Introduction
Cancer and Alzheimer's disease (AD) have become
leading causes of death worldwide (1). Previous studies have identified
relationships between cancer and AD in which the amyloid precursor
protein (APP) plays an important role. APP is a transmembrane
protein, source of β-amyloid aggregation which is one of the major
causes of AD, is expressed in various neuron cells and may be
involved in development of cells (2,3). In
cancer cells, it has been reported that APP is a primary
androgen-responsive gene, found in breast and prostate cancer; it
is also implicated in various human cancers including colon, lung,
breast, parathyroid, prostate, thyroid and breast cancers, and its
high immunoreactivity is related with poor prognoses for prostate
cancer and estrogen-receptor-positive breast cancer patients
(4–10). Therefore, APP may enhance the
growth and metastasis of prostate cancer cells by regulating the
expression of metalloproteinase and EMT-related genes (11). It has been observed that APP
increases expression and processing in pancreatic cancer. This
molecule also promotes growth in pancreatic cancer cells through
undergoing proteolytic processing to release a soluble NH2-terminal
ectodomain fragment (sAPP) (12).
Moreover, a relationship between reactive oxygen species (ROS) and
androgen receptor (AR) has been identified; as well as the
mechanisms of AR activation through oxidative stress including AR
overexpression, AR co-regulators or intracellular signal
transduction pathways, increasing of AR mutations or splice
variants, and de novo androgen synthesis. AR signaling
activated by oxidative stress may contribute to survival and
evading to apoptosis in prostate cancer cells in response to
androgen deprivation therapy (13). Prostate tumors characterized by
androgen receptor (AR) expression and signaling pathways in
processes of carcinogenesis, development, and progression (14). Conversely, androgen deprivation
therapy, which decreases androgen level, and interferes with AR
function, is gold-standard treatment of prostate cancer (15).
ROS, including superoxide
(O2−), hydrogen peroxide
(H2O2), and hydroxyl radicals (HO•) which are
produced by the partial reduction of oxygen, comprise an important
mechanism of AD. In the process of mitochondrial oxidative
phosphorylation, ROS are endogenously produced in the cells, or can
be generated exogenously from xenobiotic compounds when cellular
antioxidant defense system is overcome by increase in ROS or a
decrease in cellular antioxidant ability. The oxidative stress can
induce the damage of biomolecules (nucleic acids, proteins and
lipids) which is a leading cause of various disorders including
carcinogenesis (16),
neurodegenerative diseases (17),
atherosclerosis, diabetes (18),
and aging (19). Moreover,
oxidative stress can regulate various cellular reactions, including
AR signaling, through direct or indirect reactions (20). Oxidative stress has been shown to
be involved in the tumorigenesis and transformation of prostate
cancer (21–23) as well as in the conversion of
androgen-dependent prostate cancer into CRPC (22,24,25).
Together, these results indicated the cross-talk relation between
oxidative stress and AR signaling.
Transforming growth factor-β (TGF-β)-activated
kinase 1 (TAK1), is a serine/threonine kinase in the
mitogen-activated protein kinase (MAP3K) family. TAK1 is the
central core for various signaling pathways and it was originally
recognized as a transforming growth factor-β-activated kinase and
was demonstrated to phosphorylate and activate various downstream
target proteins and promote cancer. After stimulation by specific
ligands, IL and TGFβ receptors enhance the activation of TRAF6, and
E3 ubiquitin ligase mediates the activation of TAK1. Active TAK1
mediates the processes of cancer cells including proliferation,
survival and resistance to chemotherapy through NF-κB activation,
triggering additional signaling pathways including p38, JNK, and
acting on different transcription factors (TFs). Previous studies
demonstrated that targeting the TAK1 kinase activity dramatically
induced apoptosis and increased sensitivity to chemotherapy and
radiotherapy of cancer cells (26–29).
Glycogen synthase kinase (GSK)3 is a serine/threonine kinase that
consists of 2 genes, GSK-3α and GSK-3β. GSK-3 has been involved in
a number of human cancers, including pancreatic cancer. In
distinct, a recent study demonstrated that GSK-3α interacts with
TAK1 which stabilizes the TAK1-TAB complex. This enhances
noncanonical NF-kB activation in pancreatic cancer cells. The
suppression of GSK-3 results in a significant reduction of TAK1
levels. Different from other kinases, when dephosphorylated GSK-3β
which is active form, promotes inflammation and apoptosis. In
opposition, increased phosphorylation reduced GSK-3β activity.
GSK-3β suppression has beneficial effects on memory in other
disease models. GSK-3β controls TAK1 pathways. Suppression of
GSK-3β was neuroprotective and ameliorated stroke-induced cognitive
impairments. TAK1 is an upstream regulator of GSK-3β. Targeting
GSK-3β could be a novel therapeutic strategy for cognitive deficits
(30).
Clausena harmandiana (C. harmandiana)
or ‘Song fa’ in Thai is a medicinal plant, used for the treatment
of headaches, and illness stomachaches (31). It has been found that the roots of
C. harmandiana plant consist of carbazole and coumarins
alkaloids (32). Carbazoles and
coumarins alkaloids have been isolated and evaluated for their
pharmacological activities including, antimalarial,
anti-tuberculosis, and antifungal properties. In a previous study
by the authors, it was revealed that the major components in C.
harmandiana were heptaphylline and 7-methoxyheptaphylline
(7-MH), which exhibited anticancer activities against NCI-H187 and
KB cell lines (33). Moreover,
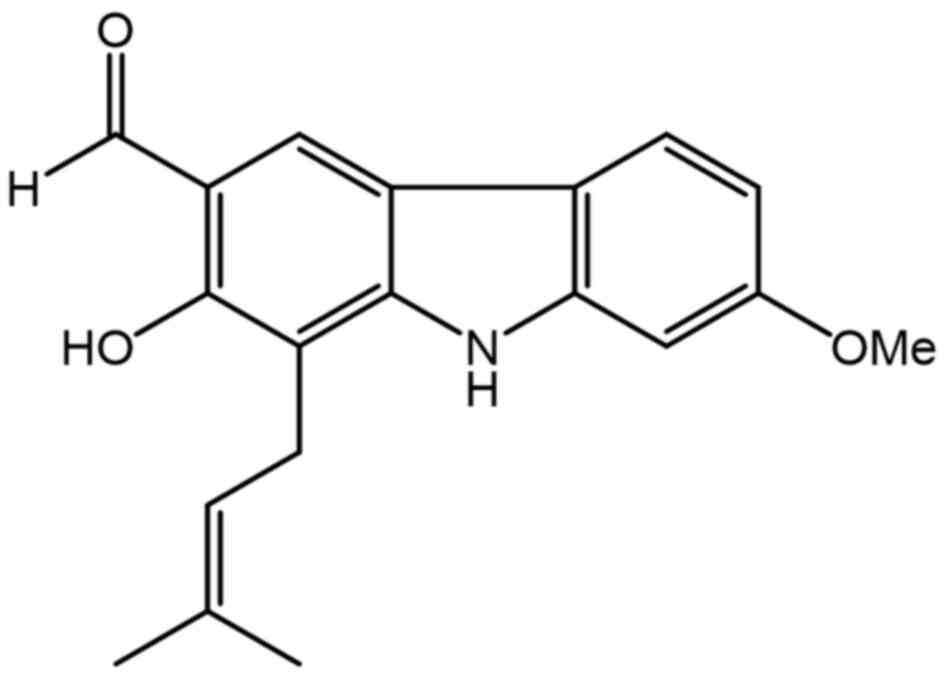
7-MH (Fig. 1) showed a
neuroprotective effect against H2O2-induced
cell death of NG108-15 cells (34). In order to search for new drug for
cancer and anti-AD prevention and treatment with high efficacy, low
toxicity, and decrease side effects, the present study investigated
the antiapoptotic effects of H2O2-induced
oxidation of 7-MH on SH-SY5Y neuroblastoma cells and the apoptotic
effects of 7-MH on SH-SY5Y neuroblastoma cells and LNCaP prostate
cancer cells. To clarify the mechanism of action of 7-MH, the
effect of 7-MH on signaling proteins involved in the TAK1-mediated
apoptosis pathway including GSK-3, MAPK13, anti-apoptotic proteins
and pro-apoptotic proteins in cancer and Alzheimer's model, was
investigated.
