Introduction
Globally, cervical cancer is the third most common
tumor and the fourth most common cause of cancer-related death in
women (1). The 5-year survival
rates for progressive cervical cancer are low, at ~55%, and these
rates range from 26.7–56% for recurrent cervical cancer (2). The majority of cervical cancer cases
are associated with certain types of high-risk types of human
papillomavirus (HPV) infection (3).
However, 3–8% of cervical cancer cases are persistently
HPV-negative (4). In addition,
patients with HPV-negative cervical cancer have a worse prognosis
than patients with HPV-positive cervical cancer (4).
Cancer recurrence and metastases are associated with
cancer dormancy, and are the leading cause of cancer-related death
in patients (5). Cancer dormancy
may explain for tumor refractoriness and the phenomenon that
initial treatment of the primary tumor cannot cure the malignancy
(6). Previous studies by the
authors confirmed that breast cancer cells seeded at metastatic
sites had a lower proliferative potential and remained quiescent
over a long period of time (7);
then, after a long time, they resumed active growth and progression
when the microenvironmental conditions of the metastatic site
shifted to support tumor expansion (8). Therefore, illuminating the mechanisms
regulating dormancy is critical for defending against disease
recurrence in patients with HPV-negative cervical cancer.
The snail family transcriptional repressor 2 (SNAI2)
gene is an evolutionarily conserved C2H2 zinc finger protein that
coordinates biological processes critical for tissue development
and tumorigenesis (9,10). It is a ubiquitous phenomenon that
SNAI2 is aberrantly expressed in human cancers and clearly
indicates poor prognosis in patients with cancer (9,11,12).
Upregulated expression of SNAI2, or its dysregulation, suggests
malignant characteristics during cancer progression (9–11,13).
In addition, SNAI2 plays a prominent role in tumor proliferation,
metastasis and angiogenesis (8).
However, the role of SNAI2 in dormancy, especially that of
HPV-negative cervical cancer cells, has not been fully elucidated.
Therefore, the present study investigated whether SNAI2
participated in HPV-negative cervical cancer cell dormancy. The
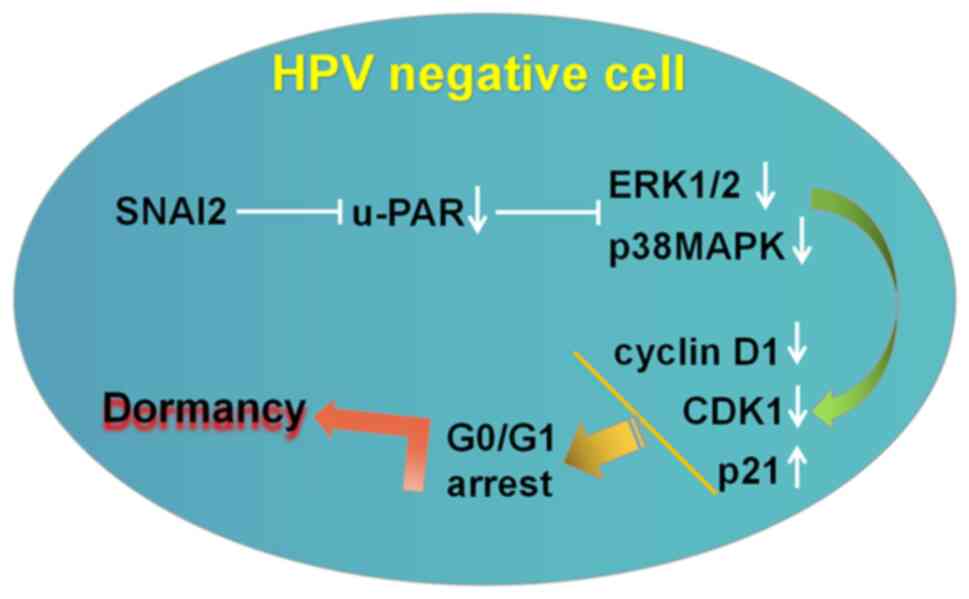
present study indicated that SNAI2 enhanced HPV-negative cervical
cancer cell dormancy, which was characterized by G0/G1 arrest, by
downregulating the expression of urokinase plasminogen activator
receptor (u-PAR), the activity of the ERK1/2 and p38MAPK signaling
pathways, and by further regulating the expression of
proliferation-associated genes (downregulation of cyclin D1 and
CDK1, upregulation of p21 expression) in vitro. Interference
with this process may provide new therapeutic strategies for
inhibiting the metastatic switch from dormancy to
proliferation.
Materials and methods
Cells and reagents
The cervical cancer C33A (HPV-negative) and HeLa
(HPV-positive) cell lines were purchased from the Type Culture
Collection of the Chinese Academy of Sciences, and maintained in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc.) with high glucose, 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.). The cells were maintained in 6-cm
plates at 37°C in an incubator with 5% CO2. The
C-33A-Wild group are untransfected C-33A cervical cancer cells.
Cell transduction
To induce upregulation of SNAI2, the coding
sequences of SNAI2 was synthesized and cloned into the GV367
overexpression plasmids by JIKAI GENE Company (https://www.genechem.com.cn/). Subsequently,
lentiviruses were produced by transfecting the 293T cells using
Lipofectamine 2000 (cat. no. 11668019; Thermo Fisher Scientific,
Inc.) with GV367-Ctrl and GV367- SNAI2 vectors. After 48 h, the
virus supernatants were harvested, filtered and concentrated.
Subsequently, the lentiviruses were added to the medium, supplied
with 5 µg/ml polybrene (cat. no. REVG0001; JIKAI GENE; http://www.genechem.com.cn/) to infect the C-33A cells
at 37°C in an incubator with 5% CO2 for 96 h.
Multiplicity of infection was 20. Finally, the cell lines with a
stable overexpression were constructed by treating the cells with
2.5 µg/ml puromycin (cat. no. HB-PU-1000; Hanbio Biotechnology Co.,
Ltd.) for 2 weeks.
