Introduction
Esophageal cancer, the sixth leading cause of
cancer-related mortality worldwide, poses a significant health
concern. Esophageal squamous cell carcinoma (ESCC) and esophageal
adenocarcinoma, which are the two major subtypes of esophageal
cancer, are epidemiologically and biologically distinct. ESCC is
particularly prevalent in populous Asian regions with a weak
alcohol reductase and acetaldehyde reductase expression, and
accounts for ~90% of all esophageal cancer cases (1). Additionally, esophageal cancer is
known to be one of the solid cancers with a very poor prognosis
despite multidisciplinary treatment. This is due to malnutrition
associated with the cancerous stenosis of the esophageal lumen,
metastases to distant organs and lymph nodes, and its invasiveness
to other organs around the esophagus (2).
In cases of esophageal cancer progression, not only
cancer cells, but also the tumor microenvironment constructed from
the extracellular matrix, fibroblasts, immune cells, blood vessels,
cytokines and chemokines, and exosomes, are considered to play a
critical role. Each of these factors greatly contributes to cancer
cell proliferation, invasiveness and metastatic potential, and the
fibrosis of the cancer stroma. In particular, investigating the
roles and mechanisms of cancer-associated fibroblasts (CAFs), which
are intratumoral fibroblasts, in the cancer microenvironment is
necessary to elucidate the pathology and develop new therapeutic
methods.
Cellular senescence has been reported to be involved
in cancer growth and progression (3). Cellular senescence, along with
apoptosis, is considered a major pathway of cancer suppression. The
senescence-associated secretory phenotype (SASP), which indicates
the secretion of inflammatory cytokines [interleukin (IL)-6, IL-1,
IL-8, TNF-α, and INF-γ], chemokines [growth-regulated protein
(GRO)-α, C-X-C motif chemokine ligand (CXCL), CC chemokine ligand],
extracellular matrix remodeling factors [matrix metalloproteases
(MMPs) and so on] and growth factors (hepatocyte growth factor,
platelet-derived growth factor AA, fibroblast growth factor,
epidermal growth factor and vesicular endothelial growth factor)
secreted from senescent CAFs, is a key factor affecting the
fibrosis of the cancer stroma, cancer a paracrine mechanism which
induces migration and invasiveness in the epithelial mesenchyme, a
hallmark of malignancy (1).
Senescent stromal cells also secrete membrane cofactor protein,
colony-stimulating factor, macrophage inflammatory protein, GRO and
CXCL, and may have an amplification activation loop to replenish
inflammatory and immune cells that also secrete pro-angiogenic
factors (vascular endothelial growth factor, IL-8 and MMPs)
(3). In fibroblasts surrounding
ovarian cancer, the chemokine GRO-1 has been reported to induce
fibroblast senescence via the RAS signaling pathway, which in turn
contributes to ovarian tumorigenesis (4). SASP is among the homeostatic
mechanisms inherent in cells to prevent carcinogenesis. In this
phenomenon, cells undergo irreversible cell cycle arrest by DNA
damage, such as telomere shortening and oncogene activation
(5).
Unlike apoptosis, senescent cells are considered to
survive for a long period of time without immediately dying. In the
mechanism of tissue repair, progenitor cells and fibroblasts
undergo cellular senescence in the body (6). The process of cellular senescence is
as follows: Telomere shortening due to mitotic lifespan and
accumulation of DNA damage due to external stress induce the
activation of growth inhibitory factors, including the p53-p21 and
p16 pathways. RB protein is constitutively activated by the
induction of the CDK inhibitors, p21 and p16. RB protein activation
inhibits the transcriptional activity of E2F, a transcription
factor required for the transition from the G1 phase to the S
phase, resulting in cell cycle arrest and cellular senescence. p16
binds to CDK4/6 and p21 binds to CDK2. This results in complete RB
activation, which alone, is insufficient (7–9).
Several factors alter the expression of genes encoding the SASP
factors. In particular, C/EBPβ and NF-κB are considered major
factors. Although cellular senescence is considered to be involved
in tissue repair and immune cell migration, it is also considered
to be involved in the promotion of carcinogenesis and the
development of age-related diseases through the action of SASP
(1).
Conversely, it has become clear that
epithelial-mesenchymal transition (EMT) is involved in the
metastasis and invasion of esophageal cancer (10). In the EMT process, epithelial cells
lose their cell polarity and cell adhesion function with
surrounding cells by responding to intracellular and extracellular
signaling pathways (11). EMT is
induced by several transcription factors and cytokines, including
transforming growth factor-β (TGF-β) and cell growth factors,
increasing the expression of these transcription factors and
inducing EMT (12,13). These SASP-associated factors are
considered to promote tumor growth by inducing EMT (3,14).
