Head and neck squamous cell carcinoma (HNSCC) is one
of the most widespread malignancies worldwide (1–3).
Statistics indicate that >54,010 oral and pharyngeal cancer
cases are diagnosed, and >10,850 individuals succumb to the
disease annually (4). Numerous risk
factors lead to the incidence of HNSCC, including smoking, alcohol
consumption, human papillomavirus infection, and genetic
disposition (5). Despite the
advanced treatment methods, HNSCC has a high recurrence rate
(6). Therefore, studying the
pathogenic mechanism in HNSCC is of great importance in providing
individualized treatment for patients.
p53, as a transcription factor, can play its role in
tumor suppression by activating the expression of numerous target
genes (7). However, p53 is one of
the most commonly mutated genes in human tumors, with mutations
detected in 65–85% of HNSCC (8,9). Most
p53 mutations in HNSCC are missense mutations, which lead to the
substitution of only one amino acid (10). The missense mutations not only
suppress the tumor suppressive role of wild-type p53 but also
provide novel functions to promote tumor recurrence and
chemoresistance, called gain-of-function (GOF) (11). A previous study revealed that the
p53 protein, the translated product of the TP53 gene, is frequently
mutated in HNSCC (12); therefore,
studying the pathogenic mechanism of mutant p53 in HNSCC is crucial
to provide more individualized treatment for patients.
A study has revealed that patients with HNSCC
carrying p53 mutations have a high risk of malignancy and a poor
prognosis (10). p53 mutation can
affect a variety of cellular processes, including drug resistance
and carcinogenesis (6). The present
study focused on the prevalence and clinical relevance of p53
mutation in HNSCC and further described how mutant p53 accumulates.
In addition, the molecular mechanisms by which GOF of mutant p53
can affect the proliferation, migration invasion, immunosuppression
and metabolic effects of HNSCC were investigated. Finally,
therapeutic strategies to abolish the tumor-promoting effects of
mutant p53 were elucidated to provide a basis for further
understanding the mechanism of mutant p53 to develop targeted
therapies.
A systematic literature search was conducted through
the electronic search engine PubMed to find eligible studies
published before March 9, 2023. The key words for the search were
‘Head and neck squamous carcinoma’, ‘mutant p53’ and
‘gain-of-function’. In addition, the references in the retrieved
articles were also manually reviewed to identify potentially
relevant studies.
p53, as a transcription factor, can play its role in
tumor suppression by activating the expression of numerous target
genes (7). The main functional
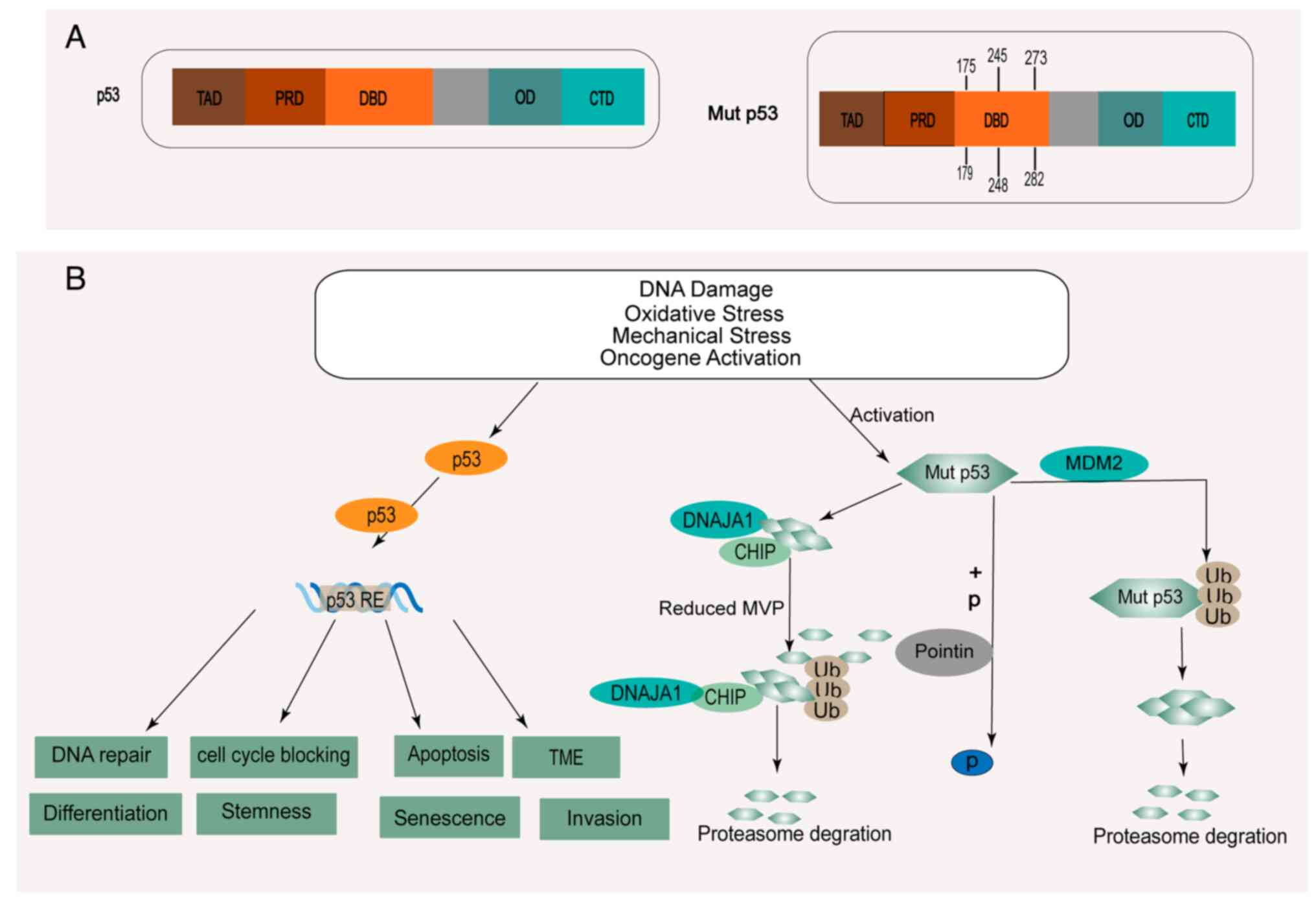
domains of full-length p53 include two transactivation domains
(TAD) at the N-terminus, a proline-rich domain (PRD), a central DNA
binding domain (DBD), an oligomerization domain (OD), and a
regulatory C-terminal domain (CTD) (7) (Fig.
1A). A missense mutation refers to a substitution of an amino
acid in the DBD (10). Most p53
mutations in HNSCC are missense mutations, and the mutation rate of
p53 is 65–85% (8,9,13).
Furthermore, the mutation rate of p53 was revealed to be as high as
30% in oral precancerous lesions (OPL) (14). In HNSCC, p53 mutations frequently
occur at amino acids R248, G245, R273, R175, H179, and R282 among
its DBD (10) (Fig. 1A). In addition, a new mutation in
exon 7 of the p53 gene has been identified in the tumors of
patients with oral squamous cell carcinoma (OSCC) (15). A missense mutation resulting in a
codon alteration from ‘AGT’ to ‘ACT’ was identified at position 719
of TP53 (15).
Clinically, TP53 mutations are associated primarily
with a low survival rate, drug resistance, and extranodal extension
in patients, which makes p53 mutation status a potential molecular
marker for predicting the clinical response of these patients
(12,20,21).
