.
Increasing evidence has revealed that ZEB1 is a key
factor for the transition between non-CSCs and CSCs in other types
of cancer, such as pancreatic and breast cancer (55,73,74). This
effect may be mediated by non-coding RNAs inhibiting the expression
levels of stemness genes. Several stemness-repressing microRNAs
(miRs) have been described (75).
Among them, miR-200 may represent the link between ZEB1 and CSCs.
It has been demonstrated that ZEB1 and miR-200 are associated with
a double-negative feedback loop: ZEB1 inhibits miR-200 expression,
which in turn suppresses the translation of ZEB1 mRNA (76). On the other hand, miR-200 also
represses the expression levels of SOX2 and KLF4 (75). Therefore, one of the limitations of
the present study was the lack of analyzing miR-200 expression
following ZEB1-knockdown. It would be interesting to determine
whether the downregulation of SOX2 by ZEB1-knockdown would result
in the loss of direct interaction between ZEB1 and the SOX2
promoter or through the lack of ZEB1 inhibition on miR-200.
Knocking down ZEB1 in the present study decreased
the expression levels of SOX2, as well as the number of colonies
formed in vitro and the proportion of holoclones, and
increased the number of paraclones. Cells that form holoclones and
express CSC markers, such as CD44 and CD133, have the ability of
self-renewal, generate cultures in non-adherent conditions and have
highly tumorigenic abilities when injected into immunodeficient
mice (77,78). On the other hand, paraclones can
proliferate, but not self-renew (77). The results in the present study are
consistent with the study by Knaack et al (79), which found higher expression levels of
ZEB1 in holoclones in pancreatic cancer cells compared with those
in paraclones. Moreover, holoclones of pancreatic cancer cells had
increased expression levels of TNFα and other pro-inflammatory
genes acting as EMT inducers compared with paraclones, which is
consistent with the higher expression levels of ZEB1 (79).
The present study also revealed that there was a
decrease in the expression levels of CD44 and CD133 in DU145 cells
following ZEB1-knockdown. Overexpression of CD44 and SOX2 in PCa
cells results in the upregulation of the SNAI1 and SNAI2
transcription factors leading to EMT (67). Overexpression of CD133 in PCa cells
increases the expression levels of other CSC markers, decreases
E-cadherin expression and enhances migration and bone metastasis
formation (80). Furthermore, the
presence of CD44 and CD133 have been identified as important
factors in the formation of prostatic spheroids, which is directly
associated with the ability of self-renewal and anoikis resistance
in PCa cells (81). In the present
study, knocking down ZEB1 decreased the number of colonies formed
in adherent conditions, as well as the number and size of the
prostatospheres generated, which is consistent with the observed
downregulation of CD44, CD133 and SOX2 expression. Pluripotency
genes are important factors in chemoresistance and apoptosis
evasion. Overexpression of SOX2 and OCT4 in gastric cancer cells
increases their resistance to oxaliplatin and fluorouracil
(82). In PCa, CD133+
cells, sorted from human 22Rv1 PCa cells, are highly resistant to
γ-radiation and docetaxel (83). In
T-cell acute lymphoblastic leukemia, CD44 enhances the activity of
ATP-binding cassette multidrug efflux transporters, inducing
resistance to doxorubicin (84). In
agreement with this, our previous study revealed that knocking down
ZEB1 in DU145 cells decreases the expression levels of multidrug
resistance-associated protein 1 and ATP-binding cassette subfamily
C member 4, and enhances their sensitivity to docetaxel (37). This suggested that ZEB1, SOX2, CD44
and CD133 may participate together to promote chemoresistance.
Overall, the results of the present study indicated that targeting
ZEB1 in PCa decreased the expression levels of CSC markers and
affected their function; thus, this may directly impact tumor
resistance and recurrence (Fig.
7).