As a drug affecting SASP, rapamycin suppresses SASP
expression by repressing NF-κB activation through the inhibition of
IL-1α translation by mammalian target of rapamycin (mTOR)C1
(15). Additionally, metformin,
which is used widely as an antidiabetic drug, has been reported to
inhibit mTOR. The inhibition of the mTOR and Stat3 pathways by
metformin suppresses the action of SASP (16).
Controlling the induction of SASP from senescent
fibroblasts in cancer tissues suppresses the paracrine effects on
cancer cells; however, the details of this mechanism remain
unclear. The present study aimed to elucidate the in vitro
SASP-mediated interactions between ESCC cells and radiation-induced
senescent CAFs in the tumor microenvironment. Additionally, the
present study aimed to determine the effect of preventing cancer
progression through the regulation of SASP by drug repositioning
using metformin.
Materials and methods
Ethics approval
The present study was approved by the Institutional
Review Board of Kanazawa University (study no. 2016-463). Written
informed consent was obtained from all patients for the use of
surgically resected specimens.
Cell line and cell culture
The human ESCC cell line, KES, has been previously
established in the authors' laboratory from endoscopic biopsy
specimens obtained from a patient carrying a well-differentiated
ESCC (17). KES cells were cultured
in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% heat-inactivated fetal bovine serum (FBS; Nichirei
Biosciences Inc.), 100 IU/ml penicillin, 100 µg/ml streptomycin
(Thermo Fisher Scientific, Inc.), and 2-mM glutamine (Nissui
Pharmaceutical Co. Ltd.) at 37°C with 5% CO2.
Establishment of the fibroblast cell
line
Normal esophageal mucosae were collected from
surgically resected specimens from patients with advanced-stage
esophageal cancer. The fibroblasts were separated and cultured
using the following method: i) Tissues were washed with serum-free
DMEM (FUJIFILM Wako Pure Chemical) containing 200 µg/ml kanamycin
(Meiji Seika Pharma Co., Ltd.) and 10 µg/ml amphotericin B
(Bristol-Myers Squibb) three times; ii) tissues were cut into 3–5
mm square sections, which were placed on 200 µl BD Matrigel Matrix
(BD Biosciences) in a 12-well plate; iii) to prevent the small
tissues from moving in each well, the tissues were covered with a
sterilized 15-mm round cover glass and cultured with DMEM
containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 50
µg/ml kanamycin (Meiji Seika Pharma Co., Ltd.) and 2.5 µg/ml
amphotericin B at 37°C in a humidified atmosphere; i) the medium
was changed every other day until the tissues were surrounded by
adherent fibroblasts; and v) when the fibroblasts reached 30–50%
confluency, they were harvested with 0.25% trypsin and 1-mM EDTA
and then re-plated in a T25 culture flask.
Reagents and antibodies
Metformin was purchased from FUJIFILM Wako Pure
Chemical and was dissolved in phosphate-buffered saline at a stock
concentration of 100 mM and stored at −20°C. The reagents and
primary antibodies used are listed in Table I.
 | Table I.Primary antibodies used for western
blot analysis. |
Table I.
Primary antibodies used for western
blot analysis.
| Primary
antibody | Supplier | Clonality | Cat. no. | Isotype | Dilution |
|---|
| Anti-ATM | Abcam | 2C1(1A1) | ab78 | Mouse monoclonal
IgG | 1:2,000 |
| Anti-C/EBPβ | Santa Cruz
Biotechnology, Inc. | H-7 | sc-7962 | Mouse monoclonal
IgG | 1:200 |
| Anti-NF-κB | Abcam | P65 | ab16502 | Rabbit polyclonal
IgG | 1:1,000 |
| Anti-p21 | Abcam | EPR362 | ab109520 | Rabbit monoclonal
IgG | 1:1,000 |
|
Anti-CDKN2A/p16INK4a | Abcam | EPR1473 | ab108349 | Rabbit monoclonal
IgG | 1:2,000 |
| Anti-β-actin | MilliporeSigma | AC-15 | A5441 | Mouse monoclonal
IgG | 1:10,000 |
|
Anti-E-cadherin | Santa Cruz
Biotechnology, Inc. | H-108 | sc-7870 | Rabbit polyclonal
IgG | 1:2,000 |
|
Anti-N-cadherin | Santa Cruz
Biotechnology, Inc. | H-63 | sc-7939 | Rabbit polyclonal
IgG | 1:1,000 |
| Anti-vimentin | Santa Cruz
Biotechnology, Inc. | V9 | sc-6260 | Mouse monoclonal
IgG | 1:2,000 |
Irradiation of fibroblasts
Cultures were irradiated using an MBR-1520R-3
(Hitachi Medical Corporation), with a power output of 125 kV and 20
mA. Forward-scattered radiation with 0.5 mm Al and 0.2 mm Cu
filters were used. Normal fibroblasts were irradiated with a single
X-ray dose at 4, 8 and 10 Gy. In subsequent experiments, the
fibroblasts irradiated with 8 Gy and cultured for 5–8 days
(IR+FB) were used as senescent cells, and non-irradiated
fibroblasts within 2–5 passages (IR−FB) were used as
normal fibroblasts.