Materials and methods
Cell culture
SH-SY5Y (neuroblastoma cell line) (CRL-2266), HepG2
(liver cancer cells) (HB-8065), HT29 (colorectal cancer cells)
(HTB-38), and 4T1 (breast cancer cells) (CRL-2539) were purchased
from the American Type Culture Collection (ATCC) and authenticated
using short tandem repeat analysis (also conducted by the ATCC).
The cells were maintained in Eagle's minimum essential medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Amresco, LLC), 100 units/ml penicillin and 100 µg/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in 5%
CO2. LNCaP cell were maintained in RPMI-1640 (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal calf
serum, 100 units/ml penicillin, and 100 µg/ml streptomycin at 37°C
in 5% CO2.
Cell cytotoxicity assay
SH-SY5Y cells, HepG2, HT29, 4T1, and LNCaP cells
were plated in 96-well microplates at 4×105 cells/wells
and then incubated for 48 h. Cells were treated with 7-MH at
different concentrations and reference compound for 24, 48 and 72
h. Then, 10 µl of MTT reagent (5 mg/ml) (Sigma-Aldrich; Merck KGaA)
was added. The cells were incubated for 2 h until purple
precipitate was visible after addition of dimethyl sulfoxide. The
absorbance at 570 nm was measured. The percentage calculation of
cell viability was carried out using the following formula: % Cell
viability=(Absorbance of treated cells ×100)/Absorbance of control
(untreated cells). Cell morphology was examined using a
phase-contrast microscope by having 36 µM doxorubicin
(Sigma-Aldrich) as positive control.
Neuroprotective effect
SH-SY5Y cells were plated in 96-well microplates at
a density 4×105 cells/wells and then incubated for 48 h.
The cells were treated with various concentrations of 7-MH or 100
µM NAC for 2 h. Then, the cells were treated with 250 µM
H2O2 for 4 h to induce oxidative stress. Cell
viability was determined by MTT colorimetry. Absorbance was
measured at 570 nm.
Fluorescence-activated cell-sorting
(FACS) analysis
Apoptosis occurs as a result of G0/G1 phase arrest.
Apoptosis as a protective mechanism ensures homeostasis of host
cells through cell shrinkage, fragmentation of cellular DNA and
formation of ‘apoptotic bodies’ subsequently leading to cell death.
For cell cycle analysis, the cells were treated with various
concentrations of 7-MH for 2 h. Then, the cells were treated with
H2O2 for 4 h to induce oxidative stress. The
cells were fixed by ethanol for 2 h at 4°C and stained with 50
mg/ml propidium iodide (PI) for 30 min in the presence of RNase
before analysis. The percentage of apoptotic cells was quantitated
using an FACScan flow cytometer with BD FACSDivaTM software (v.
6.1.3) (BD FACSAria; BD Biosciences). Late apoptotic cells were
distinguished from non-apoptotic, intact cells by their decreased
DNA content, which was determined by their low PI staining
intensity.
Preparation of cell extracts
In order to investigate the mechanisms of
interaction with the apoptotic pathway in cancer cells, the cells
were plated in six-well plates at a density of 1×106
cells/wells and then incubated for 48 h. The cells were treated
with various concentrations of 7-MH at 30 min, and cell death was
induced with H2O2 at 15 min for SH-SY5Y
cells. In HepG2, HT29, 4T1, and LNCaP cells, the cells were treated
with various concentrations of 7-MH at the indicated time.
Whole-cell lysates were prepared with lysis buffer [25 mM HEPES, pH
7.7, 0.3 mM MgCl2, 0.2 mM EDTA, 10% Triton X-100, 20 mM
β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM
phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), 10
µg/ml aprotinin, and 10 µg/ml leupeptin] (Gibco; Thermo Fisher
Scientific, Inc.). The cell lysates were collected from the
supernatant after centrifugation at 2,500 × g for 10 min 4°C.
Immunoblotting
The total protein concentration was measured by
using the Bradford dye-binding method (Bio-Rad). The cell lysates
(15 µl)were loaded and resolved by 7.5-12.5% SDS-PAGE and
transferred to an Immobilon-P-nylon membrane (MilliporeSigma). The
membrane was treated with BlockAcc (Dainippon Phamaceutical Co.,
Ltd.) and probed at room temperature for 2 h with the following
primary antibodies: anti-caspase-3 (cat. no. 9662), GSK-3 (cat. no.
5558), phospho-p38 (cat. no. 4511), p38 (cat. no. 54470), Mcl-1
(cat. no. 94296), Bcl-xL (cat. no. 2764), BAX (cat. no. 5023),
phospho-Akt (cat. no. 4060), Akt (cat. no. 4691), phospho-ERK (cat.
no. 9911), phospho-P65 (cat. no. 3031), P65 (cat. no. 3033), Bcl-2
(cat. no. 4223), survivin (cat. no. 2808), MAPK13 (cat. no. 4511),
and anti-actin antibodies (cat. no. 3700), all diluted at 1:1,000
and obtained from Cell Signaling Technology, Inc. The antibodies
were detected using horseradish peroxidase-conjugated anti-rabbit
(cat. no. 14708), anti-mouse (cat. no. 14709), and anti-goat IgG
(cat. no. 98164) secondary antibodies (1: 5,000; Cell Signaling
Technology, Inc.) and visualized using the enhanced
chemiluminescence system (Life Science Technology). Densitometric
analysis of western blot bands was performed using ImageJ software
(version IJ 1.46 r; National Institutes of Health.).
Migration and invasion assays
The cancer cell migration and invasion capacities
were determined using a Transwell assay. A total of
1×106 cells/well in serum-free DMEM were added into the
top chambers of a 24-well insert (pore size, 8-mm; Corning Life
Sciences) and incubated for 4 h to allow the cells to attach. The
cells were treated with candidone and incubated for 24 h at 37°C.
For the invasion assays, the membranes were pre-coated with
collagen for 4 h at 37°C. The lower chambers were filled with DMEM
supplemented with 10% fetal bovine serum as a chemoattractant.
After 4 h, the migratory/invasive cells were fixed with a 10%
formalin solution at room temperature for 30 min and then stained
with 0.05% crystal violet solution at room temperature for 30 min,
after which the stained cells were counted under an inverted light
microscope (magnification, ×20).
Luciferase assay
4T1 cells were transfected with a luciferase
reporter plasmid (Promega Corporation) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) under the control of NF-κB and STAT3 sites
containing a neo-resistance gene. The luciferase activity of
transfected cells was compared with 4T1 CMV control cell (the 4T1
cells stably expressing luciferase with a CMV-promoter). A stable
clone was isolated in medium containing 500 µg/ml G418 (Thermo
Fisher Scientific, Inc.). Cells were seeded in a 96-well plate and
treated with DMSO (control) and 0.1, 1, 10, or 100 µM of 7-MH for
24 h and compared with resveratrol as a positive control.
Luciferase activity was measured using the Dual-Luciferase reporter
assay system (Promega Corporation).