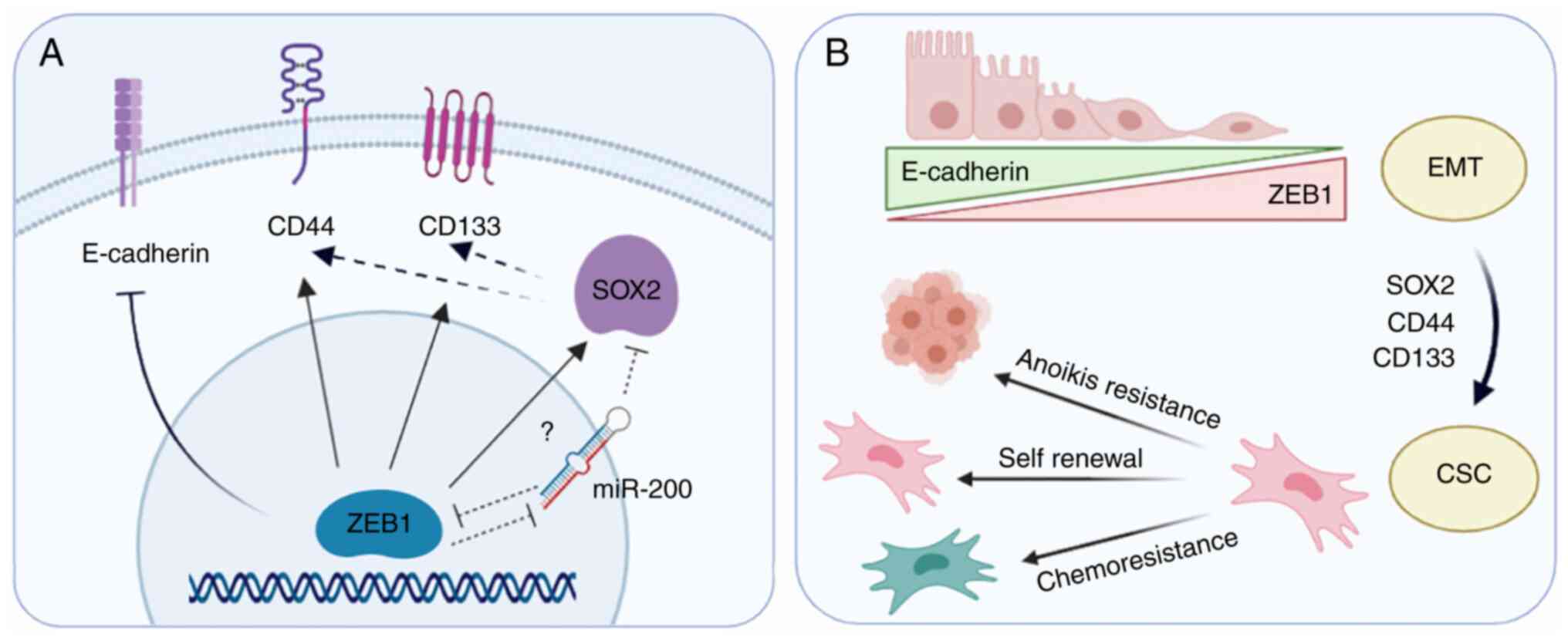 | Figure 7.Representation of the effect of ZEB1
expression on EMT and CSC markers in prostate cancer cells. (A)
ZEB1 transcription factor inhibits E-cadherin expression, inducing
EMT. ZEB1-knockdown downregulates the stemness transcription factor
SOX2 and decreases the expression levels of the prostate CSC
markers CD44 and CD133, indicating that ZEB1 may be promoting the
expression of these proteins directly (arrows) or indirectly
(dashed arrows). A possible mediator may be miR-200, as previous
studies (75,76) have shown that it is directly repressed
by ZEB1, and in turn, miR-200 directly represses SOX2. (B)
Upregulation of SOX2, CD44 and CD133 by ZEB1 may lead to a CSC
phenotype. Targeting ZEB1 with small interfering RNAs may reverse
this process, decreasing anoikis resistance, self-renewal capacity
and chemoresistance of androgen-independent prostate cancer cells.