It was revealed that the mutation rate of p53 in OPL was as high as
30%, indicating that these mutations occur at an early stage of
oral tumor development and may influence the development and
progression of OPL (14). A
previous study reported that in HNSCC, tumors with high-risk p53
mutations are more likely to develop combined resistance to
cisplatin and fluorouracil chemotherapy than tumors with low-risk
mutations or wild-type TP53 (22).
Furthermore, the anticancer effect of cisplatin differs among HNSCC
cell lines with different p53 mutation statuses. Further
investigation on the association between the mutational statuses of
p53 and cisplatin resistance in HNSCC cell lines are required to
develop more suitable therapeutic approaches. Notably, serum p53
antibody levels in HNSCC patients have important clinical
significance. Mutations in the TP53 gene could lead to the
accumulation of mutant p53 protein in cancer cells, which induces
the production of serum anti-p53 antibodies (Ap53Ab) in patients
with OSCC (23). The results of
related assays revealed that the presence of Ap53Ab may reflect p53
mutational status and the aggressive phenotype, which serves as a
valid predictive marker for OSCC in clinical practice (23). A previous relevant study reported
that the expression of fibroblast growth factor receptor 3 (FGFR3)
is highly correlated with the expression of mutant p53 in
oropharyngeal squamous cell carcinoma (24). Kaplan Meier analysis of relevant
samples showed that patients carrying high expression levels of
FGFR3 and mutant p53 had worse disease-free survival (24).
Missense mutations not only attenuate the tumor
suppressive role of wild-type p53 but also provide novel functions
to promote tumor recurrence and chemoresistance, called GOF
(11). However, only when the
mutant p53 protein remains stable and accumulates to a very high
level in tumor tissue can it perform its GOF property (25). At present, the mechanism of mutant
p53 aggregation in HNSCC is not completely clear. Previous research
has reported that the level of mutant p53 can be regulated by
posttranslational modification (ubiquitination, phosphorylation)
and molecular chaperone (Fig.
1B).
Mutant p53 protein level can be regulated by
phosphorylation modification. R2TP, a molecular chaperone complex
containing Pontin, stabilizes substrate proteins (26). Independent of the function of R2TP,
Pontin was demonstrated to have the ability to control gene
transcription factors, including p53 and mutant p53 (27). A previous study reported that Pontin
can promote robust phosphorylation of the GOF mutant p53-R248Q at
Ser15 and Ser46 by interacting independently with mutant p53-R248Q
(28).
Chaperones, such as heat shock proteins (HSPs),
interact with newly synthesized proteins to restore the correct
structure of damaged or misfolded proteins (29). A previous study revealed that MDM2
can inhibit p53 expression by mediating the ubiquitin-proteasome
pathway to reactivate a negative feedback loop to strictly regulate
p53 activity. Therefore, it is possible to reactivate the function
of wild-type p53 as a tumor suppressor after blocking the
interaction between MDM2 and p53 (30). Notably, a previous study
demonstrated that MDM2 can ubiquitinate mutant p53 and lead to its
degradation in vitro (31).
Another previous study revealed that the heat shock protein 90
(HSP90) chaperone protein can inhibit the activity of MDM2 and
CHIP, thereby enhancing the stability of mutp53 (31).
DNAJA1, a member of the HSP40 family, stabilizes
mutant p53 by competing with the ubiquitin ligase CHIP for binding
to p53, thus rendering mutant p53 more stable (29). Further study has revealed that
DnaJA1 can stabilize unfolded mutant p53 and promote mutant
p53-mediated activation of Yes-associated protein (YAP)/TAZ signal,
which can regulate Cdc42/Rac1 and promote the metastasis of HNSCC
(32,33). It was revealed that specific
reduction in the level of mevalonate-5-phosphate, a metabolic
intermediate in the sodium mevalonate pathway, can promote the
degradation of p53 conformational mutants by inhibiting the
interaction between mutants and DNAJA1 (34).
A previous study confirmed that various stress
signals, including DNA damage and oncogene activation signals, can
stabilize and activate wild-type p53 (35). Notably, previous research indicated
that different stress signals, including signals related to
oxidative stress, DNA damage related to excessive proliferation,
hyperoxia, and oncogene activation, can regulate the stability and
accumulation of mutant p53 in HNSCC, thus contributing to the
acquisition of GOF activity (32).
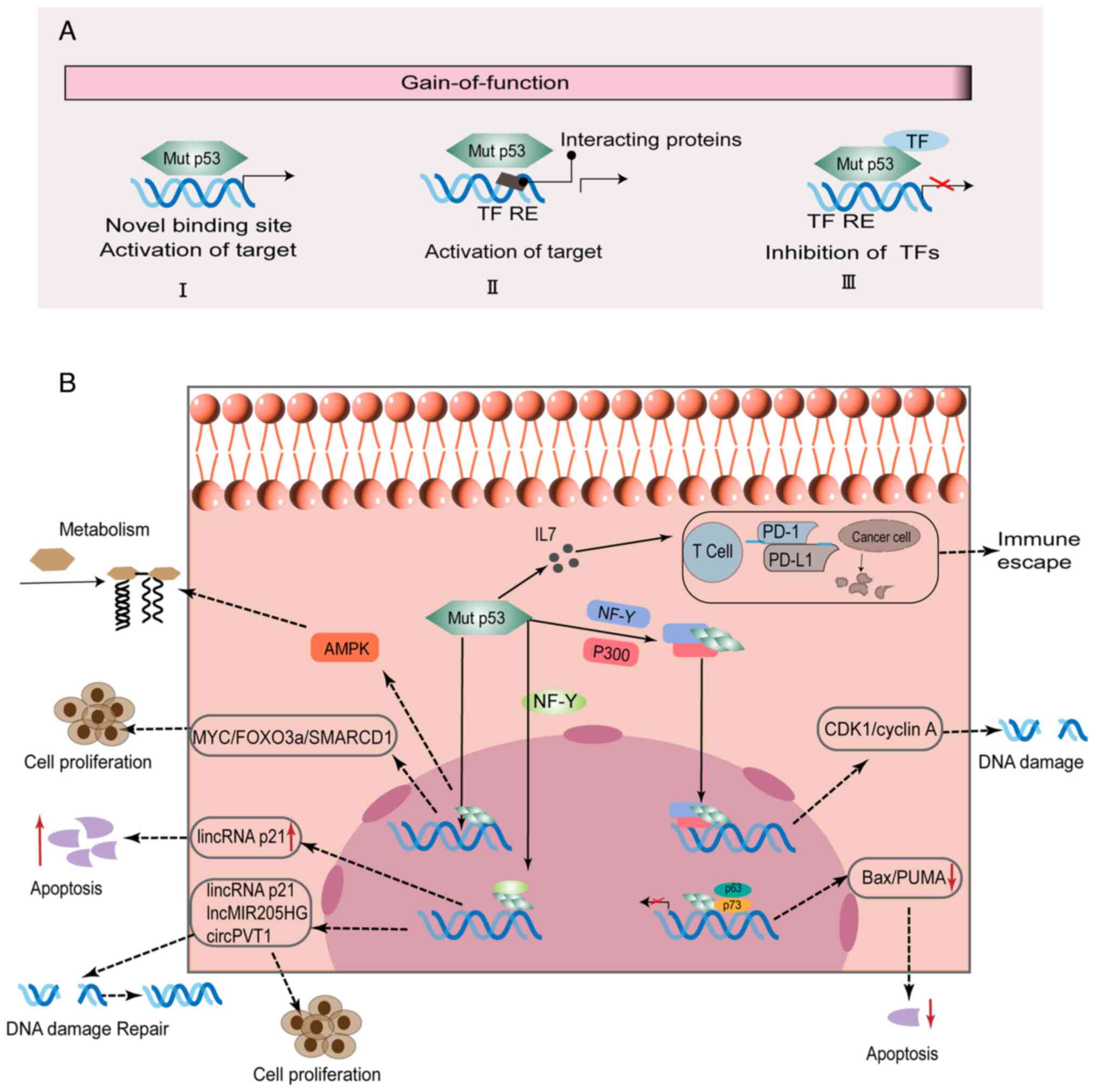
p53 mutations can promote tumor progression, enhance
metastatic potential or promote drug resistance through the effects
of GOF activity (10,36,37).