Gene expression assessed by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was extracted from cells with TRIzol®
reagent (Invitrogen; Thermo Fisher scientific, Inc.). The relative
quantity of mRNA was determined by RT-qPCR, as previously described
(8). The primer sequences were as
follows: SNAI2 sense, 5′-AGGAATCTGGCTGCTGTG-3′ and antisense,
5′-GGAGAAAATGCCTTTGGAC-3′; Cyclin D1 sense,
5′-CAGATCATCCGCAAACACG-3′ and antisense,
5′-GGGACATCACCCTCACTTAC-3′; u-PAR sense,
5′-GAGCTATCGGACTGGCTTGAA-3′ and antisense,
5′-CGGCTTCGGGAATAGGTGAC-3′; CDK1 sense,
5′-AAACTACAGGTCAAGTGGTAGCC-3′ and antisense,
5′-TCCTGCATAAGCACATCCTGA-3′ p21 sense,
5′-GGACAGCAGAGGAAGACCATGT-3′, and antisense,
5′-TGGAGTGGTAGAAATCTGTCATGC-3′; and GAPDH sense,
5′-TCATTGACCTCAACTACATGGTTT-3′ and antisense,
5′-GAAGATGGTGATGGGATTTC-3′.
Cell cycle analysis by flow
cytometry
The tumor cells (1.0×105/well) were
cultured in 6-well plates for 3 days, and were then analyzed using
propidium iodide (Molecular Probes; Invitrogen; Thermo fisher
scientific, Inc.) to discriminate non-viable cells. DNA synthesis
or the total DNA content were measured by flow cytometry using a
FACSCalibur flow cytometer (BD Accuri™ C6; BD Biosciences). FlowJo
software (version 7.6.1; FlowJo LLC) was used for analysis of the
results.
Glucose consumption detected by
oxidase assay
The tumor cells (1.0×103/well) were
cultured in 96-well plates for 2 days. The glucose consumption in
cervical cancer cells, with or without upregulation of SNAI2, was
detected by oxidase assay (cat. no. 09000238813; Shanghai Rongsheng
Biotech Co., Ltd.), according to the manufacturer's protocol.
Senescence assay by
senescence-associated beta-galactosidase (SA-beta-Gal)
staining
The cells (C-33A-Wild, C-33A-SNAI2, HeLa and
HeLa-H2O2) were plated at a density of
3×103 cells/well in 10% FBS-containing medium. The
HeLa-H2O2 group cells were prepared by
incubating the cells with 0.9908 mmol/l H2O2
at 37°C for 4 h. The senescence of tumor cells was detected by
SA-beta-Gal staining at room temperature for 15 min (cat. no.
C0602; Beyotime Institute of Biotechnology), according to the
manufacturer's protocol.
Cell Counting Kit-8 (CCK-8)
proliferation assay
The cells (1×103/well) were resuspended
in 100 µl 10% FBS-containing medium and were cultured on plates for
24 h. The CCK-8 assay (WST; Dojindo Laboratories, Inc.) was then
used to measure cell proliferation according to the manufacturer's
protocol. The cells were incubated with 10 µl CCK-8 buffer and 90
µl of DMEM at 37°C for 4 h. The OD value was then measured at 450
nm.
Western blot assay (WB)
WB was performed as previously described (8). Total protein was extracted from the
cells using RIPA buffer (Thermo Fisher Scientific, Inc.). The
protein concentration was measured using the Bradford Protein
Assays (Thermo Fisher Scientific, Inc.). A total of 40–70 µg
proteins were then separated by SDS-PAGE on 12% gels, followed by
transfer onto PVDF membranes. The PVDF membranes were blocked with
5% non-fat milk and incubated with primary antibodies [SNAI2
(1:1,000; cat. no. sc-166476; Santa Cruz Biotechnology, Inc.),
ERK1/2 (1:1,000; cat. no. sc-514302; Santa Cruz Biotechnology,
Inc.), phosphorylated (p-)ERK1/2 (1:2,000; cat. no. sc-7976; Santa
Cruz Biotechnology, Inc.), p38 (1:1,000; cat. no. 9212; Cell
Signaling Technology, Inc.), p-p38 (1:1,000; cat. no. 9211; Cell
Signaling Technology, Inc.) and β-actin (1:2,000; cat. no. 4967;
Cell Signaling Technology, Inc.)] at 4°C overnight. After washing
with PBS-Tween (Beyotime Institute of Biotechnology), the membranes
were incubated with secondary antibodies for 2 h at room
temperature. All bands were washed in TBS with 0.05% Tween-20 and
immunoblotted with peroxidase-conjugated anti-mouse or anti-rabbit
secondary antibodies, respectively. The bands were detected using
an enhanced ECL-Plus kit (cat. no. P1010-25; Applygen Technologies,
Inc.) and exposed to film. ImageJ 2019 (National Institutes of
Health) was used for densitometry. Actin protein was used for all
western blotting as a loading control.
Statistical analysis
Results were expressed as the mean ± standard
deviation of three independent experiments with a total of six
samples and interpreted by one-way ANOVA followed by Tukey's post
hoc test. P<0.05 was considered to indicate a statistically
significant difference. SPSS software (version 17.0; SPSS, Inc.)
was used for statistical analyses.