EMT, epithelial-mesenchymal transition; CSC, cancer stem cell; miR,
microRNA; ZEB1, zinc finger E-box-binding homeobox 1. |
In conclusion, knocking down ZEB1 in aggressive PCa
cells decreased the expression levels of the CSC markers CD44 and
CD133, and of the transcription factor SOX2. Additionally, compared
with the control cells, cells with ZEB1-knockdown exhibited a lower
capacity for anchorage-independent growth and self-renewal,
important characteristics for metastasis and recurrence. As a
future therapy, targeting ZEB1 may reprogram CSCs into non-CSCs,
decreasing their number within a tumor, and therefore improving the
response to therapy and prognosis of patients with advanced
PCa.
Acknowledgements
The authors would like to thank Ms. Graciela Caroca
and Ms. Catherine Gatica from the Laboratory of Cellular and
Molecular Oncology, Department of Basic and Clinical Oncology,
University of Chile (Santiago, Chile), for their technical
assistance.
Funding
The present study was supported by grants from the
FondoNacional de Ciencia y Tecnología (grant nos. 1151214 and
1201704) and U-Redes (grant no. 007/17).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GP and FLM designed and performed the experiments,
conducted the statistical analysis and wrote the manuscript. SI
participated in the design, experimental work and data analysis of
LNCaP cells. MJT analyzed the data. EAC and HRC conceived the
study, participated in its design and coordination, wrote the
manuscript and are responsible for confirming the authenticity of
the data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global Cancer Statistics 2018: GLOBOCAN
Estimates of Incidence and Mortality Worldwide for 36 Cancers in
185 Countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fakhrejahani F, Madan RA and Dahut WL:
Management options for biochemically recurrent prostate cancer.
Curr Treat Options Oncol. 18:262017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang K, Ruan H, Xu T, Liu L, Liu D, Yang
H, Zhang X and Chen K: Recent advances on the progressive mechanism
and therapy in castration-resistant prostate cancer. Onco Targets
Ther. 11:3167–3178. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mottet N, Bellmunt J, Bolla M, Briers E,
Cumberbatch MG, De Santis M, Fossati N, Gross T, Henry AM, Joniau
S, et al: EAU-ESTRO-SIOG guidelines on prostate cancer. Part 1:
Screening, diagnosis, and local treatment with curative intent. Eur
Urol. 71:618–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cornford P, Bellmunt J, Bolla M, Briers E,
De Santis M, Gross T, Henry AM, Joniau S, Lam TB, Mason MD, et al:
EAU-ESTRO-SIOG guidelines on prostate cancer. Part II: Treatment of
relapsing, metastatic, and castration-resistant prostate cancer.
Eur Urol. 71:630–642. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chandrasekar T, Yang J, Gao A and Evans
CP: Mechanisms of resistance in castration-resistant prostate
cancer (CRPC). Transl Androl Urol. 4:365–380. 2015.PubMed/NCBI
|
|
7
|
Yun EJ, Lo UG and Hsieh JT: The evolving
landscape of prostate cancer stem cell: Therapeutic implications
and future challenges. Asian J Urol. 3:203–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj
K, Zhang D, Liu B, Jeter C, Calhoun-Davis T, et al: Defining a
population of stem-like human prostate cancer cells that can
generate and propagate castration-resistant prostate cancer. Clin
Cancer Res. 22:4505–4516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng Q and Tang DG: Androgen receptor and
prostate cancer stem cells: Biological mechanisms and clinical
implications. Endocr Relat Cancer. 22:T209–T220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Di Zazzo E, Galasso G, Giovannelli P, Di
Donato M, Di Santi A, Cernera G, Rossi V, Abbondanza C, Moncharmont
B, Sinisi AA, et al: Prostate cancer stem cells: The role of
androgen and estrogen receptors. Oncotarget. 7:193–208. 2015.