Mechanistically, mutated p53 proteins can perform complex and
important functions by interacting with other transcription factors
and cofactors or directly binding to relevant target genes
(Fig. 2A). The molecular mechanisms
by which p53 mutations exert GOF effects in HNSCC are presented in
Table I and Fig. 2B.
Mutant p53 can promote HNSCC proliferation and
invasion by interacting with other transcription factors, including
nuclear transcription factor Y (NF-Y), p63 and p73 (38,39).
It was demonstrated that mutant p53 can bind to NF-Y-targeted
promoters, recruit P300, and contribute to histone acetylation
after DNA damage (40). The
resulting complexes containing p53 mutants (P151S, R175H, G245C and
R282W) and nuclear transcription factor Y subunit α (NF-YA) can
transcriptionally regulate lincRNA-p21, which inhibits G1 arrest in
HNSCC cells (39). Moreover, mutant
p53 can interact with p73, which inhibits the expression of
apoptotic target genes [p21/p53 upregulated modulator of apoptosis
(PUMA) and Bcl-2-associated X protein (Bax)], thus giving rise to
chemoresistance (38). In HNSCC
with mutations or inactivation of p53, the imbalance between p63
and p73 may have particular importance for apoptosis and drug
resistance. Research has shown that ΔNp63α is overexpressed mainly
with TAp73 in HNSCC with p53 mutations (41). Tumor necrosis factor-α can promote
the nuclear translocation of p63 and c-Rel, which affects the
translocation of TAp73 to the cytoplasm (42–44).
In HNSCC, mutant p53 participates in transcriptional
regulation of target genes, including MYC, SMARCD1, and AMPK, by
binding their DBD (45–47). Studies have shown that mutant p53
can alter metabolism pathways, including reactive oxygen species,
autophagy, and lipid metabolism pathways (48,49). A
previous study revealed that AMPK can sense energy stress to
stimulate the transmission of relevant information and regulate
metabolic homeostasis (50). Mutant
p53 can participate in metabolic reprogramming by affecting related
energy conduction-related protein kinases to perform its GOF
(45). Under energy stress, p53 GOF
mutants (P151S, R282W, G245C, and R175H) preferentially inhibit
AMPK activation, thereby enhancing metabolism and cell invasive
growth, unlike wild-type p53 (45).
A study by Tanaka et al revealed that GOF mutant p53 G245D
could reduce the phosphorylation of FOXO3a mediated by AMPK,
leading to proliferation in HNSCC (51). MYC is an essential target of
tumorigenicity mediated by mutant p53 and the simultaneous
expression of mutant p53 and MYC proteins is a more accurate
predictor of the clinical outcome of HNSCC than the expression of
either alone (52). In OSCC, it was
confirmed that mutant p53 291R can transcriptionally activate
SMARCD1, and overexpression of SMARCD1 enhances tumorigenic
characteristics, including cell viability and the ability to form
colonies (47,53).
Mutant p53 is able to regulate the expression of
specific non-coding RNAs (ncRNAs), including microRNAs (miRNAs),
long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) by
binding to DNA regions (39,54–57).
Therefore, p53 mutation may alter the wtp53/ncRNA networks to
promote cancer (58).
LncRNAs are RNAs that are >200 nucleotides in
length and have no translation capability (54,59).
Functionally, lncRNAs can sponge miRNAs and competitively interact
with target mRNAs to interfere with the role of miRNAs (60). Research has indicated that lncRNAs
are correlated with tumor development, lymph node metastasis,
advanced clinical stage, and poor prognosis in OSCC (61). Mutant p53 and NF-YA complexes can
promote lincRNA-p21 expression, which inhibits STAT3-regulated
downstream genes (MYC and cyclin D1), thereby suppressing cell
proliferation in HNSCC (39).
Moreover, elevated expression of lincRNA-p21 regulated by mutant
p53 and NF-YA complexes can significantly promote the cleavage of
PARP and caspase-3, which in turn promotes apoptosis in HNSCC cells
(39). Another study has reported
that NF-Y and E2F1 can recruit mutant p53 to the MIR205HG promoter
and significantly upregulate the expression of lncMIR205HG and
miR-205-5p (54). Notably,
lncMIR205HG was revealed to sponge miR-590-3p, and then increased
the expression of cyclin B, CDK1 and YAP, promoting the
proliferation of HNSCC cells (54).
Furthermore, studies have revealed that the function
of mutant p53 proteins can be enhanced via regulation of miRNA
expression in numerous cancers, such as non-small cell lung
carcinoma (NSCLC), breast cancer, and HNSCC (62–64). A
previous study demonstrated that the p53 R175H mutant can induce
miR-128-2 expression to exert an antiapoptotic effect in response
to anticancer drug therapy in NSCLC (64). A study by Masciarelli et al
revealed that the association of mutant p53 with the ZEB-1
transcriptional suppressor protein complex could regulate the
activity of the miR-223 promoter and inhibit its transcriptional
response, which leads to the acquisition of drug resistance in
breast cancer (63). Mutant TP53
was found to suppress the activity of the miR-27a promoter, thereby
promoting the survival of patients with HNSCC (55). In HNSCC, mutant p53 can maintain the
high expression level of miR-205-5p, which could reduce the
expression of BRCA1 and Rad17, resulting in abnormal DNA repair
activity, thus promoting the proliferation of HNSCC cells (56). In addition, TP53 mutation-associated
miRNAs (miR-17-3p, miR-21-3p, miR-21-5p) have become recognized as
influential prognostic factors in HNSCC treatment (65).
CircRNAs are endogenous RNAs with important roles in
regulating gene expression (66,67).
Functionally, they can play multiple roles in regulating
alternative splicing and miRNA expression (68,69). A
previous study reported that circPVT1 was enriched in tumors
expressing mutant p53 protein compared with normal tissues, based
on sequencing data from HNSCC tissue samples (57). The study reported that the
transcription factor complex (mutant p53/YAP/TEAD)
transcriptionally enhanced the expression of circPVT1, which
regulated the expression of miR-497-5p and its target genes,
thereby promoting the proliferation in HNSCC cells (57).