Results
Establishment of HPV-negative cervical
cancer cells with stable expression of SNAI2
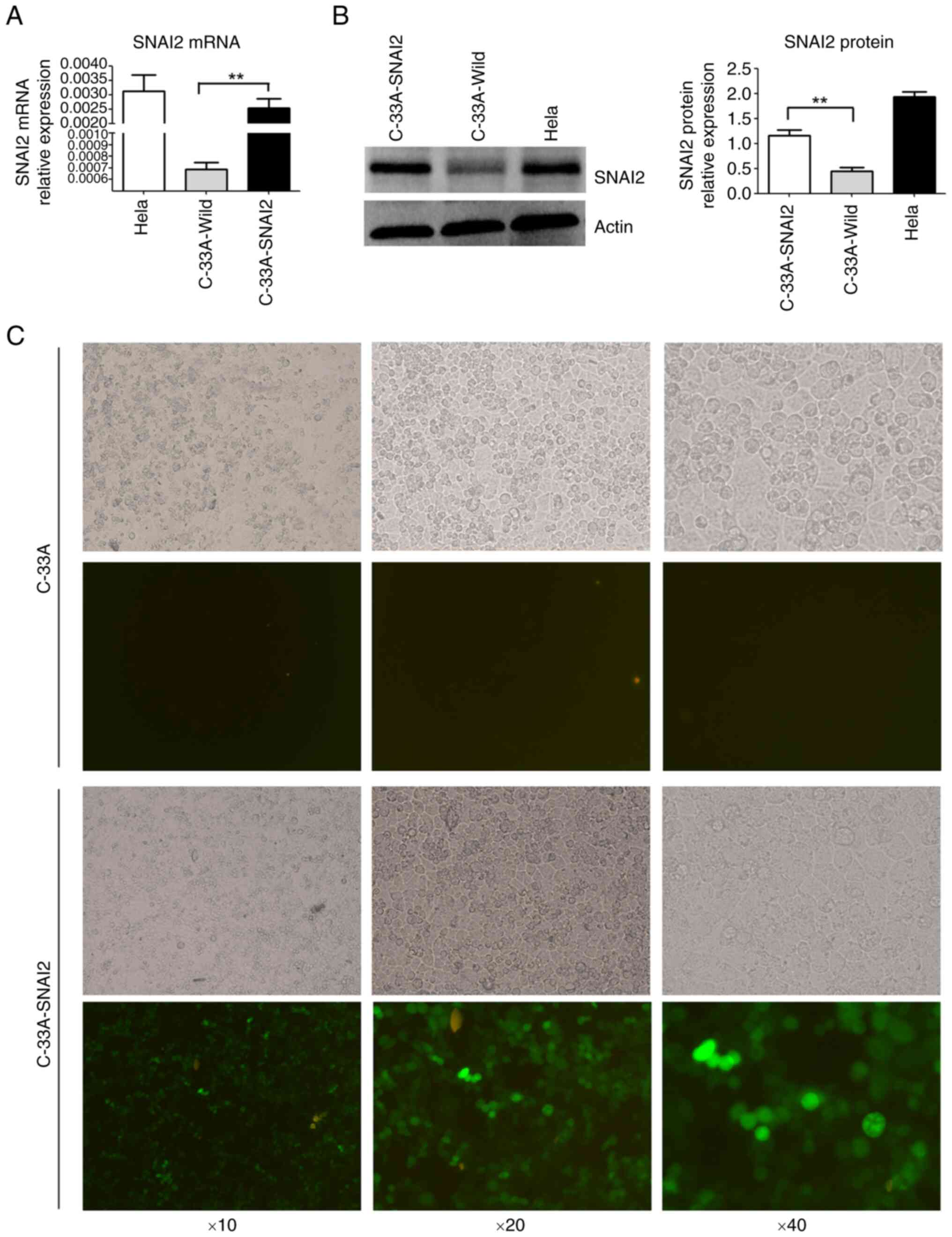
SNAI2 expression was detected using RT-qPCR and WB
in cervical cancer cell lines. A high level of SNAI2 expression was
detected in the HPV-positive cervical carcinoma HeLa cell line, but
almost no SNAI2 expression was detected in the HPV-negative
cervical carcinoma C-33A cell line (Fig. 1A and B).
To further investigate the function of SNAI2 in
HPV-negative cervical cancer cells, exogenous SNAI2 was stably
overexpressed in C-33A cells (C-33A-SNAI2) using GV367-SNAI2
lentiviral particles, and expression was determined using RT-qPCR,
WB and fluorescence microscopy (Fig.
1C). The results showed that the mRNA and protein expression
levels of SNAI2 were high in C-33A-SNAI2 cells (Fig. 1).
SNAI2 promotes HPV-negative cervical
cancer cell dormancy in vitro
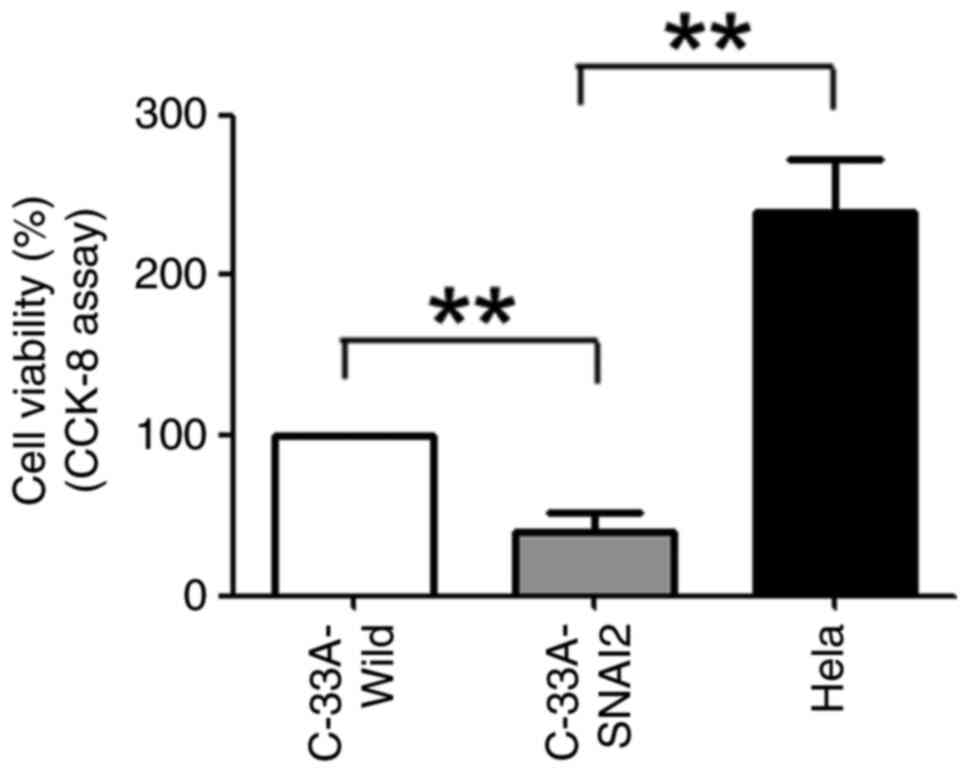
To investigate whether SNAI2 expression promoted the
dormancy of HPV-negative cervical cancer cells, C-33A-Wild,
C-33A-SNAI2 and HeLa cells were first cultured for 24 h, and cell
proliferation was tested using the CCK-8 method. The results
revealed that the viability of C-33A-SNAI2 cells was significantly
lower than that of their respective control cells (C-33A-Wild and
HeLa), indicating that upregulation of the SNAI2 gene suppressed
the proliferation of HPV-negative cervical cancer C-33A cells
(Fig. 2).