View Article : Google Scholar
|
|
11
|
Ojo D, Lin X, Wong N, Gu Y and Tang D:
Prostate cancer stem-like cells contribute to the development of
castration-resistant prostate cancer. Cancers (Basel). 7:2290–2308.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peitzsch C, Tyutyunnykova A, Pantel K and
Dubrovska A: Cancer stem cells: The root of tumor recurrence and
metastases. Semin Cancer Biol. 44:10–24. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsao T, Beretov J, Ni J, Bai X, Bucci J,
Graham P and Li Y: Cancer stem cells in prostate cancer
radioresistance. Cancer Lett. 465:94–104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Contreras HR, López-Moncada F and
Castellón EA: Cancer stem cell and mesenchymal cell cooperative
actions in metastasis progression and hormone resistance in
prostate cancer: Potential role of androgen and
gonadotropin-releasing hormone receptors. Int J Oncol.
56:1075–1082. 2020.PubMed/NCBI
|
|
15
|
Castellón EA, Valenzuela R, Lillo J,
Castillo V, Contreras HR, Gallegos I, Mercado A and Huidobro C:
Molecular signature of cancer stem cells isolated from prostate
carcinoma and expression of stem markers in different Gleason
grades and metastasis. Biol Res. 45:294–305. 2012. View Article : Google Scholar
|
|
16
|
Castillo V, Valenzuela R, Huidobro C,
Contreras HR and Castellon EA: Functional characteristics of cancer
stem cells and their role in drug resistance of prostate cancer.
Int J Oncol. 45:985–994. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carnero A, Garcia-Mayea Y, Mir C, Lorente
J, Rubio IT and LLeonart ME: The cancer stem-cell signaling network
and resistance to therapy. Cancer Treat Rev. 49:25–36. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Najafi M, Mortezaee K and Majidpoor J:
Cancer stem cell (CSC) resistance drivers. Life Sci.
234:1167812019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steinbichler TB, Dudás J, Skvortsov S,
Ganswindt U, Riechelmann H and Skvortsova II: Therapy resistance
mediated by cancer stem cells. Semin Cancer Biol. 53:156–167. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mitra A, Mishra L and Li S: EMT, CTCs and
CSCs in tumor relapse and drug-resistance. Oncotarget.
6:10699–10710. 2015. View Article : Google Scholar
|
|
21
|
Leão R, Domingos C, Figueiredo A, Hamilton
R, Tabori U and Castelo-Branco P: Cancer stem cells in prostate
cancer: Implications for targeted therapy. Urol Int. 99:125–136.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Packer JR and Maitland NJ: The molecular
and cellular origin of human prostate cancer. Biochim Biophys Acta.
1863:1238–1260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun Y, Wang BE, Leong KG, Yue P, Li L,
Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, et al: Androgen
deprivation causes epithelial-mesenchymal transition in the
prostate: Implications for androgen-deprivation therapy. Cancer
Res. 72:527–36. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuşoğlu A and Biray Avcı Ç: Cancer stem
cells: A brief review of the current status. Gene. 681:80–85. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adamowicz J, Pakravan K, Bakhshinejad B,
Drewa T and Babashah S: Prostate cancer stem cells: From theory to
practice. Scand J Urol. 51:95–106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lan L, Luo Y, Cui D, Shi BY, Deng W, Huo
LL, Chen HL, Zhang GY and Deng LL: Epithelial-mesenchymal
transition triggers cancer stem cell generation in human thyroid
cancer cells. Int J Oncol. 43:113–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eun K, Ham SW and Kim H: Cancer stem cell
heterogeneity: Origin and new perspectives on CSC targeting. BMB
Rep. 50:117–125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nieto MA, Huang RYYJ, Jackson RAA and
Thiery JPP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang J, Tian XJ and Xing J: Signal
Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and
Their Crosstalks. J Clin Med. 5:412016. View Article : Google Scholar
|
|
31
|
Sánchez-Tilló E, Liu Y, De Barrios O,
Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A
and Postigo A: EMT-activating transcription factors in cancer:
Beyond EMT and tumor invasiveness. Cell Mol Life Sci. 69:3429–3456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Goossens S, Vandamme N, Van Vlierberghe P
and Berx G: EMT transcription factors in cancer development
re-evaluated: Beyond EMT and MET. Biochim Biophys Acta Rev Cancer.