Assessment of cellular senescence in
irradiated fibroblasts using SA-β-gal staining
SA-β-gal staining was used to count the number of
senescent cells in each condition after 4, 8 and 10 days (Fig. S1). The cells were stained with
SA-β-Gal using the senescence kit (OZ Bioscience) according to the
manufacturer's protocol. The cells were incubated with staining
solution at 37°C overnight, and SA-β-Gal-positive cells were
counted. The number of dead and viable cells was measured using an
EVE automatic cell counter (AR Brown Co., Ltd.). Using this result,
the radiation dose and number of days after irradiation required
for senescence were determined. A microscope (BX-50, Olympus
Corporation) was used for cell observation.
The irradiated fibroblasts were supplemented with 5
mM metformin for the following defined periods: i) The control (no
addition); and ii) 8 days after irradiation. A total of 5 mM
metformin was added to the non-irradiated fibroblasts. In the
irradiated fibroblast group, irradiation was performed at 72 h
after fibroblast seeding. In each group, the number of surviving
cells and percentage of cells positive for SA-β-gal staining at 11
days after seeding were determined.
Collection of fibroblast culture
supernatant
IR+FB and IR−FB were each
cultured in a confluent state in a six-well plate. The medium was
replaced (RPMI-1640 + 10% FBS ± 5 mM metformin) and cultured at
37°C with 5% CO2 for 48 h. The medium was replaced with
serum-free RPMI-1640 and cultured at 37°C with 5% CO2
for 24 h. The culture medium was recovered, centrifuged at a
temperature of 4°C at 358.08 × g for 10 min, and the supernatants
were obtained, which were as follows: i) IR−FB with
metformin (IR−Met+FB); ii) IR−FB
without metformin (IR−Met−FB); iii)
IR+FB with metformin (IR+Met+FB);
and iv) IR+FB without metformin
(IR+Met−FB). The harvested culture
supernatant was added to the KES cells and cell proliferation,
migration and invasion, and the promotion of EMT were evaluated
using immunocytochemistry, western blot analysis, cell
proliferation assay, scratch wound assay and Matrigel invasion
assay.
Immunocytochemistry
KES cells were cultured on Lab-Tec chamber slides
(Nalge Nunc International Corporation). The cells were cultured in
a serum-free medium for 24 h and then incubated with supernatant
separated from the fibroblasts for 48 h. The cells were blocked
with 1.5% bovine serum albumin and incubated with a mouse
mono-clonal antibody against vimentin (sc-6260, 1:100 dilution;
Santa Cruz Biotechnology Inc.) or a rabbit poly-clonal antibody
against E-cadherin (sc-7870, 1:100 dilution; Santa Cruz
Biotechnology, Inc.) and mouse monoclonal antibody against
β-catenin (A5441, 1:100 dilution; Santa Cruz Biotechnology Inc.)
overnight at a temperature of at 4°C. Immunoreactivity was
visualized by incubating the cells with anti-rabbit IgG conjugated
with Alexa Fluor 488 (A11008) or 594 (A11012) (Molecular Probes;
Thermo Fisher Scientific, Inc.) for 1 h at 26°C. The cells were
counterstained with bisbenzimide (Hoechst 33258, 100 ng/ml;
MilliporeSigma) to visualize the nuclei. Finally, the cells were
examined under an immunofluorescence microscope (BX50/BX-FLA,
Olympus Corporation).