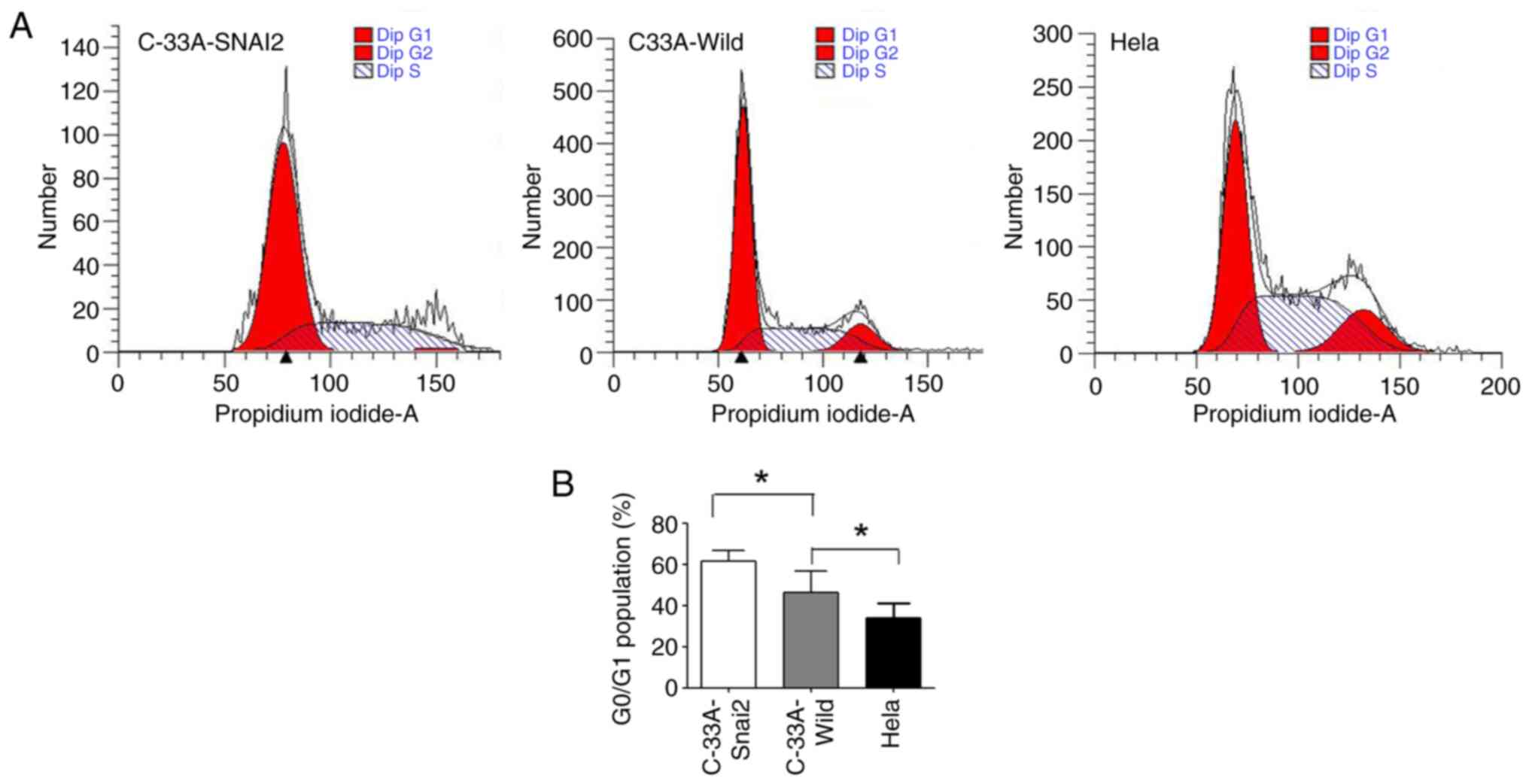
It was further investigated whether the upregulation
of SNAI2 affected the cell cycle regulation in HPV-negative
cervical cancer C-33A cells. The present results revealed that a
high percentage of HPV-negative cervical cancer C-33A cells
remained in the G0/G1 phase, with a smaller S and G2/M cell
population, when SNAI2 expression was upregulated, suggesting that
the cells remained arrested at G0 phase (Fig. 3). In addition, HPV-positive cervical
cancer HeLa cells maintained the highest viability, with the
smallest population in the G0/G1 phase. In conclusion, SNAI2 may
administrate difference function in HPV-positive and HPV-negative
cervical cancer cells.
All of these results demonstrated that
overexpression of the SNAI2 gene inhibited the proliferation of
HPV-negative cervical cancer cells by inducing cell cycle arrest
during the G0/G1 phase of the cell cycle.
Senescence plays a fundamental role in some
age-related diseases, such as osteoarthritis, glaucoma, diabetes
and cancer (14). The critical
characteristic for senescence is cell cycle arrest; and senescent
cells exhibit permanent proliferation arrest with increased
expression of cell cycle inhibitors (15), meaning that senescent cells cannot
restart proliferation after release from cell cycle arrest
(14,15).