1868:584–591. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang P, Sun Y and Ma L: ZEB1: At the
crossroads of epithelial-mesenchymal transition, metastasis and
therapy resistance. Cell Cycle. 14:481–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lazarova D and Bordonaro M: ZEB1 mediates
drug resistance and EMT in p300-deficient CRC. J Cancer.
8:1453–1459. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y,
Zhang J, Yuan J, Wang M, Chen D, Sun Y, et al: ATM-mediated
stabilization of ZEB1 promotes DNA damage response and
radioresistance through CHK1. Nat Cell Biol. 16:864–875. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo C, Ma J, Deng G, Qu Y, Yin L, Li Y,
Han Y, Cai C, Shen H and Zeng S: ZEB1 promotes oxaliplatin
resistance through the induction of epithelial-mesenchymal
transition in colon cancer cells. J Cancer. 8:3555–3566. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Orellana-serradell O, Herrera D, Castellón
EA and Contreras HR: The transcription factor ZEB1 promotes
chemoresistance in prostate cancer cell lines. Asian J Androl.
21:460–467. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Orellana-Serradell O, Herrera D, Castellón
EA and Contreras HR: The transcription factor ZEB1 promotes an
aggressive phenotype in prostate cancer cell lines. Asian J Androl.
20:294–299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Farfán N, Ocarez N, Castellón EA, Mejía N,
de Herreros AG and Contreras HR: The transcriptional factor ZEB1
represses Syndecan 1 expression in prostate cancer. Sci Rep.
8:114672018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stone KR, Mickey DD, Wunderli H, Mickey GH
and Paulson DF: Isolation of a human prostate carcinoma cell line
(DU 145). Int J Cancer. 21:274–281. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Horoszewicz JS, Leong SS, Kawinski E, Karr
JP, Rosenthal H, Chu TM, Mirand EA and Murphy GP: LNCaP model of
human prostatic carcinoma. Cancer Res. 43:1809–1818.
1983.PubMed/NCBI
|
|
42
|
Krasnov GS, Kudryavtseva AV, Snezhkina AV,
Lakunina VA, Beniaminov AD, Melnikova NV and Dmitriev AA:
Pan-cancer analysis of TCGA data revealed promising reference genes
for qPCR normalization. Front Genet. 10:972019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Barrandon Y and Green H: Three clonal
types of keratinocyte with different capacities for multiplication.
Proc Natl Acad Sci USA. 84:2302–2306. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Acikgoz E, Guven U, Duzagac F, Uslu R,
Kara M, Soner BC and Oktem G: Enhanced G2/M arrest, caspase related
apoptosis and reduced E-cadherin dependent intercellular adhesion
by trabectedin in prostate cancer stem cells. PLoS One.
10:e01410902015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang S: Anchorage-independent growth of
prostate cancer stem cells. Methods Mol Biol. 568:151–160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sobel RE and Sadar MD: Cell lines used in
prostate cancer research: A compendium of old and new lines-Part 1.
J Urol. 173:342–359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yu Z, Pestellc TG, Lisantic MP and Pestell
RG: Cancer Stem Cells. Int J Biochem Cell Biol. 44:2144–2151. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Johnston MD, Maini PK, Jonathan Chapman S,
Edwards CM and Bodmer WF: On the proportion of cancer stem cells in
a tumour. J Theor Biol. 266:708–711. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Collins AT, Berry PA, Hyde C, Stower MJ
and Maitland NJ: Prospective identification of tumorigenic prostate
cancer stem cells. Cancer Res. 65:10946–10951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ajani JA, Song S, Hochster HS and
Steinberg IB: Cancer stem cells: The promise and the potential.
Semin Oncol. 42 (Suppl 1):S3–S17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jolly MK and Celià-Terrassa T: Dynamics of
phenotypic heterogeneity during EMT and stemness in cancer
progression. J Clin Med. 8:15422019. View Article : Google Scholar
|
|
53
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hanrahan K, O'Neill A, Prencipe M, Bugler
J, Murphy L, Fabre A, Puhr M, Culig Z, Murphy K and Watson RW: The
role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in
mediating docetaxel-resistant prostate cancer. Mol Oncol.