Mutant p53 can alter the extracellular matrix
microenvironment through extracellular vesicles (EVs) to exert GOF
effects (70). EVs can transfer
important bioactive molecules (protein, DNA, mRNAs, and ncRNAs)
between cells (71). Through this
process, they can affect the tumor microenvironment and alter the
related response of recipient cells, which promotes tumor growth,
metastasis, and drug resistance (71). A previous study revealed that mutant
p53 can be transported between cells through EVs to alter the tumor
microenvironment, which could trigger immunosuppression (71). A previous study has shown that
mutant p53 proteins are expressed in pancreatic, lung, and colon
cancer cell lines; these proteins can be selectively packaged into
EVs and then affect the reprogramming of the tumor microenvironment
(72). It has been shown that
exosomal miR-1246 can transfer the mutant p53 protein product from
cancer cells to neighboring cancer cells and macrophages, leading
to alterations in the tumor microenvironment (73). Immunosuppressant molecule,
programmed cell death protein-1 (PD-1)-blocking antibody has been
utilized in the clinical trial treatment of patients with HNSCC,
improving the survival rate of patients with advanced HNSCC
(74). Furthermore, a previous
study reported that p53 R172H regulates the upregulation of the
interleukin 17 (IL17) signaling pathway in the tumor
microenvironment and depletes CD8+ cells, thereby
abolishing the immunotherapeutic effect of anti-PD-1 antibody in
OSCC (19).
Therapies targeting mutant p53 are very promising
for a wide range of human tumors since almost 50% of tumors carry
mutant p53 (75). The main
strategies include normalizing the activity of wild-type p53,
inhibiting new protein-protein interactions of factors related to
the response of mutant p53, exploiting synthetic lethal
vulnerabilities, inducing selective degradation, and administering
immunotherapy (Figs. 3 and 4).
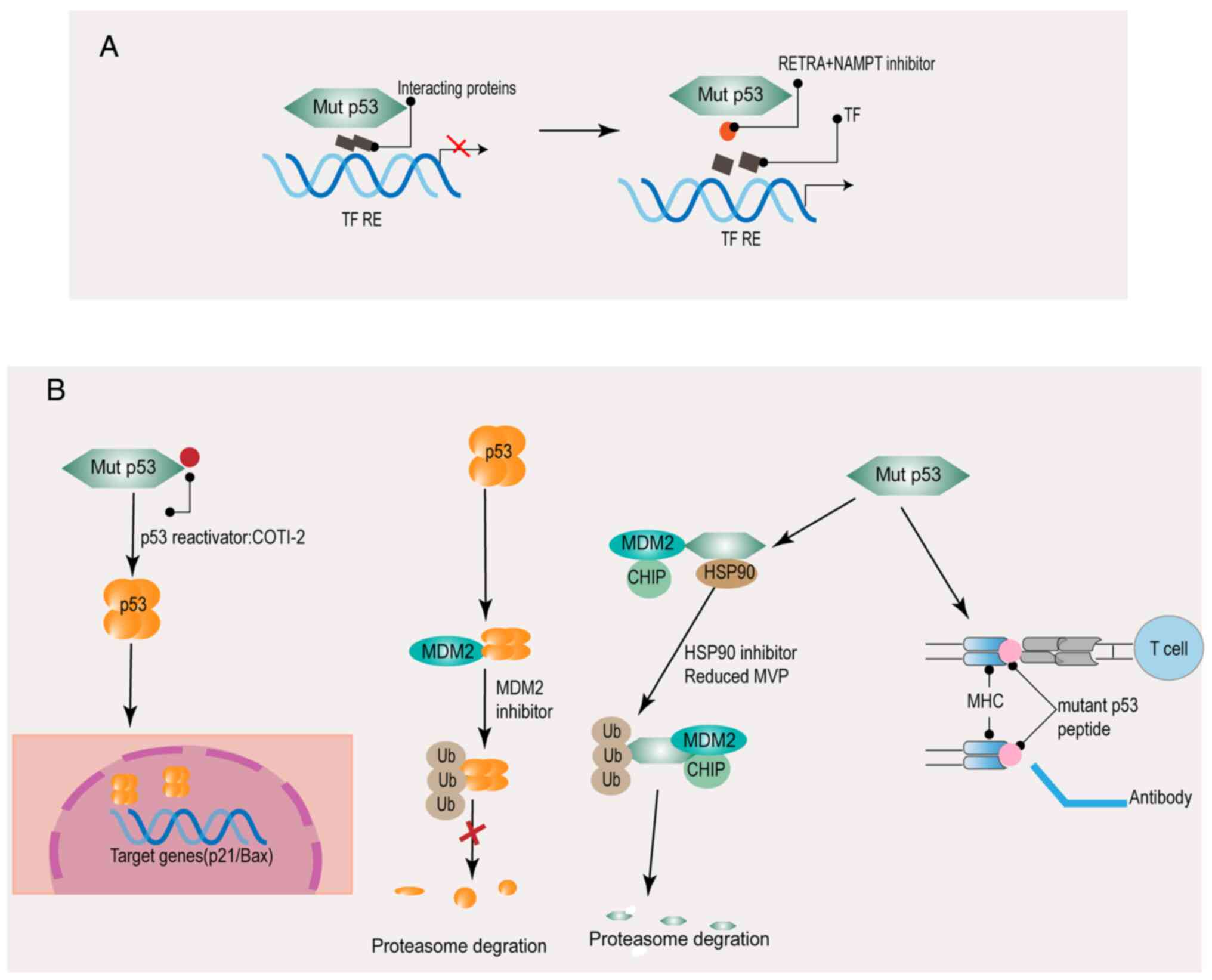
The function of mutant p53 is abolished by
preventing the interaction between mutant p53 and related proteins
(Fig. 3A). The drug RETRA and
NSC59984 (p53 pathway activator) reactivate p73 by blocking the
biological interaction between mutant p53 and p73 or promoting the
degradation of mutant p53 (76).
Nicotinamide phosphoribosyl transfer (NAMPT) can regulate the
aggregation of mutant p53, as determined by comparison of the gene
expression profiles of several regulatory factors in HNSCC cells
(77). Furthermore, combination
treatment with NAMPT inhibitor and a p73 activator can inhibit the
proliferation of HNSCC cells with p53 GOF mutations (77). Another essential drug is PI3K
inhibitor. Mechanistically, mutant p53 facilitates the binding of
MYC to its target promoter, thus enhancing MYC-mediated
carcinogenesis (46). PI3K
inhibitors eliminate the GOF effect of mutant p53 by preventing the
interaction between MYC, mutant p53, YAP proteins with MYC target
promoter (46).
The purpose of restoring the function of wild-type
p53 is to restore the natural construction of the DBD (Fig. 3B). It has been reported that several
compounds can reactivate wild-type p53 to restore the p53-induced
biological functions; these drugs are either cysteine-targeting
compounds or Zn2+ agents (78,79). A
previous study revealed that significant p53 reactivation was
observed in HNSCC cells with mutant p53 treated with a p53
reactivator (80). In combination
therapy, the p53-reactivation molecule enhanced the antitumor
activity of cisplatin, 5-fluorouracil, and paclitaxel against HNSCC
cells (80). Another method
developed was the use of Zn2+ agents to restore the
wild-type conformation of p53. The p53 structure contains a zinc
ion, an essential cofactor, which stabilizes the DBD to support the
role of p53 in inhibiting carcinogenesis (81,82).
The Zn2+ binding ability of mutant p53 is easily lost
(82). The clinical application of
the Zn2+ pharmaceutical agents is represented by COTI-2.
COTI-2 is a novel associated third-generation thiosemicarbazone
that binds to the misfolded mutant conformation of the p53 protein
to induce conformational changes (83,84). A
previous study reported that COTI-2 could normalize the expression
of wild-type p53 target genes and restore the DNA binding ability
of GOF p53 mutant proteins in HNSCC (84). COTI-2 may bring new promise for the
treatment of patients with HNSCC carrying p53 mutations.