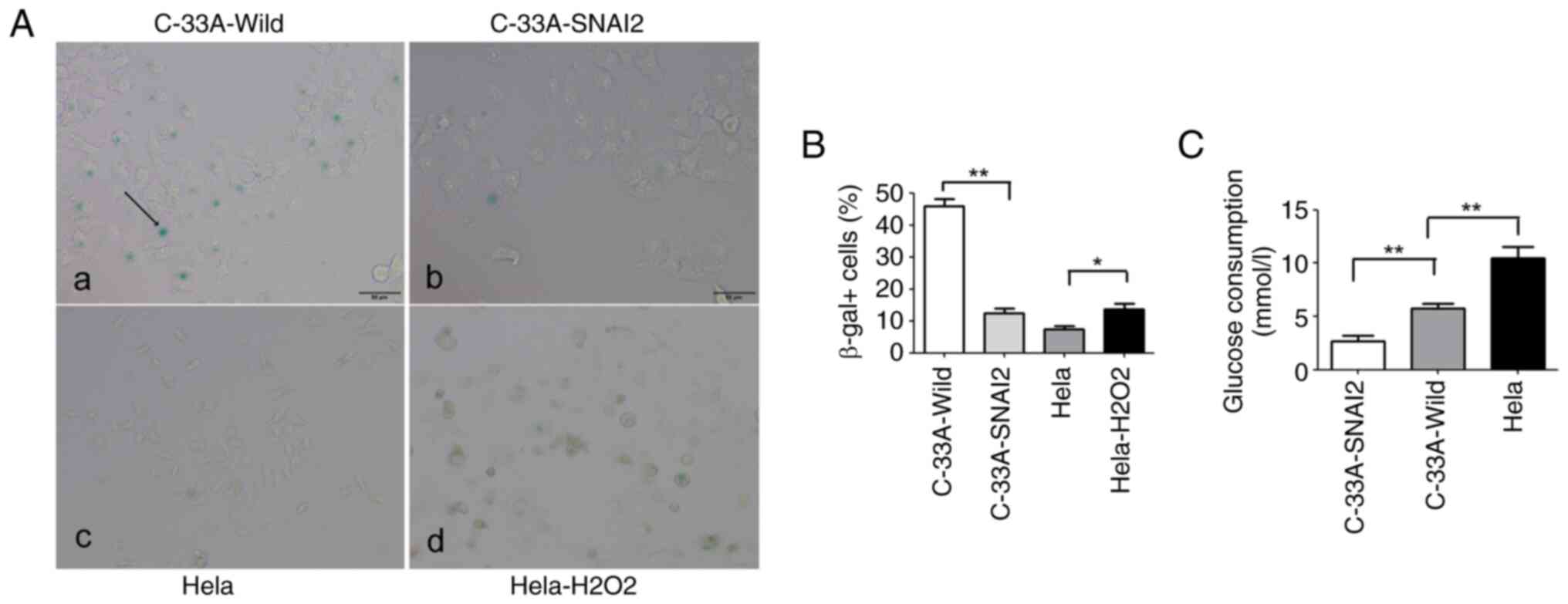
To further examine whether SNAI2-mediated C-33A cell
cycle arrest was due to the promotion of cell entry into dormancy
or senescence, tumor cells were stained for expression of the
senescence marker SA-beta-Gal. It was observed that C-33A-SNAI2
cells lacked the typical morphological signs of senescence
(including cell enlargement, flattening and long cytoplasmic
projections), and the percentage of cells positive for the
senescence marker SA-beta-Gal was significantly decreased in
C-33A-SNAI2 cells, compared with that in C-33A-Wild cells,
indicating that the upregulation of SNAI2 was required for the
proliferating-to-dormant switch of HPV-negative cervical cancer
C-33A cells via the induction of G0/G1 arrest, but not the
enhancement of cell senescence (Fig. 4A
and B).
Glucose plays an important role in the survival and
proliferation of tumor cells, and it was previously demonstrated
that the dependence of cancer cells on glucose promotes survival
and cellular proliferation (16).
To further identify whether the upregulation of the SNAI2 gene was
involved in regulating glucose consumption in C-33A cells, glucose
consumption of tumor cells was assessed using oxidase assays. The
present results revealed that C-33A-SNAI2 cells had lower glucose
consumption than C-33A-Wild and HeLa tumor cells (Fig. 4C).
SNAI2 regulates the expression of
proliferation-related genes by suppressing the ERK/p38 pathway
ratio
SNAI2 is involved in mediating the proliferation,
metastasis and angiogenesis of tumors (17). SNAI2 enhances the kinase activity of
cyclin D1/CDK4/CDK6, which is a central mediator in the transition
from the G1 to the S phase, to further promote the switch to tumor
cell proliferation (18).
Therefore, SNAI2 may participate in the proliferating-to-dormant
switch of tumor cells.
The u-PAR is identified in tumor cells with a high
potential for invasion, metastasis, dormancy and proliferation,
upon downregulation of which, the p38SAPK pathway is induced, and a
G0/G1 arrest representing tumor cell dormancy is initiated
(19).