11:251–265. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Krebs AM, Mitschke J, Lasierra Losada M,
Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D,
Reichardt W, Bronsert P, et al: The EMT-activator Zeb1 is a key
factor for cell plasticity and promotes metastasis in pancreatic
cancer. Nat Cell Biol. 19:518–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang S and Cui W: Sox2, a key factor in
the regulation of pluripotency and neural differentiation. World J
Stem Cells. 6:305–311. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Adachi K, Suemori H, Yasuda SY, Nakatsuji
N and Kawase E: Role of SOX2 in maintaining pluripotency of human
embryonic stem cells. Genes Cells. 15:455–470. 2010.PubMed/NCBI
|
|
58
|
Ghaleb AM and Yang VW: Krüppel-like factor
4 (KLF4): What we currently know. Gene. 611:27–137. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang P, Andrianakos R, Yang Y, Liu C and
Lu W: Kruppel-like factor 4 (Klf4) prevents embryonic stem (ES)
cell differentiation by regulating Nanog gene expression. J Biol
Chem. 285:9180–9189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
An Z, Liu P, Zheng J, Si C, Li T, Chen Y,
Ma T, Zhang MQ, Zhou Q and Ding S: Sox2 and Klf4 as the functional
core in pluripotency induction without exogenous Oct4. Cell Rep.
29:1986–2000,e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mamun MA, Mannoor K, Cao J, Qadri F and
Song X: SOX2 in cancer stemness: Tumor malignancy and therapeutic
potentials. J Mol Cell Biol. 12:85–98. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Russo MV, Esposito S, Tupone MG, Manzoli
L, Airoldi I, Pompa P, Cindolo L, Schips L, Sorrentino C and Di
Carlo E: SOX2 boosts major tumor progression genes in prostate
cancer and is a functional biomarker of lymph node metastasis.
Oncotarget. 7:12372–12385. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mu P, Zhang Z, Benelli M, Karthaus WR,
Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al: SOX2
promotes lineage plasticity and antiandrogen resistance in TP53-and
RB1-deficient prostate cancer. Science. 355:84–88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu X, Qiao B, Zhao T, Hu F, Lam AK and
Tao Q: Sox2 promotes tumor aggressiveness and
epithelial-mesenchymal transition in tongue squamous cell
carcinoma. Int J Mol Med. 42:1418–1426. 2018.PubMed/NCBI
|
|
65
|
Gao H, Teng C, Huang W, Peng J and Wang C:
SOX2 promotes the epithelial to mesenchymal transition of
esophageal squamous cells by modulating slug expression through the
activation of STAT3/HIF-α signaling. Int J Mol Sci. 16:21643–21657.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Herreros-Villanueva M, Zhang JS, Koenig A,
Abel EV, Smyrk TC, Bamlet WR, de Narvajas AA, Gomez TS, Simeone DM,
Bujanda L, et al: SOX2 promotes dedifferentiation and imparts stem
cell-like features to pancreatic cancer cells. Oncogenesis.
2:e612013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Srinivasan D, Senbanjo L, Majumdar S,
Franklin RB and Chellaiah MA: Androgen receptor expression reduces
stemness characteristics of prostate cancer cells (PC3) by
repression of CD44 and SOX2. J Cell Biochem. 120:2413–2428. 2019.
View Article : Google Scholar
|
|
68
|
Zhou W, Lv R, Qi W, Wu D, Xu Y, Liu W, Mou
Y and Wang L: Snail contributes to the maintenance of stem
cell-like phenotype cells in human pancreatic cancer. PLoS One.