Animal model
Female BALB/c mice (6 weeks old) were separated into
groups of 6–7 mice at temperature and humidity of 23±2°C and
50±10%, respectively, and were administered food and water ad
libitum. The experiment was carried out according to the
standard guidelines of the National Institutes of Health and was
approved (approval no. A2017INM-7) by the Animal Care and Use
Committee of the University of Toyama (Toyama, Thailand). The lung
metastasis model was performed by culturing mouse mammary carcinoma
4T1 cells with the luciferase gene (4T1-Luc2 cells), and then the
cells were harvested and resuspended in cold phosphate-buffered
saline (PBS). The cell suspensions (5×105/50 µl/mouse)
were inoculated in mice by intravenous injection (the number of
mice was n=6 each group and repeated twice so at least 24 mice as
total from 30 prepared mice). After 6 days, D-luciferin (150 mg/kg)
was injected, and 20 min later, the lungs were harvested from the
mice. Lung luminescence was then determined using an imaging system
(IVIS Spectrum; Caliper Life Science). The method of euthanasia was
cervical dislocation and death was confirmed before disposal of the
animal by evaluating consciousness including lack of a heartbeat,
lack of respiration, lack of corneal reflex and presence of rigor
mortis. In accordance with the Guide for the Care and use of
Laboratory Animals of the National Institutes of Health, the humane
endpoint was set based on the percentage of body weight loss (20%
body-weight reduction).
Molecular docking study
The TAK1 kinase template was prepared from 4GS6 and
validated by redocking with the irreversible inhibitor
(5Z)-7-oxozeaenol. All hydrogens were added, water molecules were
deleted, and Gasteiger charges were assigned to the TAK1 kinase
template and all ligands by using AutoDockTools (ADT). The AutoGrid
was used to generate grid maps. The grids were designated to
include the active site of TAK1 kinase. The grid box dimensions
were defined as 100×100×100 Å, and the grid spacing was set to
0.375 Å. All ligands were docked using the Lamarckian genetic
algorithm via the Autodock 4.2.6 auxiliary program. The Lamarckian
genetic algorithm protocol was set to a population size of 150
individuals with 150 ligand orientation runs. Additionally, the
energy evaluation was 1,000,000, which was as the maximum number of
evaluations. The docking complex poses were analyzed for their
interactions by using BIOVIA Discovery Studio 2017.
Statistical analysis
The statistical technique used for the analysis was
a one-way analysis of variance (ANOVA) followed by Tukey's post hoc
test for comparison between 2 groups and between 3 or more groups.
The data were analyzed using SPSS software (version 24; IBM Corp.).
The analysis was performed in triplicate, and the values are
presented as the mean ± SDs. P<0.05 was considered to indicate a
statistically significant difference.
Results
The neuroprotective effect of 7-MH on
H2O2-induced apoptosis in SH-SY5Y cells via
GSK-3
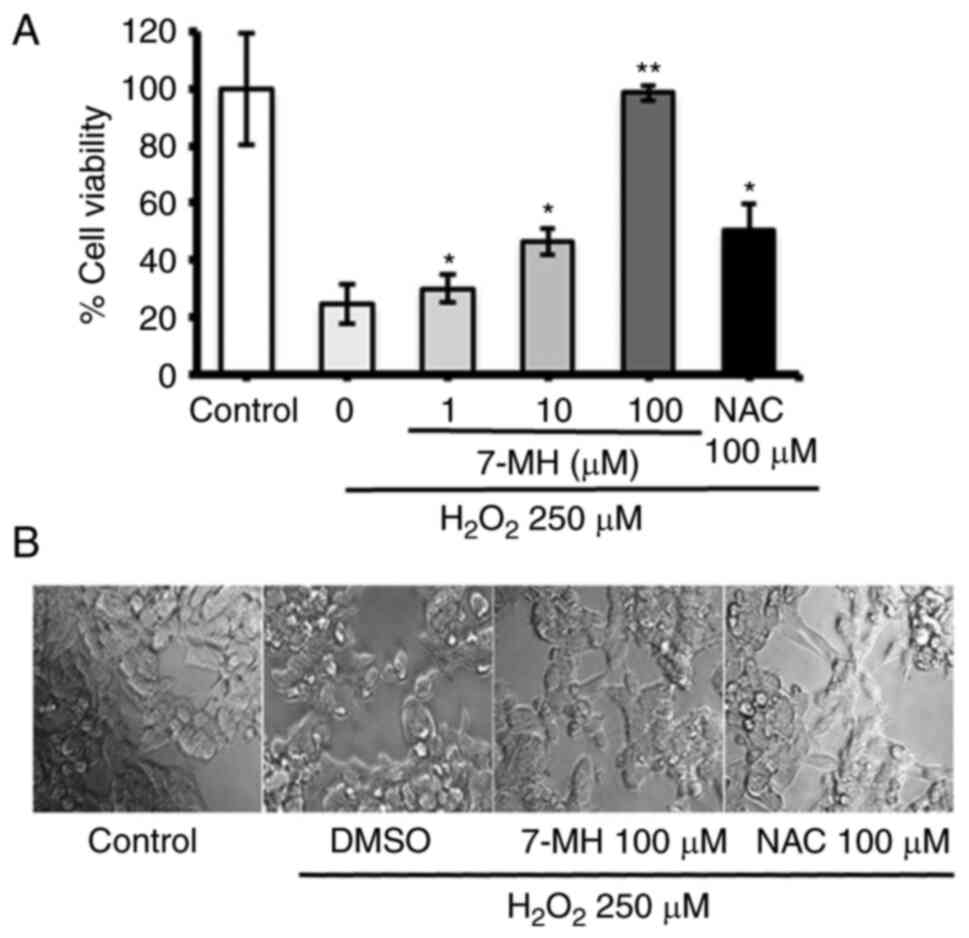
To investigate the effect of 7-MH on
H2O2-induced neuronal cell death, neuronal
cells were treated with various concentrations of 7-MH or 100 µM
NAC (reference compound) for 2 h before switching to 250 µM H2O2
for 4 h. Cell viability was assessed using MTT assay. The results
showed significantly increased cell viability when compared with
H2O2-insulted samples. The values obtained
with 7-MH treatment at a concentration of 100 µM showed a stronger
neuroprotective effect than that achieved by NAC treatment
(Fig. 2A). A decrease in
morphologically-confirmed cell death was observed as a result of
7-MH, compared with 100 µM NAC (Fig.
2B). In consistency with previous findings, 7-MH showed a
neuroprotective effect on NG108-15 cells (mouse neuroblastoma and
rat glioma cell lines) (35).
To verify the 7-MH inhibition of apoptosis by
H2O2, the cells were labeled with PI and
analyzed using flow cytometry (There was a limitation to stain with
Annexin V). The results revealed the DNA content histograms
obtained after the PI staining of cells that had been treated with
various concentrations of 7-MH for 2 h and had been eliminated by
treatment with an H2O2 concentration of 250
µM for 4 h. When the cells were incubated in the medium alone
(control), a single peak of nuclei with diploid DNA content was
observed. By contrast, when the cells were incubated in
H2O2 alone (negative control), an increase in
apoptotic cells in sub-G0/G1 was observed.
When the cells were incubated in NAC and H2O2
(positive control), the results were similar to those of the
control group. In the presence of 7-MH, inhibiting apoptosis with
H2O2, apoptotic cells with increased DNA
content were distinguishable. A characteristic hypodiploid DNA
content peak, which shows sub-G0/G1 apoptotic
populations, was observed following the treatment of neuronal cells
with 7-MH in a concentration-dependent manner (Fig. 3).