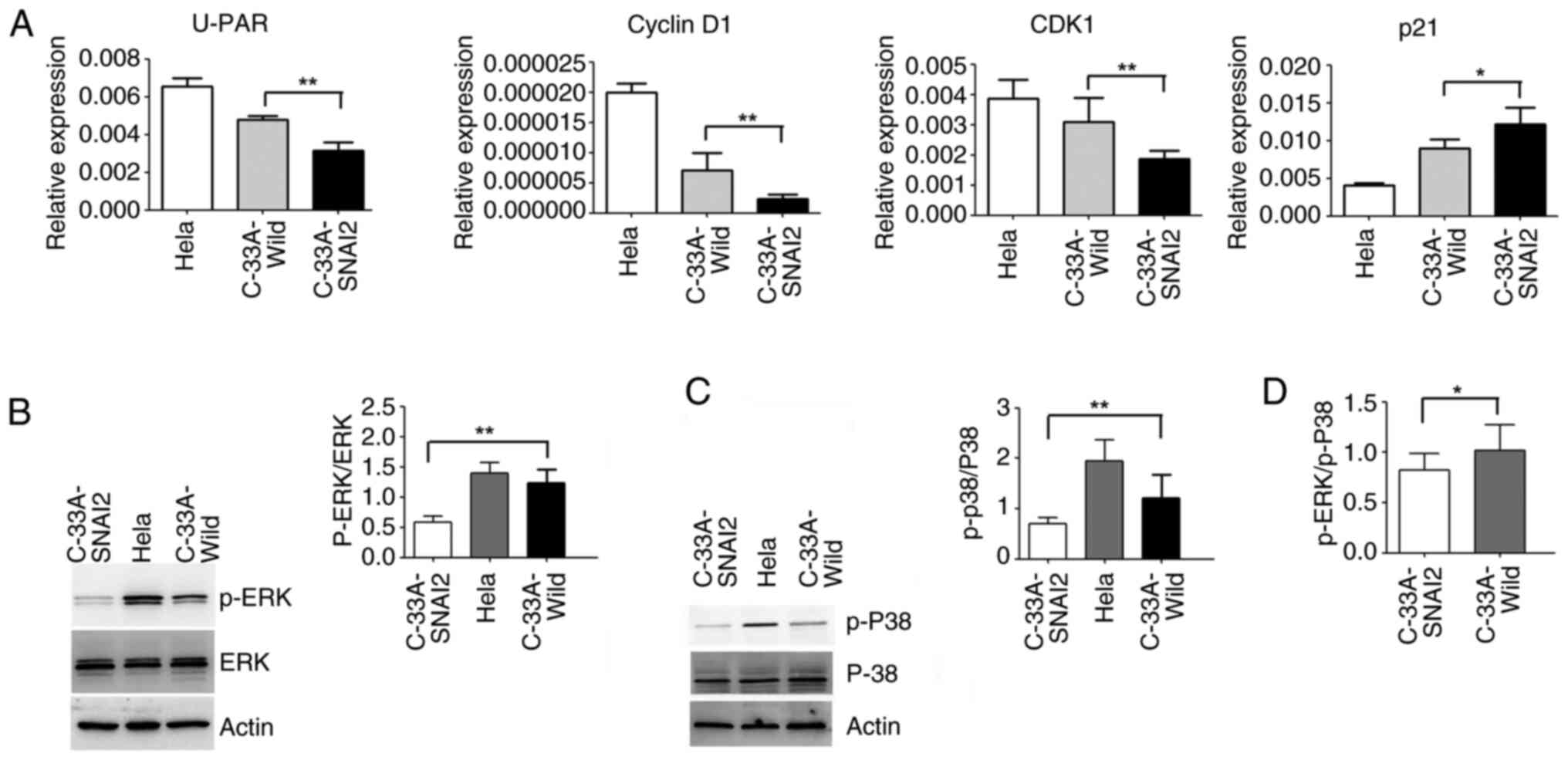
To confirm the role of SNAI2 in the
proliferation-to-dormancy switch of HPV-negative cervical cancer
cells, the dormant tumor cells were analyzed using RT-qPCR and WB.
As demonstrated in Fig. 5A, the
upregulation of SNAI2 could enhance the increase in p21 expression,
and the decrease in CDK1, u-PAR and cyclin D1 expression in C-33A
cells, compared with that of C-33A-Wild cells.
A previous study reported that the ERK activity
level and ERK/p38 activity balance are valid general predictors of
tumorigenicity and dormancy, and a high ERK/p38 ratio allows for
cell cycle progression and tumor growth (20). Disruption of this complex caused an
inversion in the ERK/p38 ratio toward p38 signaling; a change that
results in cell cycle arrest and dormancy in breast cancer,
prostate cancer, melanoma and fibrosarcoma cell lines (20).
To further clarify the mechanisms underlying the
determination of HPV-negative cervical cancer C-33A cell dormancy,
the activation of ERK and p38MAPK was detected using WB. The
results verified that the sustained activation of p-ERK and
p-p38MAPK was significantly decreased when SNAI2 expression was
upregulated in C-33A cells (Fig. 5B and
C). The p-ERK/p-p38 ratio in C-33A-SNAI2 cells also revealed a
downward trend compared with that of C-33A cells (Fig. 5D).
Discussion
Tumor cell dormancy is defined as the temporary
mitotic G0/G1 arrest of tumor growth (21). In tumor mass dormancy, the number of
cancer cells remains balanced between cell division and apoptosis
(21). These dormant tumor cells go
undetected for long periods, several years or even decades, and may
explain prolonged asymptomatic residual disease and treatment
resistance. Unfortunately, dormant tumor cells may eventually
result in cancer recurrence and death (22). However, the mechanisms underlying
the reactivation of dormant tumor cells to a proliferative state
are not yet known.
Notably, aberrant SNAI2 expression is a widespread
phenomenon in human cancers, which can predict poor prognosis in
patients with cancer (23). In
addition, it has been found to have different effects on tumor
growth, recurrence and metastasis in various carcinomas (24). SNAI2 can enhance tumor cell
proliferation and growth in lung cancer and glioblastoma (23), increase tumor cell recurrence and
metastasis in breast cancer (8),
and promote the development of ovarian cancer through regulating
ferroptosis (25).
However, SNAI2 acts as an oncogene or tumor
suppressor during cervical carcinogenesis (24,26).
Cui et al (24) reported
that SNAI2 can suppress the proliferation of HPV-negative cervical
cancer cells in vitro and tumor formation in vivo. In
HPV-positive cervical Siha and HeLa cells with high tripartite
motif containing 62 expression, cell proliferation is inhibited
when SNAI2 expression is downregulated (26). The aforementioned studies showed
that SNAI2 are diverse and context-dependent, reminiscent of the
various cancer types and heterogeneity of the tumor cells. Notably,
the present study demonstrated that upregulation of SNAI2
expression could inhibit proliferation, and promote cell dormancy
in HPV-negative cervical cancer C-33A cells. Overall proliferative
capacity and glucose consumption were significantly decreased, and
G0/G1 phase arrest was detected when SNAI2 was highly expressed in
HPV-negative cervical cancer C-33A cells. At the same time,
C-33A-SNAI2 cells lacked the typical morphological signs of
senescence, and the percentage of cells positive for the senescence
marker SA-beta-Gal was significantly decreased in the C-33A-SNAI2
group. Briefly, SNAI2 could enhance HPV-negative cervical cancer
C-33A cell dormancy by inducing G0/G1 arrest, but not senescence,
which is not only decreased cell proliferation capacity and tumor
formation of cervical cancer cells (24).