Inducing the degradation of mutant p53 is another
strategy, which therapeutically targets mutant p53 (Fig. 3B). HSP90, a chaperone molecule, is
capable of inactivating p53 ubiquitin ligase MDM2 (85). Therefore, HSP90 inhibitor treatment
can destabilize mutant p53, thereby increasing tumor cell apoptosis
in HNSCC (85).
Research has identified anti-p53 antibodies in
cancer patients (including patients with HNSCC), and it has also
revealed that p53 mutants were able to be recognized by antibodies
and T cell receptors, thus vaccines for the mutant p53 gene were
evaluated and assessed in clinical trials for the treatment of
various types of cancer, including patients with HNSCC (86,87)
(Fig. 3B).
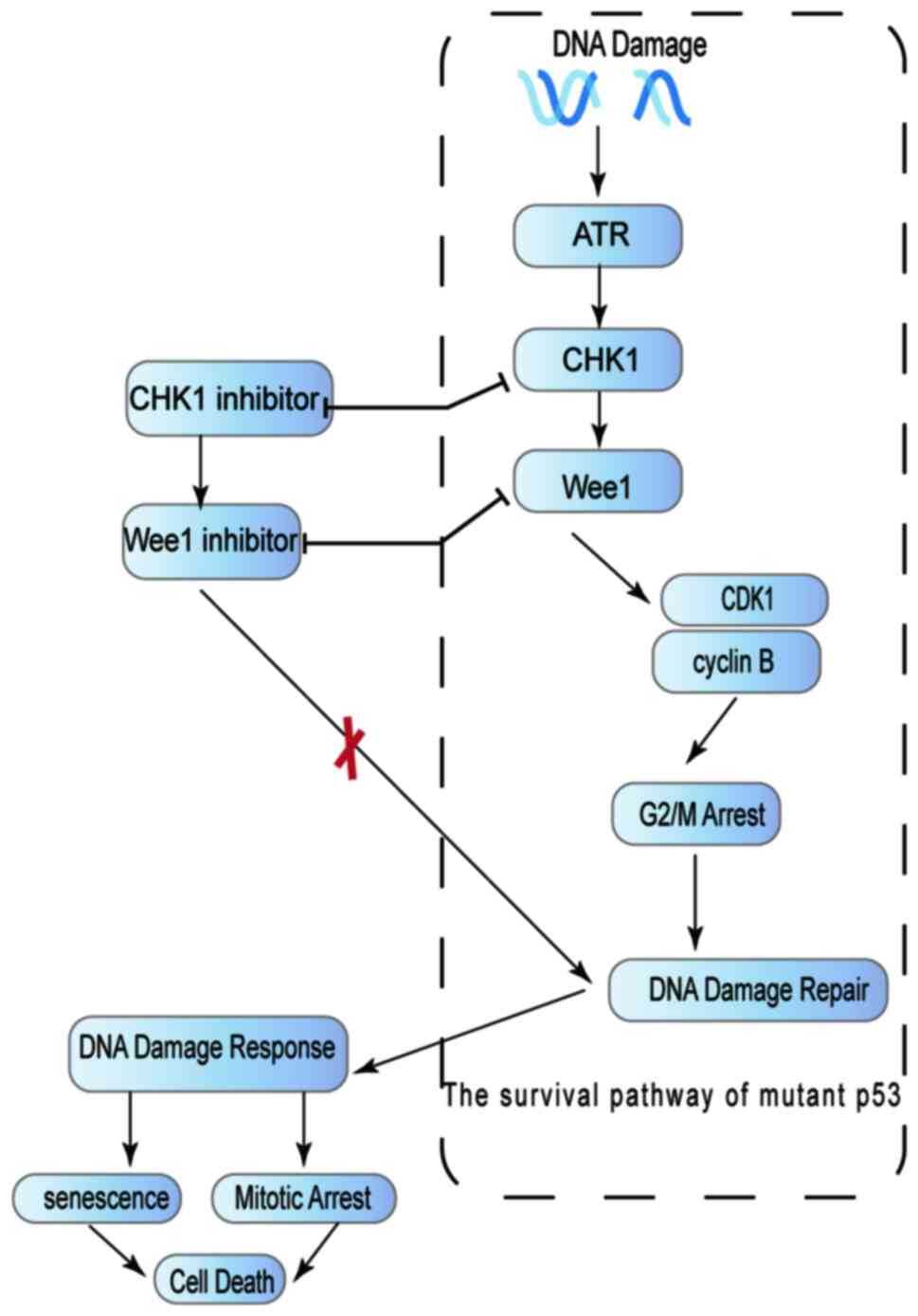
An additional strategy for the management of mutant
p53 is the exploitation of synthetic lethality vulnerabilities,
which causes mutant p53 to lose its ability to promote cell
survival (Fig. 4). Tumors in which
wild-type p53 function cannot be normalized, must rely on the
activation of S and G2 checkpoints (ATR, CHK1, MK2, Wee1, etc.) to
mediate the repair of DNA damage; thus, these tumor cells are more
sensitive to ablation of the G2 checkpoints (88,89).
Inhibition of the kinases involved in the G2/M checkpoint, such as
CHK1 and Wee1, can cause p53 mutants to lose their ability to
promote cell survival (90). The
use of abrogation of G2 checkpoints, Wee-1 kinase inhibition and
CHK1 inhibition, can significantly induce sensitivity to cisplatin
treatment by affecting HNSCC cells expressing high-risk p53
mutations (91,92). The Wee-1 inhibitor, MK-1775, was
demonstrated to render tumor cells chemosensitive in p53-deficient
tumors (93). A clinical trial has
shown that in patients with HNSCC, combined treatment with MK-1775,
cisplatin, and docetaxel effectively inhibited the function of
mutant p53 and increased the synthetic lethality of mutant p53
(94).
p53 is one of the most commonly mutated genes in
human tumors, with mutations detected in 65–85% of HNSCC cases,
highlighting the critical role of p53 in inhibiting tumorigenesis.
It is challenging to directly target the mutant p53 protein. This
ability is highly dependent on the unique structure of the protein,
rendering targeted drug development more complex. In addition, with
the accumulating research on the role of ncRNAs, the functions of
ncRNAs are becoming better appreciated. Mutant p53 can regulate
related ncRNAs through transcriptional or posttranscriptional
mechanisms; thus, targeted inhibition of the related ncRNAs-mutant
p53 network can enhance the synthetic lethality. In the future, the
challenge of studying p53 will be at the molecular and cellular
levels. With an in-depth understanding of p53, the aim will be to
translate this knowledge into clinical application. Notably, the
study of p53 mutations has historically been conducted mainly in
cell lines and mouse models, which may cause interspecies
differences in p53 sequences and signaling pathways. The
development of human tumor-like organs that closely reproduce the
tumor conditions has offered considerable advantages in
understanding the function of p53 and assessing treatment schemes
in a more definitive manner.
Not applicable.
The present study was supported by the 2021 Youth Innovation
Talent Introduction and Education Program of Shandong Province
Universities.
Not applicable.
ML and CH contributed to the selection of the
studies. Images were completed by XC and DS, and the table was
created by NS, WZ, XZ and YY. The first draft of the manuscript was
written by ML, and all authors revised previous versions of the
manuscript. All authors read and approved the final manuscript.
Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Johnson DE, Burtness B, Leemans CR, Lui
VWY, Bauman JE and Grandis JR: Head and neck squamous cell
carcinoma. Nat Rev Dis Primers. 6:922020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ford PJ and Rich AM: Tobacco use and oral
health. Addiction. 116:3531–3540. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Auguste A, Joachim C, Deloumeaux J, Gaete
S, Michineau L, Herrmann-Storck C, Duflo S and Luce D: Head and
neck cancer risk factors in the French West Indies. BMC Cancer.
21:10712021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hedberg ML, Goh G, Chiosea SI, Bauman JE,
Freilino ML, Zeng Y, Wang L, Diergaarde BB, Gooding WE, Lui VW, et
al: Genetic landscape of metastatic and recurrent head and neck
squamous cell carcinoma. J Clin Invest. 126:16062016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gleber-Netto FO, Zhao M, Trivedi S, Wang
J, Jasser S, McDowell C, Kadara H, Zhang J, Wang J, William WN Jr,
et al: Distinct pattern of TP53 mutations in human immunodeficiency
virus-related head and neck squamous cell carcinoma. Cancer.
124:84–94. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou G, Liu Z and Myers JN: TP53 mutations
in head and neck squamous cell carcinoma and their impact on
disease progression and treatment response. J Cell Biochem.
117:2682–2692. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sabapathy K and Lane DP: Therapeutic
targeting of p53: All mutants are equal, but some mutants are more
equal than others. Nat Rev Clin Oncol. 15:13–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deneka AY, Baca Y, Serebriiskii IG,
Nicolas E, Parker MI, Nguyen TT, Xiu J, Korn WM, Demeure MJ,
Wise-Draper T, et al: Association of TP53 and CDKN2A mutation
profile with tumor mutation burden in head and neck cancer. Clin
Cancer Res. 28:1925–1937. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cancer Genome Atlas Network, .
Comprehensive genomic characterization of head and neck squamous
cell carcinomas. Nature. 517:576–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ogmundsdóttir HM, Björnsson J and Holbrook
WP: Role of TP53 in the progression of pre-malignant and malignant
oral mucosal lesions. A follow-up study of 144 patients. J Oral
Pathol Med. 38:565–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saleem S, Abbasi ZA, Hameed A, Qureshi NR,
Khan MA and Azhar A: Novel p53 codon 240 Ser > Thr coding region
mutation in the patients of oral squamous cell carcinoma (OSCC).
Tumour Biol. 35:7945–7950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakazawa S, Sakata KI, Liang S, Yoshikawa
K, Iizasa H, Tada M, Hamada JI, Kashiwazaki H, Kitagawa Y and
Yamazaki Y: Dominant-negative p53 mutant R248Q increases the motile
and invasive activities of oral squamous cell carcinoma cells.
Biomed Res. 40:37–49. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Enaka M, Nakanishi M and Muragaki Y: The
gain-of-function mutation p53R248W suppresses cell proliferation
and invasion of oral squamous cell carcinoma through the
down-regulation of keratin 17. Am J Pathol. 191:555–566. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sano D, Xie TX, Ow TJ, Zhao M, Pickering
CR, Zhou G, Sandulache VC, Wheeler DA, Gibbs RA, Caulin C and Myers
JN: Disruptive TP53 mutation is associated with aggressive disease
characteristics in an orthotopic murine model of oral tongue
cancer. Clin Cancer Res. 17:6658–6670. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Hu Y, Escamilla-Rivera V, Gonzalez
CL, Tang L, Wang B, El-Naggar AK, Myers JN and Caulin C: Epithelial
mutant p53 promotes resistance to anti-PD-1-mediated oral cancer
immunoprevention in carcinogen-induced mouse models. Cancers
(Basel). 13:14712021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gleber-Netto FO, Neskey D, Costa AFM,
Kataria P, Rao X, Wang J, Kowalski LP, Pickering CR, Dias-Neto E
and Myers JN: Functionally impactful TP53 mutations are associated
with increased risk of extranodal extension in clinically advanced
oral squamous cell carcinoma. Cancer. 126:4498–4510. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee HJ, Kang YH, Lee JS, Byun JH, Kim UK,
Jang SJ, Rho GJ and Park BW: Positive expression of NANOG, mutant
p53, and CD44 is directly associated with clinicopathological
features and poor prognosis of oral squamous cell carcinoma. BMC
Oral Health. 15:1532015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perrone F, Bossi P, Cortelazzi B, Locati
L, Quattrone P, Pierotti MA, Pilotti S and Licitra L: TP53
mutations and pathologic complete response to neoadjuvant cisplatin
and fluorouracil chemotherapy in resected oral cavity squamous cell
carcinoma. J Clin Oncol. 28:761–766. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gohara S, Yoshida R, Kawahara K, Sakata J,
Arita H, Nakashima H, Kawaguchi S, Nagao Y, Yamana K, Nagata M, et
al: Re-evaluating the clinical significance of serum p53 antibody
levels in patients with oral cancer in Japanese clinical practice.
Mol Clin Oncol. 15:2092021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nannapaneni S, Griffith CC, Magliocca KR,
Chen W, Lyu X, Chen Z, Wang D, Wang X, Shin DM, Chen ZG and Saba
NF: Co-expression of fibroblast growth factor receptor 3 with
mutant p53, and its association with worse outcome in oropharyngeal
squamous cell carcinoma. PLoS One. 16:e02474982021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yue X, Zhao Y, Xu Y, Zheng M, Feng Z and
Hu W: Mutant p53 in cancer: Accumulation, gain-of-function, and
therapy. J Mol Biol. 429:1595–1606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kakihara Y and Houry WA: The R2TP complex:
Discovery and functions. Biochim Biophys Acta. 1823:101–107. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao YQ and Houry WA: The role of pontin
and reptin in cellular physiology and cancer etiology. Front Mol
Biosci. 4:582017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kiguchi T, Kakihara Y, Yamazaki M, Katsura
K, Izumi K, Tanuma JI, Saku T, Takagi R and Saeki M: Identification
and characterization of R2TP in the development of oral squamous
cell carcinoma. Biochem Biophys Res Commun. 548:161–166. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parrales A, Ranjan A, Iyer SV, Padhye S,
Weir SJ, Roy A and Iwakuma T: DNAJA1 controls the fate of misfolded
mutant p53 through the mevalonate pathway. Nat Cell Biol.
18:1233–1243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zheng T, Wang J, Zhao Y, Zhang C, Lin M,
Wang X, Yu H, Liu L, Feng Z and Hu W: Spliced MDM2 isoforms promote
mutant p53 accumulation and gain-of-function in tumorigenesis. Nat
Commun. 4:29962013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li D, Marchenko ND, Schulz R, Fischer V,
Velasco-Hernandez T, Talos F and Moll UM: Functional inactivation
of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization
of mutant p53 in human cancer cells. Mol Cancer Res. 9:577–588.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mantovani F, Collavin L and Del Sal G:
Mutant p53 as a guardian of the cancer cell. Cell Death Differ.