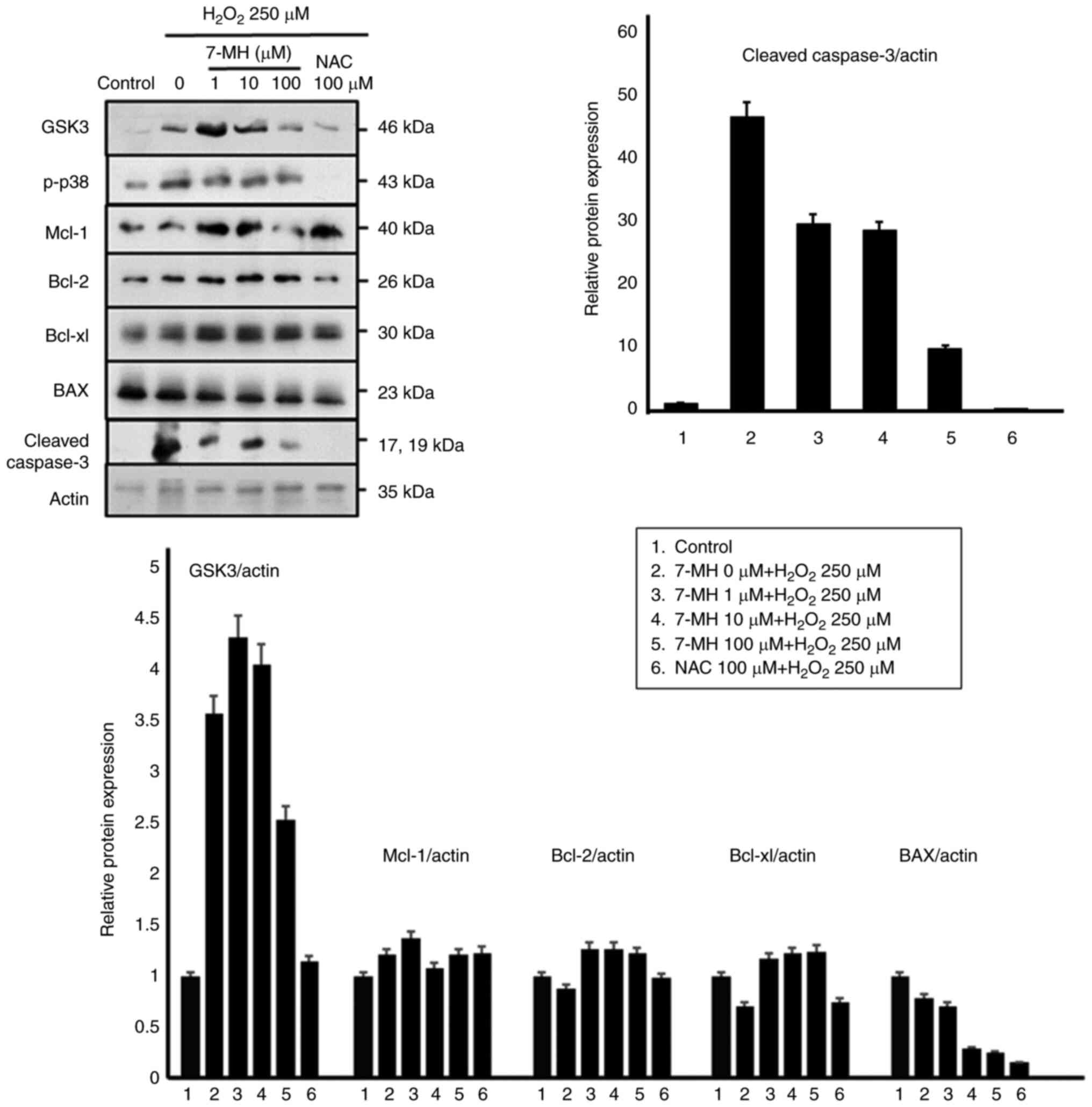
To further evaluate the protective molecular
mechanisms of 7-MH in neuronal cells, the cells were treated with
various concentrations of 7-MH for 2 h, and cell death was induced
with H2O2 for 4 h. Key proteins involved in
apoptosis regulation were examined, including GSK-3, p-p38, Mcl-1,
Bcl-2, and BAX, using an immunoblot assay. As demonstrated in
Fig. 4, 7-MH markedly inhibited
p-p38, BAX, and cleaved caspase-3 compared with the control; and
induced Mcl-1, Bcl-2, and Bcl-xL in a concentration-dependent
manner. The results indicated that 7-MH efficiently inhibits the
apoptotic effect of H2O2 induced in neuronal
cells.
7-MH induces apoptosis in SH-SY5Y
cells via GSK-3
In order to elucidate the molecular mechanism of
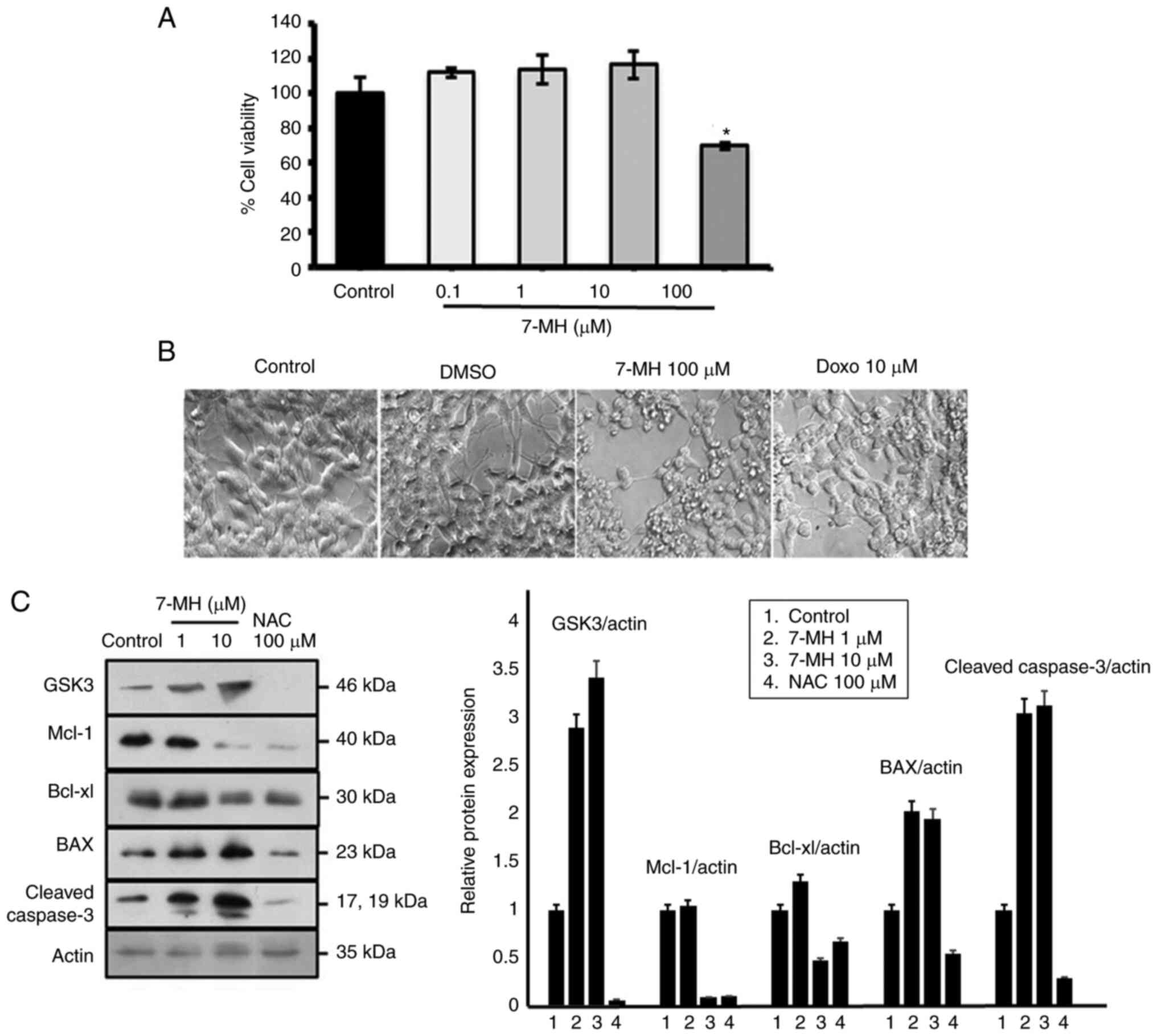
cancer cellapoptosis, the GSK-3 signaling pathways were assessed.