A previous study showed that SNAI2 was a trigger in
the regulation of breast cancer dormancy by regulating cyclin
D1/CDK4/CDK6 activity (18), which
involved the transition from G1 to S phase, to further promote the
switch to tumor cell proliferation (20), thus indicating that cyclins may
participate in the SNAI2-mediated proliferation-to-dormant switch
of cervical cancer cells. Cyclins are a key factor in cell cycle
turnover and initiate cell proliferation potential (27). As an inhibitory protein of CDKs, p21
mediates p53-dependent cell cycle arrest and inhibits CDK1 activity
by combining the complex formed of cyclin and CDK1. p21 directly
inhibits DNA synthesis, arrests the cell cycle in the G1 phase,
leads to the disappearance of cyclin biology via competitive
inhibition of the cyclin/CDK complex, and negatively regulates cell
proliferation (28,29). It was further demonstrated that
upregulation of SNAI2 could downregulate cyclin D1 and CDK1
expression, and upregulate p21 expression, further enhancing tumor
dormancy in HPV-negative cervical cancer cells.
Increasing evidence has suggested that u-PAR is a
critical determinant of the shift between the proliferation and
dormancy of solitary tumor cells (21,30),
the blockade of which could induce apoptosis in brain tumor cells;
however, the induction of which could protract Hep3 tumor cell
dormancy via G0/G1 arrest in vivo (27,30).
u-PAR expression and activation generate a high level of ERK
activity and low level of p38 activity, which are necessary for the
in vivo proliferation of cancer cells. The high ERK activity
feeds into a positive feedback loop that transactivates u-PAR
expression, and high levels of u-PAR maintain high ERK activity by
activating α5β1 (20). Notably, the
ERKMAPK/p38SAPK activity ratio predicts
whether cells proliferate or enter a state of dormancy (20); a high ratio favors tumor growth,
whereas a low ratio promotes tumor growth arrest (dormancy). ERK is
negatively regulated by p38MAPK in breast cancer, prostate cancer,
melanoma and fibrosarcoma cell lines (31), with u-PAR being a crucial modulator
of this process (21). However,
whether the ERK and p38 pathway participates in regulating
SNAI2-mediated dormancy in HPV-negative cervical cancer cells
remains to be determined.
The present study identified that upregulation of
SNAI2 could downregulate u-PAR expression, to further enhance the
decreased activity of p-ERK and p-p38MAPK in HPV-negative cervical
cancer cells. Notably, the p-ERK/p-p38MAPK activity ratio in
C-33A-SNAI2 cells was also decreased compared with in the control
group (C-33A cells) (Fig. 6). The
present findings are consistent with the conclusions of Julio et
al (20), which revealed that
high ERK1/2 and p38 MAPK activity coexist in promoting the
proliferative activity of melanoma M24MET cells (32,33),
due to several isoforms of p38MAPK, α, γ and δ (24). By contrast, SNAI2 could
trans-suppress the expression of Akt1/p-Akt1 by binding to E-box
motifs in the promoter of the Akt1 gene and then inhibit the cell
proliferation and tumor formation, not promote dormancy of cervical
cancer cells, by upregulating p21/p27 and/or downregulating the
activity of the Wnt/β-catenin signaling pathway (24). Altogether, SNAI2 is more specific to
a particular type of cancer or malignant phenotype (34).
In conclusion, cancer recurrence and metastasis are
the leading cause of cervical cancer deaths. Because the
upregulation of SNAI2 is required for promoting HPV-negative
cervical cancer cell dormancy, interfering the u-PAR expression and
the activity of the ERK/p38 signaling pathway may represent a
viable strategy for the treatment of patients with HPV-negative
cervical cancer, providing novel insight into their treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Nature Science
Foundation of China (grant nos. 81403163 and 81402404), the Science
and Technology Research Project of Hubei Provincial Education
Department (grant no. D20201204), the Yi Chang Scientific and
Technological Bureau (grant no. A22-2-017), the Hubei Provincial
Natural Science Foundation (grant nos. 2022CFB037 and 2024AFB832)
and the Hubei Provincial Administration of Traditional Chinese
Medicine (grant no. ZY2023M039).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YHZ and QL contributed to the conceptualization and
design of the present study. YZL, YX SLW and QH contributed to
performing the experiments and the data analyses. YHZ, QL, YX and
SLW wrote the original draft. YHZ, QL and YX contributed to the
interpretation of the results in the present study. All authors
read and approved the final manuscript. YHZ and YX confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
u-PAR
|
urokinase plasminogen activator
receptor
|
|
CCK-8
|
Cell Counting Kit-8
|
References
|
1
|
Tao P, Sun L, Sun Y, Wang Y, Yang Y, Yang
B and Li F: ISG15 is associated with cervical cancer development.
Oncol Rep. 24:3802022.
|
|
2
|
Melan K, Janky E, Macni J, Ulric-Gervaise
S, Dorival MJ, Veronique-Baudin J and Joachim C: Epidemiology and
survival of cervical cancer in the French West-Indies: Data from
the martinique cancer registry (2002–2011). Glob Health Action.
10:13373412017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Castro-Muñoz LJ, Manzo-Merino J,
Muñoz-Bello JO, Olmedo-Nieva L, Cedro-Tanda A, Alfaro-Ruiz LA,
Hidalgo-Miranda A, Madrid-Marina V and Lizano M: The human
papillomavirus (HPV) E1 protein regulates the expression of
cellular genes involved in immune response. Sci Rep. 9:136202019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee JE, Chung Y, Rhee S and Kim TH: Untold
story of human cervical cancers: HPV-negative cervical cancer. BMB
Rep. 55:429–438. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang HF, Wang SS, Huang MC, Liang XH, Tang
YJ and Tang YL: Targeting immune-mediated dormancy: A promising
treatment of cancer. Front Oncol. 9:4982019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goss PE and Chambers AF: Does tumour
dormancy offer a therapeutic target? Nat Rev Cancer. 10:871–877.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou YH, Liao SJ, Li D, Luo J, Wei JJ, Yan
B, Sun R, Shu Y, Wang Q, Zhang GM and Feng ZH: TLR4
ligand/H2O2 enhances TGF-β1 signaling to
induce metastatic potential of non-invasive breast cancer cells by
activating non-Smad pathways. PLoS One. 8:e659062013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou Y, Liu Q, Dai X, Yan Y, Yang Y, Li H,
Zhou X, Gao W, Li X and Xi Z: Transdifferentiation of type II
alveolar epithelial cells induces reactivation of dormant tumor
cells by enhancing TGF-β1/SNAI2 signaling. Oncol Rep. 39:1874–1882.