9:e874092014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Deep G, Jain AK, Ramteke A, Ting H,
Vijendra KC, Gangar SC, Agarwal C and Agarwal R: SNAI1 is critical
for the aggressiveness of prostate cancer cells with low
E-cadherin. Mol Cancer. 13:372014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Celià-terrassa T, Meca-cortés Ó, Mateo F,
Martínez de Paz A, Rubio N, Arnal-Estapé A, Ell BJ, Bermudo R, Díaz
A, Guerra-Rebollo M, et al: Epithelial-mesenchymal transition can
suppress major attributes of human epithelial. J Clin Invest.
122:1849–1868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Anose BM and Sanders MM: Androgen receptor
regulates transcription of the ZEB1 transcription factor. Int J
Endocrinol. 2011:9039182011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mooney SM, Parsana P, Hernandez JR, Liu X,
Verdone JE, Torga G, Harberg CA and Pienta KJ: The presence of
androgen receptor elements regulates ZEB1 expression in the absence
of androgen receptor. J Cell Biochem. 116:115–23. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Chaffer CL, Marjanovic ND, Lee T, Bell G,
Kleer CG, Reinhardt F, D'Alessio AC, Young RA and Weinberg RA:
Poised chromatin at the ZEB1 promoter enables breast cancer cell
plasticity and enhances tumorigenicity. Cell. 154:61–74. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhou C, Jiang H, Zhang Z, Zhang G, Wang H,
Zhang Q, Sun P, Xiang R and Yang S: ZEB1 confers stem cell-like
properties in breast cancer by targeting neurogenin-3. Oncotarget.
8:54388–54401. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yu Z and Pestell RG: MicroRNAs and Cancer
Stem Cells. MicroRNAs in Cancer Translational Research. William
C.S.C: Springer; pp. 373–398. 2011, View Article : Google Scholar
|
|
76
|
Brabletz S, Bajdak K, Meidhof S, Burk U,
Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J
and Brabletz T: The ZEB1/miR-200 feedback loop controls Notch
signalling in cancer cells. EMBO J. 30:770–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Tan L, Sui X, Deng H and Ding M: Holoclone
forming cells from pancreatic cancer cells enrich tumor initiating
cells and represent a novel model for study of cancer stem cells.
PLoS One. 6:e233832011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhang L, Jiao M, Li L, Wu D, Wu K, Li X,
Zhu G, Dang Q, Wang X, Hsieh JT and He D: Tumorspheres derived from
prostate cancer cells possess chemoresistant and cancer stem cell
properties. J Cancer Res Clin Oncol. 138:675–686. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Knaack H, Lenk L, Philipp LM, Miarka L,
Rahn S, Viol F, Hauser C, Egberts JH, Gundlach JP, Will O, et al:
Liver metastasis of pancreatic cancer: The hepatic microenvironment
impacts differentiation and self-renewal capacity of pancreatic
ductal epithelial cells. Oncotarget. 9:31771–31786. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sohn HM, Kim B, Park M, Ko YJ, Moon YH,
Sun JM, Jeong BC, Kim YW and Lim W: Effect of CD133 overexpression
on bone metastasis in prostate cancer cell line LNCaP. Oncol Lett.
18:1189–1198. 2019.PubMed/NCBI
|
|
81
|
Bisson I and Prowse DM: WNT signaling
regulates self-renewal and differentiation of prostate cancer cells
with stem cell characteristics. Cell Res. 19:683–697. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chen B, Zhu Z, Li L, Ye W, Zeng J, Gao J,
Wang S, Zhang L and Huang Z: Effect of overexpression of oct4 and
sox2 genes on the biological and oncological characteristics of
gastric cancer cells. Onco Targets Ther. 12:4667–4682. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kanwal R, Shukla S, Walker E and Gupta S:
Acquisition of tumorigenic potential and therapeutic resistance in
CD133+ subpopulation of prostate cancer cells exhibiting stem-cell
like characteristics. Cancer Lett. 430:25–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hoofd C, Wang X, Lam S, Jenkins C, Wood B,
Giambra V and Weng AP: CD44 promotes chemoresistance in T-ALL by
increased drug efflux. Exp Hematol. 44:166–171.e17. 2016.
View Article : Google Scholar : PubMed/NCBI
|