Neuroblastoma cells were treated with various concentrations of
7-MH for 24 h. Cell viability was assessed using MTT assays. These
results revealed that 7-MH at a concentration of 100 µM
significantly induced cancer cell death with morphological changes,
including cell rounding, shrinkage, and detachment (Fig. 5A and B). 7-MH activated the
cleaving of caspase-3 by increasing the level of Bax and decreasing
the levels of Mcl-1 and Bcl-xl, which are regulated by GSK-3
(Fig. 5C). This indicated that
7-MH induced apoptosis in SH-SY5Y cells via the GSK-3 pathway.
7-MH induces cell death and inhibits
migration and invasion of HT29 cancer cells
To test the effect of 7-MH on cancer migration and
invasion, HT29 and HepG2 cancer cells were treated with various
concentrations of 7-MH for 24 h. Cell viability was assessed using
an MTT assay. The results showed that 7-MH at a concentration of
100 µM significantly induced cancer cell death in a time-dependent
manner, due to its effect on HT29 being more potent than on HepG2
cells (Fig. 6A). The morphological
changes, including cell rounding, shrinkage, and detachment, were
observed in cells treated with 100 µM of 7-MH compared with 36 µM
doxorubicin as a positive control (Fig. 6B). Moreover, 7-MH at concentrations
of 1 and 10 µM inhibited the migration and invasion of HT29 cancer
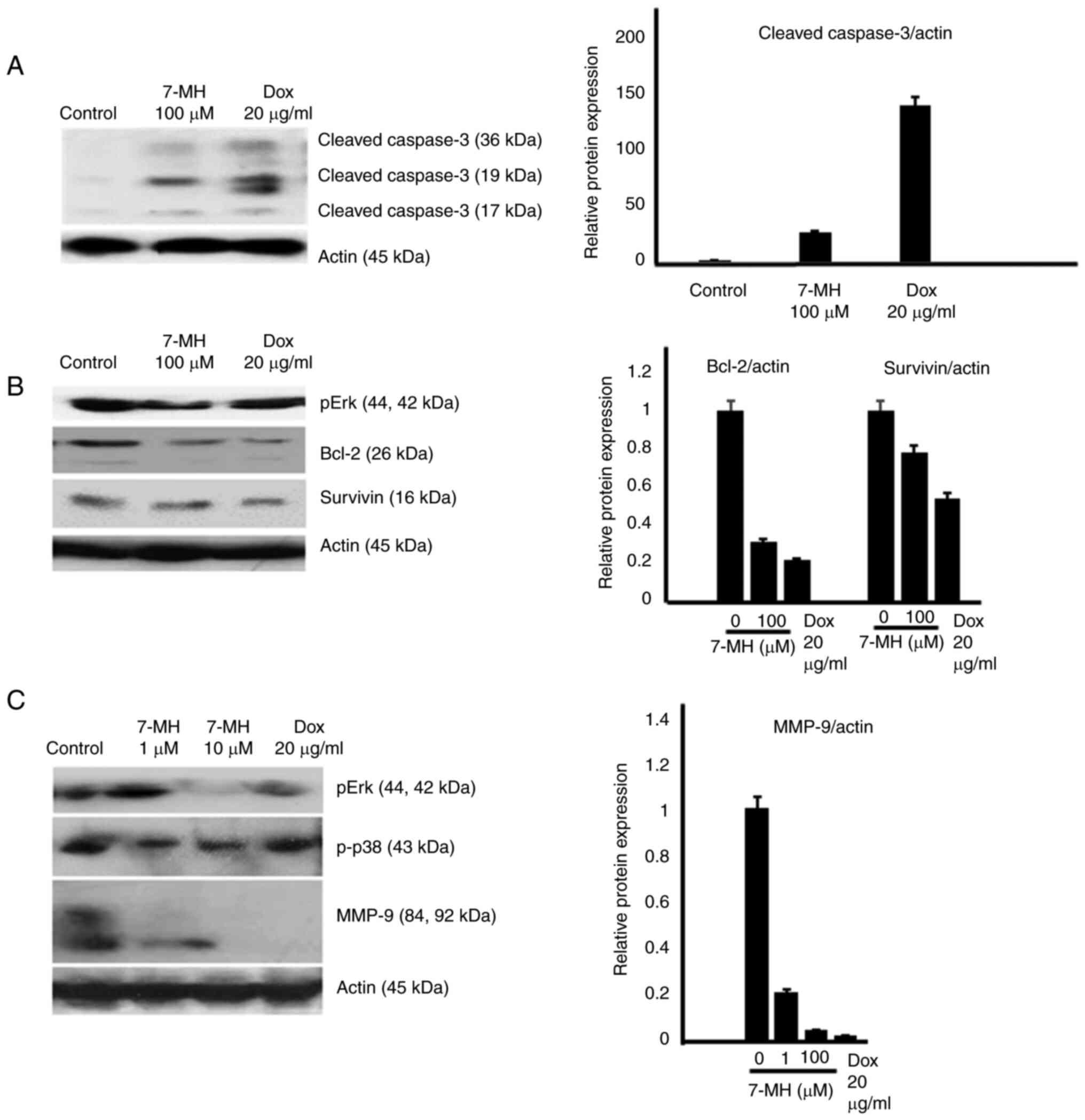
cells (Fig. 7A and B). The western
blot results revealed that 7-MH activated the cleaving of caspase-3
(marker of apoptosis) and decreased the levels of phospho-Erk,
phospho-p38, Bcl-2, survivin, and matrix metalloproteinase-9
(marker of metastasis) (Fig.
8A-C). The results indicated that 7-MH-induced cell death,
inhibited migration, and invasion of HT29 cancer cells.
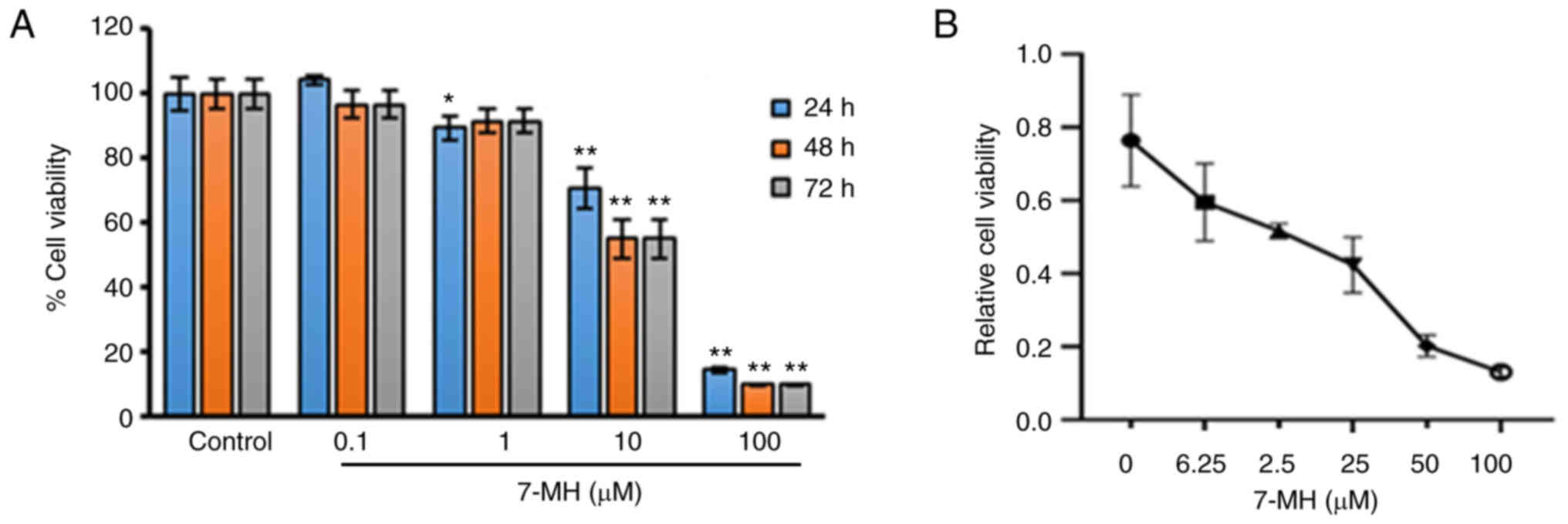
Effects of 7-MH on the viability of
LNCaP cells
To examine the effect of 7-MH on the viability of
LNCaP cells, the cells were treated with various concentrations of
7-MH for 24, 48 and 72 h, and the cell viability was examined using
MTT assay. The result showed that 7-MH significantly inhibited cell
growth at concentrations of 1, 10, and 100 µM for 24 h; and at
concentrations of 10 and 100 µM for 48 h and 72 h (Fig. 9A). To confirm the effects of 7-MH
on LNCaP cell proliferation, cells were treated with various
concentrations of 7-MH for 24 h. 7-MH was observed to significantly
inhibit cell growth in a dose dependent manner (Fig. 9B). In previous studies, 7-MH was
observed to be a carbazole isolated from the roots of C.