26:199–212. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaida A, Yamamoto S, Parrales A, Young ED,
Ranjan A, Alalem MA, Morita KI, Oikawa Y, Harada H, Ikeda T, et al:
DNAJA1 promotes cancer metastasis through interaction with mutant
p53. Oncogene. 40:5013–5025. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Parrales A, Thoenen E and Iwakuma T: The
interplay between mutant p53 and the mevalonate pathway. Cell Death
Differ. 25:460–470. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Levine AJ: The many faces of p53:
Something for everyone. J Mol Cell Biol. 11:524–530. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Poeta ML, Manola J, Goldwasser MA,
Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D,
Saunders J, et al: TP53 mutations and survival in squamous-cell
carcinoma of the head and neck. N Engl J Med. 357:2552–2561. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wolf ER, McAtarsney CP, Bredhold KE, Kline
AM and Mayo LD: Mutant and wild-type p53 form complexes with p73
upon phosphorylation by the kinase JNK. Sci Signal.
11:eaao41702018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin S, Yang X, Li J, Yang W, Ma H and
Zhang Z: p53-targeted lincRNA-p21 acts as a tumor suppressor by
inhibiting JAK2/STAT3 signaling pathways in head and neck squamous
cell carcinoma. Mol Cancer. 18:382019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Di Agostino S, Strano S, Emiliozzi V,
Zerbini V, Mottolese M, Sacchi A, Blandino G and Piaggio G: Gain of
function of mutant p53: The mutant p53/NF-Y protein complex reveals
an aberrant transcriptional mechanism of cell cycle regulation.
Cancer Cell. 10:191–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deyoung MP and Ellisen LW: p63 and p73 in
human cancer: Defining the network. Oncogene. 26:5169–5183. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lu H, Yang X, Duggal P, Allen CT, Yan B,
Cohen J, Nottingham L, Romano RA, Sinha S, King KE, et al: TNF-α
promotes c-REL/ΔNp63α interaction and TAp73 dissociation from key
genes that mediate growth arrest and apoptosis in head and neck
cancer. Cancer Res. 71:6867–6877. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Younes F, Quartey EL, Kiguwa S and
Partridge M: Expression of TNF and the 55-kDa TNF receptor in
epidermis, oral mucosa, lichen planus and squamous cell carcinoma.
Oral Dis. 2:25–31. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Osman AA, Neskey DM, Katsonis P, Patel AA,
Ward AM, Hsu TK, Hicks SC, McDonald TO, Ow TJ, Alves MO, et al:
Evolutionary action score of TP53 coding variants is predictive of
platinum response in head and neck cancer patients. Cancer Res.
75:1205–1215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou G, Wang J, Zhao M, Xie TX, Tanaka N,
Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, et al:
Gain-of-function mutant p53 promotes cell growth and cancer cell
metabolism via inhibition of AMPK activation. Mol Cell. 54:960–974.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ganci F, Pulito C, Valsoni S, Sacconi A,
Turco C, Vahabi M, Manciocco V, Mazza EMC, Meens J, Karamboulas C,
et al: PI3K inhibitors curtail MYC-dependent mutant p53
gain-of-function in head and neck squamous cell carcinoma. Clin
Cancer Res. 26:2956–2971. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Adduri RSR, George SA, Kavadipula P and
Bashyam MD: SMARCD1 is a transcriptional target of specific
non-hotspot mutant p53 forms. J Cell Physiol. 235:4559–4570. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Berkers CR, Maddocks OD, Cheung EC, Mor I
and Vousden KH: Metabolic regulation by p53 family members. Cell
Metab. 18:617–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Goldstein I and Rotter V: Regulation of
lipid metabolism by p53-fighting two villains with one sword.
Trends Endocrinol Metab. 23:567–575. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tanaka N, Zhao M, Tang L, Patel AA, Xi Q,
Van HT, Takahashi H, Osman AA, Zhang J, Wang J, et al:
Gain-of-function mutant p53 promotes the oncogenic potential of
head and neck squamous cell carcinoma cells by targeting the
transcription factors FOXO3a and FOXM1. Oncogene. 37:1279–1292.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Waitzberg AF, Nonogaki S, Nishimoto IN,
Kowalski LP, Miguel RE, Brentani RR and Brentani MM: Clinical
significance of c-myc and p53 expression in head and neck squamous
cell carcinomas. Cancer Detect Prev. 28:178–186. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu B, Liu P, Li J and Lu H: c-MYC
depletion potentiates cisplatin-induced apoptosis in head and neck
squamous cell carcinoma: Involvement of TSP-1 up-regulation. Ann
Oncol. 21:670–672. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Di Agostino S, Valenti F, Sacconi A,
Fontemaggi G, Pallocca M, Pulito C, Ganci F, Muti P, Strano S and
Blandino G: Long non-coding MIR205HG depletes Hsa-miR-590-3p
leading to unrestrained proliferation in head and neck squamous
cell carcinoma. Theranostics. 8:1850–1868. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chari NS, Ivan C, Le X, Li J, Mijiti A,
Patel AA, Osman AA, Peterson CB, Williams MD, Pickering CR, et al:
Disruption of TP63-miR-27a* feedback loop by mutant TP53 in head
and neck cancer. J Natl Cancer Inst. 112:266–277. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Valenti F, Sacconi A, Ganci F, Grasso G,
Strano S, Blandino G and Di Agostino S: The miR-205-5p/BRCA1/RAD17
axis promotes genomic instability in head and neck squamous cell
carcinomas. Cancers (Basel). 11:13472019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Verduci L, Ferraiuolo M, Sacconi A, Ganci
F, Vitale J, Colombo T, Paci P, Strano S, Macino G, Rajewsky N and
Blandino G: The oncogenic role of circPVT1 in head and neck
squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD
transcription-competent complex. Genome Biol. 18:2372017.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sargolzaei J, Etemadi T and Alyasin A: The
P53/microRNA network: A potential tumor suppressor with a role in
anticancer therapy. Pharmacol Res. 160:1051792020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bridges MC, Daulagala AC and Kourtidis A:
LNCcation: lncRNA localization and function. J Cell Biol.
220:e2020090452021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Guglas K, Bogaczyńska M, Kolenda T, Ryś M,
Teresiak A, Bliźniak R, Łasińska I, Mackiewicz J and Lamperska K:
lncRNA in HNSCC: Challenges and potential. Contemp Oncol (Pozn).
21:259–266. 2017.PubMed/NCBI
|
|
62
|
Liao JM, Cao B, Zhou X and Lu H: New
insights into p53 functions through its target microRNAs. J Mol
Cell Biol. 6:206–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Masciarelli S, Fontemaggi G, Di Agostino
S, Donzelli S, Carcarino E, Strano S and Blandino G:
Gain-of-function mutant p53 downregulates miR-223 contributing to
chemoresistance of cultured tumor cells. Oncogene. 33:1601–1608.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Donzelli S, Fontemaggi G, Fazi F, Di
Agostino S, Padula F, Biagioni F, Muti P, Strano S and Blandino G:
MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing
mutant p53 gain of function. Cell Death Differ. 19:1038–1048. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ganci F, Sacconi A, Bossel Ben-Moshe N,
Manciocco V, Sperduti I, Strigari L, Covello R, Benevolo M,
Pescarmona E, Domany E, et al: Expression of TP53
mutation-associated microRNAs predicts clinical outcome in head and
neck squamous cell carcinoma patients. Ann Oncol. 24:3082–3088.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jeck WR and Sharpless NE: Detecting and
characterizing circular RNAs. Nat Biotechnol. 32:453–461. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hsu MT and Coca-Prados M: Electron
microscopic evidence for the circular form of RNA in the cytoplasm
of eukaryotic cells. Nature. 280:339–340. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ashwal-Fluss R, Meyer M, Pamudurti NR,
Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N and
Kadener S: circRNA biogenesis competes with pre-mRNA splicing. Mol
Cell. 56:55–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL
and Yang L: Complementary sequence-mediated exon circularization.