2018.PubMed/NCBI
|
|
9
|
Zhou W, Gross KM and Kuperwasser C:
Molecular regulation of Snai2 in development and disease. J Cell
Sci. 132:jcs2351272019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coll-Bonfill N, Peinado VI, Pisano MV,
Párrizas M, Blanco I, Evers M, Engelmann JC, Garcı́a-Lucio J,
Tura-Ceide O, Meister G, et al: Slug is increased in vascular
remodeling and induces a smooth muscle cell proliferative
phenotype. PLoS One. 11:e01594602016.11 View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu X, Feng Q, Zhang Y, Zheng P and Cui N:
Absence of EpCAM in cervical cancer cells is involved in
sluginduced epithelialmesenchymal transition. Cancer Cell Int.
21:1632021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Calcinotto A, Kohli J, Zagato E,
Pellegrini L, Demaria M and Alimonti A: Cellular senescence: Aging,
cancer, and injury. Physiol Rev. 99:1047–1078. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Leontieva OV, Lenzo F, Demidenko ZN and
Blagosklonny MV: Hyper-mitogenic drive coexists with mitotic
incompetence in senescent cells. Cell Cycle. 11:4642–4649. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martuscello RT, Vedam-Mai V, McCarthy DJ,
Schmoll ME, Jundi MA, Louviere CD, Griffith BG, Skinner CL, Suslov
O, Deleyrolle LP and Reynolds BA: A supplemented high-fat
low-carbohydrate diet for the treatment of glioblastoma. Clin
Cancer Res. 22:2482–2495. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shih JY and Yang PC: The EMT regulator
SNAI2 and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Casimiro MC, Velasco-Velázquez M,
Aguirre-Alvarado C and Pestell RG: Overview of cyclins D1 function
in cancer and the CDK inhibitor landscape: Past and present. Expert
Opin Investig Drugs. 23:295–304. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Allgayer H: Translational research on
u-PAR. Eur J Cancer. 46:1241–1251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aguirre-Ghiso JA, Estrada Y, Liu D and
Ossowski L: ERKMAPK activity as a determinant of tumor growth and
dormancy; regulation by p38SAPK. Cancer Res. 63:1684–1695.
2003.PubMed/NCBI
|
|
21
|
Tamamouna V, Pavlou E, Neophytou CM,
Papageorgis P and Costeas P: Regulation of metastatic tumor
dormancy and emerging opportunities for therapeutic intervention.
Int J Mol. 23:139312022. View Article : Google Scholar
|
|
22
|
Gomis RR and Gawrzak S: Tumor cell
dormancy. Mol Oncol. 11:62–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang YP, Wang MZ, Luo YR, Shen Y and Wei
ZX: Lentivirus-mediated shRNA interference targeting SLUG inhibits
lung cancer growth and metastasis. Asian Pac J Cancer Prev.
13:4947–4951. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cui N, Yang WT and Zheng PS: Slug inhibits
the proliferation and tumor formation of human cervical cancer
cells by up-regulating the p21/p27 proteins and down regulating the
activity of the Wnt/β-catenin signaling pathway via the
trans-suppression Akt1/p-Akt1 expression. Oncotarget.
7:26152–26167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin Y, Chen L, Li L, Huang G, Huang H and
Tang C: SNAI2 promotes the develop- ment of ovarian cancer through
regulating ferroptosis. Bioengineered. 13:6451–6463. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu TY, Chen J, Shang CL, Shen HW, Huang
JM, Liang YC, Wang W, Zhao YH, Liu D, Shu M, et al: Tripartite
motif containing 62 is a novel prognostic marker and suppresses
tumor metastasis via c-Jun/Slug signaling-mediated
epithelial-mesenchymal transition in cervical cancer. J Exp Clin
Cancer Res. 35:1702016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Allgayer H and Aguirre-Ghiso JA: The
urokinase receptor (u-PAR)-a link between tumor cell dormancy and
minimal residual disease in bone marrow? APMIS. 116:602–614. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Y, Wang S, Qian W, Ji D, Wang Q,
Zhang Z, Wang S, Ji B, Fu Z and Sun Y: uc.338 targets p21 and
cyclin D1 via PI3K/AKT pathway activation to promote cell
proliferation in colorectal cancer. Oncol Rep. 40:1119–1128.
2018.PubMed/NCBI
|
|
29
|
Fu T, Liang A and Liu Y: [Role of P21 in
resistance of lung cancer]. Zhongguo Fei Ai Za Zhi. 23:597–602.
2020.(In Chinese). PubMed/NCBI
|
|
30
|
Aguirre-Ghiso JA, Liu D, Mignatti A,
Kovalski K and Ossowski L: Urokinase receptor and fibronectin
regulate the ERKMAPK to p38MAPK activity ratios that determine
carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell.
12:863–879. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Horák P, Kreisingerová K, Réda J,
Ondrušová L, Balko J, Vachtenheim J Jr, Žáková P and Vachtenheim J:
The hedgehog/GLI signaling pathway activates transcription of slug
(Snail2) in melanoma cells. Oncol Rep. 49:752023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
33
|
Kim HJ, Hong I, Roh S, Kim S, Kim H, Oh S,
Ahn TS, Kang DH, Baek MJ and Jeong D: High expression of LY6E is an
independent prognostic factor of colorectal cancer patients. Oncol
Rep. 49:802023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu R, Zhao Y, Su S, Kwabil A, Njoku PC,
Yu H and Li X: Unveiling cancer dormancy: Intrinsic mechanisms and
extrinsic forces. Cancer Lett. 591:2168992024. View Article : Google Scholar : PubMed/NCBI
|