harmandiana, which exhibited cytotoxicity against NCI-H187
(human small-cell lung cancer cells), KB (human epidermoid
carcinoma of oral cavity cell lines) and HT29 (human colorectal
adenocarcinoma cell line) cells (36–38).
To understand the mechanism by which 7-MH induced
cell death, multiple potential signaling pathways that have been
demonstrated to be engaged in 7-MH-induced apoptosis were screened.
Western blotting showed that 7-MH incubation leads to activation of
Akt and p38, whereas the expression of p65, pERK, and MAPK13 was
inhibited in a time-dependent manner (Fig. 10). This result indicated that 7-MH
induced apoptosis by inducing Akt and p38 activation and inhibiting
the p65, pERK, and MAPK13 pathways.
7-MH inhibits the proliferation and
metastasis of 4T1 cancer cells
7-MH significantly reduced the viability of 4T1
cells when compared with resveratrol as a positive control
(Fig. 11). The effect of 7-MH on
the activation of NF-κB and STAT3 was examined in 4T1 cells stably
transfected with an NF-κB- and STAT3-dependent reporter plasmid.
Cells were treated with 7-MH for 6 h. It was found that 7-MH
inhibited NF-κB and STAT3 activation (Fig. 12).
In 4T1 cancer cell metastasis, the Transwell assay
showed that 10 µM of 7-MH significantly inhibited 4T1 cancer cell
migration (Fig. 13). In
vivo assay showed that 7-MH reduced the luminescence signal of
4T1-Luc2 cell metastasis in the lungs of mice (Fig. 14).
The interaction between 7-MH and TAK1
kinase
To understand the binding interactions between 7-MH
and TAK1 kinase, a molecular docking study utilizing the Autodock
4.2.6 program was performed. The 4GS6 PDB code, which is bound with
the irreversible inhibitor (5Z)-7-oxozeaenol, was used as the TAK1
template. The binding modes and interaction diagrams of 7-MH bound
to TAK1 kinase are represented in Fig. 15. The results of docking revealed
that 7-MH exhibited multiple binding sites with TAK1 kinase, which
are likely to be Lys63, Met104, Tyr106, Ala107, Leu163, Pro160,
Cys174, ASP175, Val42, Val50, Val90, Ala61, Gly43, Gly45, Gly110
and Ser111; and its binding energy (ΔG) is-7.72 kcal/mol.
Discussion
In summary, the 7-MH substance is not toxic to
neurons, and it can prevent nerve cell death induced by hydrogen
peroxide. The Annexin V and PI combined staining is commonly used
for studying the cell cycle. In the present study, there was a
limitation to add Annexin V. However, previous studies showed that
the PI staining is an also acceptable method to evaluate cell cycle
(39–41). Therefore, the cell cycle was
investigated by PI staining using flow cytometry. In a previous
study by the authors, it was found that 7-MH induced expression of
death receptor 5 which plays a role in cell death and is expressed
only in cancer cells, but not normal cells, therefore 7-MH induced
cell death only in cancer (data not shown). This molecule can
inhibit the expression of GSK-3, pp38, BAX, and cleaved caspase-3
proteins, which are apoptosis-related proteins. Moreover, 7-MH
increases the expression of proteins that have roles in inhibiting
apoptosis, including Bcl-2 and Bcl-xL. It has been showed in
previous study that 7-MH is toxic to prostate cancer cells, which
can inhibit the expression of the proteins pp65, pERK, and MAPK13.
It has been reported that the carbazole from C. harmandiana
induced apoptosis of HT29 cells (42). The present study showed that 7-MH
at 100 µM significantly induces HT-29, Hep-G2 cell, 4T1, and LNCaP
cell death (with no significant cytotoxic effects on normal colon
cells). Moreover, an inhibitory effect on the migration and
invasion of HT29 and 4T1 cancer cells in a concentration and
time-dependent manner was observed. Furthermore, it was revealed
that 7-MH inhibits cancer proliferation by inhibiting antiapoptotic
proteins (Bcl-2, Bcl-xL and survivin) via the MAPK/Erk pathway
(Erk1/2), and inhibits the migration and invasion of HT-29, in
relation to metastasis of cancer, via MMP-9 inhibition. TAK1, a
serine/threonine kinase, acts as a crucial mediator between
survival and cell death in TNF-α-mediated signaling. It is an
evolutionarily conserved member of the MAP3K family (43). The structure of TAK1 composes of a
N terminal (residues 1–104) and C terminal (residues 111–303)
domain which are linked together with the hinge region
(Met104-Ser111). Lys63 is a key catalytic residue in the active
site of TAK1. Asp175 is catalytically important for phosphate
transfer to substrate molecules. The hinge region provides an
opening for the ATP binding pocket. The purine ring of ATP binds at
the hinge region via hydrogen bond forming with Glu105 and Ala107.
The ATP also forms hydrogen bond with Asp175 in the DFG motif. The
ribose 3′-O of ATP from hydrogen bond with Pro160 (44). The results of docking revealed that
7-MH occupied the ATP-binding pocket of TAK1. 7-MH bound to amino
acid residues critical for kinase function: Met104, Tyr106, and
Ala107 (hinge region); Lys63 and ASP175 (catalytic amino acid);
Pro160 (the binding site of the ribose 3′-O of ATP). Furthermore,
7-MH bound to other amino acid residues including Leu163, Cys174,
Val42, Val50, Val90, Ala61, Gly43, Gly45, Gly110 and Ser111.
Carbazole ring of 7-MH bound the ATP-binding pocket of TAK1
through: Pi-Pi stacked interaction with Tyr106; Pi-sigma
interactions with Val50 and Leu163; Pi-alkyl interactions with
Val42, Ala61 and Ala 107; and Pi-sulfur interaction with Met104.
The aldehyde and hydroxy substituents on positions 2 and 3,
respectively, form hydrogen bonds with ASP175 in the DFG motif.
This residue is considered to interact with Lys63 through polar
interactions and is catalytically important for phosphate transfer
to substrate molecules (45).
Moreover, a prenyl group at the position 8 of carbazole ring form
hydrophobic interaction with Pro160 which is the key residue target
of the ribose 3′-O of ATP. Thus, the docking results confirmed that
7-MH was located in the ATP-binding site, thereby interfering with
TAK1 function. In the future study, the interaction between TAK1
and 7-MH will be investigated since in the present study there was
a limitation for evaluating TAK1 activity.