Cell. 159:134–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Novo D, Heath N, Mitchell L, Caligiuri G,
MacFarlane A, Reijmer D, Charlton L, Knight J, Calka M, McGhee E,
et al: Mutant p53s generate pro-invasive niches by influencing
exosome podocalyxin levels. Nat Commun. 9:50692018. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Azmi AS, Bao B and Sarkar FH: Exosomes in
cancer development, metastasis, and drug resistance: A
comprehensive review. Cancer Metastasis Rev. 32:623–642. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bhatta B, Luz I, Krueger C, Teo FX, Lane
DP, Sabapathy K and Cooks T: Cancer cells shuttle extracellular
vesicles containing oncogenic mutant p53 proteins to the tumor
microenvironment. Cancers (Basel). 13:29852021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cooks T, Pateras IS, Jenkins LM, Patel KM,
Robles AI, Morris J, Forshew T, Appella E, Gorgoulis VG and Harris
CC: Mutant p53 cancers reprogram macrophages to tumor supporting
macrophages via exosomal miR-1246. Nat Commun. 9:7712018.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Burtness B, Harrington KJ, Greil R,
Soulières D, Tahara M, de Castro G Jr, Psyrri A, Basté N, Neupane
P, Bratland Å, et al: Pembrolizumab alone or with chemotherapy
versus cetuximab with chemotherapy for recurrent or metastatic
squamous cell carcinoma of the head and neck (KEYNOTE-048): A
randomised, open-label, phase 3 study. Lancet. 394:1915–1928. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Soussi T and Wiman KG: Shaping genetic
alterations in human cancer: The p53 mutation paradigm. Cancer
Cell. 12:303–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kravchenko JE, Ilyinskaya GV, Komarov PG,
Agapova LS, Kochetkov DV, Strom E, Frolova EI, Kovriga I, Gudkov
AV, Feinstein E and Chumakov PM: Small-molecule RETRA suppresses
mutant p53-bearing cancer cells through a p73-dependent salvage
pathway. Proc Natl Acad Sci USA. 105:6302–6307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cai BH, Bai ZY, Lien CF, Yu SJ, Lu RY, Wu
MH, Wu WC, Chen CC and Hsu YC: NAMPT inhibitor and P73 activator
represses P53 R175H mutated HNSCC cell proliferation in a
synergistic manner. Biomolecules. 12:4382022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bykov VJ, Zhang Q, Zhang M, Ceder S,
Abrahmsen L and Wiman KG: Targeting of mutant p53 and the cellular
redox balance by APR-246 as a strategy for efficient cancer
therapy. Front Oncol. 6:212016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Puca R, Nardinocchi L, Porru M, Simon AJ,
Rechavi G, Leonetti C, Givol D and D'Orazi G: Restoring p53 active
conformation by zinc increases the response of mutant p53 tumor
cells to anticancer drugs. Cell Cycle. 10:1679–1689. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Roh JL, Kang SK, Minn I, Califano JA,
Sidransky D and Koch WM: p53-reactivating small molecules induce
apoptosis and enhance chemotherapeutic cytotoxicity in head and
neck squamous cell carcinoma. Oral Oncol. 47:8–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hainaut P and Milner J: A structural role
for metal ions in the ‘wild-type’ conformation of the tumor
suppressor protein p53. Cancer Res. 53:1739–1742. 1993.PubMed/NCBI
|
|
82
|
Butler JS and Loh SN: Structure, function,
and aggregation of the zinc-free form of the p53 DNA binding
domain. Biochemistry. 42:2396–2403. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Maleki Vareki S, Salim KY, Danter WR and
Koropatnick J: Novel anti-cancer drug COTI-2 synergizes with
therapeutic agents and does not induce resistance or exhibit
cross-resistance in human cancer cell lines. PLoS One.
13:e01917662018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Salim KY, Maleki Vareki S, Danter WR and
Koropatnick J: COTI-2, a novel small molecule that is active
against multiple human cancer cell lines in vitro and in vivo.
Oncotarget. 7:41363–41379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Alexandrova EM, Yallowitz AR, Li D, Xu S,
Schulz R, Proia DA, Lozano G, Dobbelstein M and Moll UM: Improving
survival by exploiting tumour dependence on stabilized mutant p53
for treatment. Nature. 523:352–356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hsiue EH, Wright KM, Douglass J, Hwang MS,
Mog BJ, Pearlman AH, Paul S, DiNapoli SR, Konig MF, Wang Q, et al:
Targeting a neoantigen derived from a common TP53 mutation.
Science. 371:eabc86972021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Khan AS, Ahmad S, Ullah Z, Haq M, Farooq
MU and Khan M: Serum p53 antibodies detection in oral squamous cell
carcinoma, oral potentially malignant disorders and healthy
individuals: A multicentre study. J Pak Med Assoc. 71:2364–2368.
2021.PubMed/NCBI
|
|
88
|
Wang Q, Fan S, Eastman A, Worland PJ,
Sausville EA and O'Connor PM: UCN-01: A potent abrogator of G2
checkpoint function in cancer cells with disrupted p53. J Natl
Cancer Inst. 88:956–965. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Suganuma M, Kawabe T, Hori H, Funabiki T
and Okamoto T: Sensitization of cancer cells to DNA damage-induced
cell death by specific cell cycle G2 checkpoint abrogation. Cancer
Res. 59:5887–5891. 1999.PubMed/NCBI
|
|
90
|
Leijen S, Beijnen JH and Schellens JHM:
Abrogation of the G2 checkpoint by inhibition of Wee-1 kinase
results in sensitization of p53-deficient tumor cells to
DNA-damaging agents. Curr Clin Pharmacol. 5:186–191. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Osman AA, Monroe MM, Ortega Alves MV,
Patel AA, Katsonis P, Fitzgerald AL, Neskey DM, Frederick MJ, Woo
SH, Caulin C, et al: Wee-1 kinase inhibition overcomes cisplatin
resistance associated with high-risk TP53 mutations in head and
neck cancer through mitotic arrest followed by senescence. Mol
Cancer Ther. 14:608–619. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gadhikar MA, Sciuto MR, Alves MV,
Pickering CR, Osman AA, Neskey DM, Zhao M, Fitzgerald AL, Myers JN
and Frederick MJ: Chk1/2 inhibition overcomes the cisplatin
resistance of head and neck cancer cells secondary to the loss of
functional p53. Mol Cancer Ther. 12:1860–1873. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bridges KA, Hirai H, Buser CA, Brooks C,
Liu H, Buchholz TA, Molkentine JM, Mason KA and Meyn RE: MK-1775, a
novel Wee1 kinase inhibitor, radiosensitizes p53-defective human
tumor cells. Clin Cancer Res. 17:5638–5648. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Méndez E, Rodriguez CP, Kao MC, Raju S,
Diab A, Harbison RA, Konnick EQ, Mugundu GM, Santana-Davila R,
Martins R, et al: A phase I clinical trial of AZD1775 in
combination with neoadjuvant weekly docetaxel and cisplatin before
definitive therapy in head and neck squamous cell carcinoma. Clin
Cancer Res. 24:2740–2748. 2018. View Article : Google Scholar : PubMed/NCBI
|