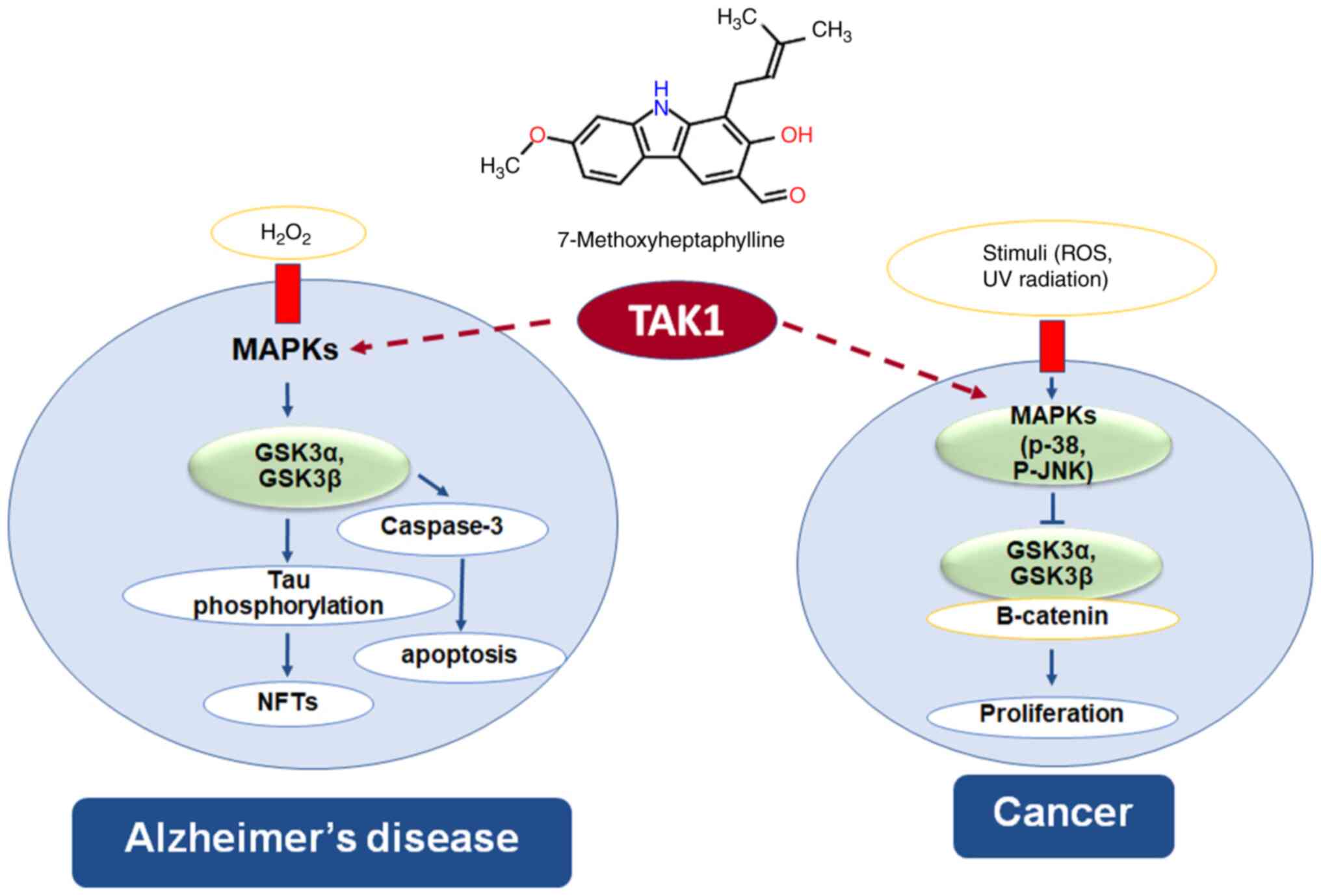
GSK-3 is a protein serine/threonine kinase, plays a
central role in cellular processes and regulation of disease
progression including cancer and AD. In oxidative stress hypothesis
of AD, hydrogen peroxide induces neuronal cell death through the
MAPKs/GSK-3-mediated apoptosis signaling pathway and GSK-3 also
phosphorylates Tau protein to generate neurofibrillary tangles. In
cancer cells, β-catenin act as oncoprotein which causes cell
proliferation; GSK-3 inhibits β-catenin by phosphorylating
β-catenin molecule resulting in degradation of β-catenin (46,47).
TAK1 and TAK1-binding protein 1 play an important role in cell
apoptosis, migration and invasion through the MAPKs, and NF-κB
signal transduction pathways (48). The present study showed that
7-MH has binding site on TAK1 kinase, indicating that 7-MH has
neuroprotective effect and anticancer activity via the TAK1 pathway
(Fig. 16).
Acknowledgements
Not applicable.
Funding
The present study was supported by the Thailand Research Fund
(grant no. TRG5780035), the National Research Council of Thailand
and Thailand Research Fund (grant no. DBG6080006), National
Science, Research and Innovation Fund, Research Program, Khon Kaen
University and the Ubon Ratchathani University of Thailand.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PW, CB, CY, YH, PLD and SC conceived the present
study. PW, MT, PT and CB developed the methodology and performed
formal analysis and investigation. PW and CY provided resources. PW
and SC conducted data curation. PW and CB prepared the original
draft, wrote, reviewed and edited the manuscript. PW performed
project administration. All authors have read and approved the
final version of the manuscript. PW and CB confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
The animal studies were conducted according to the
standard guidelines of the National Institutes of Health and were
approved (approval no. A2017INM-7) by the Animal Care and Use
Committee of the University of Toyama (Toyama, Thailand).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roe CM, Behrens MI, Xiong C, Miller JP and
Morris JC: Alzheimer disease and cancer. Neurology. 64:895–898.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mattson MP: Cellular actions of
beta-amyloid precursor protein and its soluble and fibrillogenic
derivatives. Physiol Rev. 77:1081–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Luca M, Colciaghi F, Pastorino L,
Borroni B, Padovani A and Cattabeni F: Platelets as a peripheral
district where to study pathogenetic mechanisms of alzheimer
disease: The case of amyloid precursor protein. Eur J Pharmacol.
405:277–283. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Itoh H, Kataoka H, Koita H, Nabeshima K,
Inoue T, Kangawa K and Koono M: Establishment of a new human cancer
cell line secreting protease nexin-II/amyloid beta protein
precursor derived from squamous-cell carcinoma of lung. Int J
Cancer. 49:436–443. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meng JY, Kataoka H, Itoh H and Koono M:
Amyloid beta protein precursor is involved in the growth of human
colon carcinoma cell in vitro and in vivo. Int J Cancer. 92:31–39.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Woods NK and Padmanabhan J: Inhibition of
amyloid precursor protein processing enhances gemcitabine-mediated
cytotoxicity in pancreatic cancer cells. J Biol Chem.
288:30114–30124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haven CJ, Howell VM, Eilers PH, Dunne R,
Takahashi M, van Puijenbroek M, Furge K, Kievit J, Tan MH, Fleuren
GJ, et al: Gene expression of parathyroid tumors: Molecular
subclassification and identification of the potential malignant
phenotype. Cancer Res. 64:7405–7411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krause K, Karger S, Sheu SY, Aigner T,
Kursawe R, Gimm O, Schmid KW, Dralle H and Fuhrer D: Evidence for a
role of the amyloid precursor protein in thyroid carcinogenesis. J
Endocrinol. 198:291–299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takayama K, Tsutsumi S, Suzuki T,
Horie-Inoue K, Ikeda K, Kaneshiro K, Fujimura T, Kumagai J, Urano
T, Sakaki Y, et al: Amyloid precursor protein is a primary androgen
target gene that promotes prostate cancer growth. Cancer Res.
69:137–142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takagi K, Ito S, Miyazaki T, Miki Y,
Shibahara Y, Ishida T, Watanabe M, Inoue S, Sasano H and Suzuki T:
Amyloid precursor protein in human breast cancer: An
androgen-induced gene associated with cell proliferation. Cancer
Sci. 104:1532–1538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyazaki T, Ikeda K, Horie-Inoue K and
Inoue S: Amyloid precursor protein regulates migration and
metalloproteinase gene expression in prostate cancer cells. Biochem
Biophys Res Commun. 452:828–833. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hansel DE, Rahman A, Wehner S, Herzog V,
Yeo CJ and Maitra A: Increased expression and processing of the
Alzheimer amyloid precursor protein in pancreatic cancer may
influence cellular proliferation. Cancer Res. 63:7032–7037.
2003.PubMed/NCBI
|
|
13
|
Shiota M, Yokomizo A and Naito S:
Pro-survival and anti-apoptotic properties of androgen receptor
signaling by oxidative stress promote treatment resistance in
prostate cancer. Endocr Relat Cancer. 19:R243–R253. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Basu S and Tindall DJ: Androgen action in
prostate cancer. Horm Cancer. 1:223–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miyamoto H, Messing EM and Chang C:
Androgen deprivation therapy for prostate cancer: Current status
and future prospects. Prostate. 61:332–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Andersen JK: Oxidative stress in
neurodegeneration: Cause or consequence? Nat Med. 10
(Suppl):S18–S25. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paravicini TM and Touyz RM: Redox
signaling in hypertension. Cardiovasc Res. 71:247–258. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haigis MC and Yankner BA: The aging stress
response. Mol Cell. 40:333–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bostwick DG, Alexander EE, Singh R, Shan
A, Qian J, Santella RM, Oberley LW, Yan T, Zhong W and Jiang X:
Antioxidant enzyme expression and reactive oxygen species damage in
prostatic intraepithelial neoplasia and cancer. Cancer. 89:123–134.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sharifi N, Hurt EM, Thomas SB and Farrar
WL: Effects of manganese superoxide dismutase silencing on androgen
receptor function and gene regulation: Implications for
castration-resistant prostate cancer. Clin Cancer Res.
14:6073–6080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khandrika L, Kumar B, Koul S, Maroni P and
Koul HK: Oxidative stress in prostate cancer. Cancer Lett.
282:125–136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shiota M, Yokomizo A, Tada Y, Inokuchi J,
Kashiwagi E, Masubuchi D, Eto M, Uchiumi T and Naito S: Castration
resistance of prostate cancer cells caused by castration-induced
oxidative stress through Twist1 and androgen receptor
overexpression. Oncogene. 29:237–250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shiota M, Yokomizo A and Naito S:
Oxidative stress and androgen receptor signaling in the development
and progression of castration-resistant prostate cancer. Free Radic
Biol Med. 51:1320–1328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Santoro R, Zanotto M, Simionato F,
Zecchetto C, Merz V, Cavallini C, Piro G, Sabbadini F, Boschi F,
Scarpa A and Melisi D: Modulating TAK1 expression inhibits YAP and
TAZ oncogenic functions in pancreatic cancer. Mol Cancer Ther.
19:247–257. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bang D, Wilson W, Ryan M, Yeh JJ and
Baldwin AS: GSK-3α promotes oncogenic KRAS function in pancreatic
cancer via TAK1-TAB stabilization and regulation of noncanonical
NF-κB. Cancer Discov. 3:690–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Santoro R, Carbone C, Piro G, Chiao PJ and
Melisi D: TAK-ing aim at chemoresistance: The emerging role of
MAP3K7 as a target for cancer therapy. Drug Resist Updat. 33–35.
36–42. 2017.PubMed/NCBI
|
|
29
|
Xia S, Ji L, Tao L, Pan Y, Lin Z, Wan Z,
Pan H, Zhao J, Cai L, Xu J and Cai X: TAK1 is a novel target in
hepatocellular carcinoma and contributes to sorafenib resistance.
Cell Mol Gastroenterol Hepatol. 12:1121–1143. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Venna VR, Benashski SE, Chauhan A and
McCullough LD: Inhibition of glycogen synthase kinase-3β enhances
cognitive recovery after stroke: The role of TAK1. Learn Mem.
22:336–343. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aouacheria A, Néel B, Bouaziz Z, Dominique
R, Walchshofer N, Paris J, Fillion H and Gillet G: Carbazolequinone
induction of caspase-dependent cell death in Src-overexpressing
cells. Biochem Pharmacol. 64:1605–1616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thongthoom T, Songsiang U, Phaosiri C and
Yenjai C: Biological activity of chemical constituents from
Clausena harmandiana. Arch Pharm Res. 33:675–680. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Songsiang U, Thongthoom T, Boonyarat C and
Yenjai C: Claurailas A-D, cytotoxic carbazole alkaloids from the
roots of Clausena harmandiana. Nat J Prod. 74:208–212. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thiratmatrakul S, Yenjai C, Waiwut P,
Vajragupta O, Reubroycharoen P, Tohda M and Boonyarat C: Synthesis,
biological evaluation and molecular modeling study of novel
tacrine-carbazole hybrids as potential multifunctional agents for
the treatment of Alzheimer's disease. Eur J Med Chem. 75:21–30.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rosini M, Simoni E, Bartolini M, Cavalli
A, Ceccarini L, Pascu N, McClymont DW, Tarozzi A, Bolognesi ML,
Minarini A, et al: Inhibition of acetylcholinesterase, beta-amyloid
aggregation, and NMDA receptors in Alzheimer's disease: A promising
direction for the multi-target-directed ligands gold rush. J Med
Chem. 51:4381–4384. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kozurkova M, Hamulakova S, Gazova Z,
Paulikova H and Kristian P: Neuroactive multifunctional tacrine
congeners with cholinesterase, anti-amyloid aggregation and
neuroprotective properties. Pharmaceuticals (Basel). 4:382–418.
2011. View Article : Google Scholar
|
|
37
|
Wangboonskul J and Yenjai C: Antioxidant
activity and cytotoxicity against cholangiocarcinoma of carbazoles
and coumarins from Clausena harmandiana. Sci Asia. 38:75–81.
2012. View Article : Google Scholar
|
|
38
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shen Y, Vignali P and Wang R: Rapid
profiling cell cycle by flow cytometry using concurrent staining of
DNA and mitotic markers. Bio Protoc. 7:e25172017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu L, Lu Y, Martinez J, Bi Y, Lian G,
Wang T, Milasta S, Wang J, Yang M, Liu G, et al: Proinflammatory
signal suppresses proliferation and shifts macrophage metabolism
from Myc-dependent to HIF1α-dependent. Proc Natl Acad Sci USA.
113:1564–1569. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Riccardi C and Nicoletti I: Analysis of
apoptosis by propidium iodide staining and flow cytometry. Nat
Protoc. 1:1458–1461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Boonyarat C, Yenjai C, Vajragupta O and
Waiwut P: Heptaphylline induces apoptosis in human colon
adenocarcinoma cells through bid and Akt/NF-κB (p65) pathways.
Asian Pac J Cancer Prev. 15:10483–10487. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Totzke J, Scarneo SA, Yang KW and Haystead
TAJ: TAK1: A potent tumour necrosis factor inhibitor for the
treatment of inflammatory diseases. Open Biol 10: 200099,
2020.
Brown K, Vial SCM, Dedi N, Long JM, Dunster NJ and Cheetham
GMT: Structural basis for the interaction of TAK1 kinase with its
activating protein TAB1. J Mol Biol. 354:1013–1020. 2005.PubMed/NCBI
|
|
45
|
Mancinelli R, Carpino G, Petrungaro S,
Mammola CL, Tomaipitinca L, Filippini A, Facchiano A, Ziparo E and
Giampietri C: Multifaceted roles of GSK-3 in cancer and
autophagy-related diseases. Oxid Med Cell Longev. 2017:46294952017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rippin I and Eldar-Finkelman H: Mechanisms
and therapeutic implications of GSK-3 in treating
neurodegeneration. Cells. 10:2622021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|