Western blot analysis
KES cells were cultured in a serum-free medium for
24 h and then incubated with supernatant separated from fibroblasts
for 72 h. IR+FB and IR−FB were each cultured
in a serum-free medium for 24 h and then incubated at 37°C with
various concentrations (0–10 mM) of metformin for 5 days. First,
cells were lysed in RIPA buffer (Wako Pure Chemical Industries,
Ltd.) containing a protease and phosphatase inhibitor cocktail
(MilliporeSigma). The protein concentration of each sample was
measured using a BCA protein assay kit (Thermo Fisher Scientific,
Inc.). The cell lysates were prepared in denatured sodium dodecyl
sulfate (SDS) sample buffer and subjected to SDS-polyacrylamide gel
electrophoresis. The proteins (20 µg) from each sample were loaded
on a 10% polyacrylamide gel (Bio-Rad Laboratories, Inc.). The
separated proteins were transferred to a polyvinylidene difluoride
(PVDF) membrane (Bio-Rad Laboratories, Inc.) and blocked with a
commercial gradient buffer (PVDF blocking reagent; Can Get Signal,
Toyobo Life Science) for 1 h at 26°C. The membranes were incubated
with the primary antibody overnight at 4°C. The primary antibodies
used are listed in Table I. After
washing, the membranes were incubated with horseradish
peroxidase-labeled anti-mouse (NA931) or anti-rabbit IgG (NA934)
(1:10,000 dilution; Cytiva) or anti-goat IgG (sc-2354, 1:10,000
dilution; Santa Cruz Biotechnology, Inc.) for 1 h at 26°C.
Antibody-antigen complexes were detected using the ECL Plus Kit
(Cytiva) and the Light Capture System (Atto Co., Ltd.). The
intensity of the protein bands was quantified using ImageJ software
ver 1.51k (National Institutes of Health).
Cell proliferation assay
The proliferation of fibroblasts or KES cell was
determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT; MilliporeSigmah) assays. The cells were
seeded in a 96-well culture plate at a density of 4×103
cells/200 µl/well and incubated for 24 h at 37°C to confirm cell
colonization. In the experiments with fibroblasts, fibroblasts were
treated with various concentrations (0.1–10 mM) of metformin for 48
and 72 h. In the experiments with KES cells, the KES cells were
cultured in a serum-free medium for 24 h. The KES cells were then
incubated with supernatants separated from the fibroblasts at
various concentrations for 24, 48 and 72 h. The medium was removed
and crystallized formazan dye in the cells was solubilized by
addition of dimethyl sulfoxide. The absorbance was measured at 535
nm and the effect on cell proliferation in proportion to the
control was investigated by comparing the cell density of the
drug-treated cells with that of the untreated controls. In
experiments with fibroblasts, the MTT assay was used to evaluate
the toxicity of metformin on cells and to determine the metformin
concentration to be used. All experiments were repeated at least
three times.
Scratch wound assay
The scratch wound assay was used for the assessment
of the migratory ability of the cancer cells. The KES cells were
seeded in six-well plates at a density of 1.2×105 and
incubated overnight at 37°C until reaching confluency (100%). The
cells were scratched with a 1,000-µl pipette tip and washed twice
with serum-free RPMI-1640 to remove the exfoliated cells. The
medium was then replaced with the respective supernatants prepared.
Wound closure rates were assessed and images were obtained after
24, 48, and 72 h. Each value was derived from three fixed points. A
microscope (BX-50, Olympus Corporation) was used for cell
observation.
Matrigel invasion assay
The Matrigel invasion assay was used to examine the
invasiveness of the cancer cells. The KES cells were cultured in
RPMI-1640 medium without FBS for 24 h. The cells were collected by
trypsinization and resuspended in a serum-free medium at
1×105 cells/ml. Altogether, 2.5×104 KES cells
were cultured in the upper chamber of a 24-well Transwell insert
(Corning Life Sciences) that was coated with 50 µl Matrigel (1:10
dilution in serum-free medium; BD Biosciences). IR+FB or
IR−FB with RPMI-1640 with or without 5 mM metformin were
added to the lower chamber. The cells were incubated at 37°C for 24
h. The cells that migrated through the Matrigel and membranes with
a pore size of 8 µm were fixed in 100% methanol (FUJIFILM Wako Pure
Chemical) for 1 min, stained with by hematoxylin (FUJIFILM Wako
Pure Chemical) for 2 min at 26°C and counted under an optical
microscope (BX-50, Olympus Corporation). All experiments were
carried out with three repetitions.
Statistical analysis
The data were analyzed using two-way ANOVA followed
by the Bonferroni post hoc test, one-way ANOVA followed by Tukey's
post hoc test, and repeated measures two-way ANOVA followed by the
Bonferroni post hoc test as appropriate. The results are expressed
as the mean ± standard deviation. All analyses were performed using
SPSS software (IBM Statistics ver. 25; IBM Corp.). A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Induction of cellular senescence of
fibroblasts by radiation
Normal fibroblasts were exposed to various doses of
radiation, and SA-β-gal staining was used over time to confirm
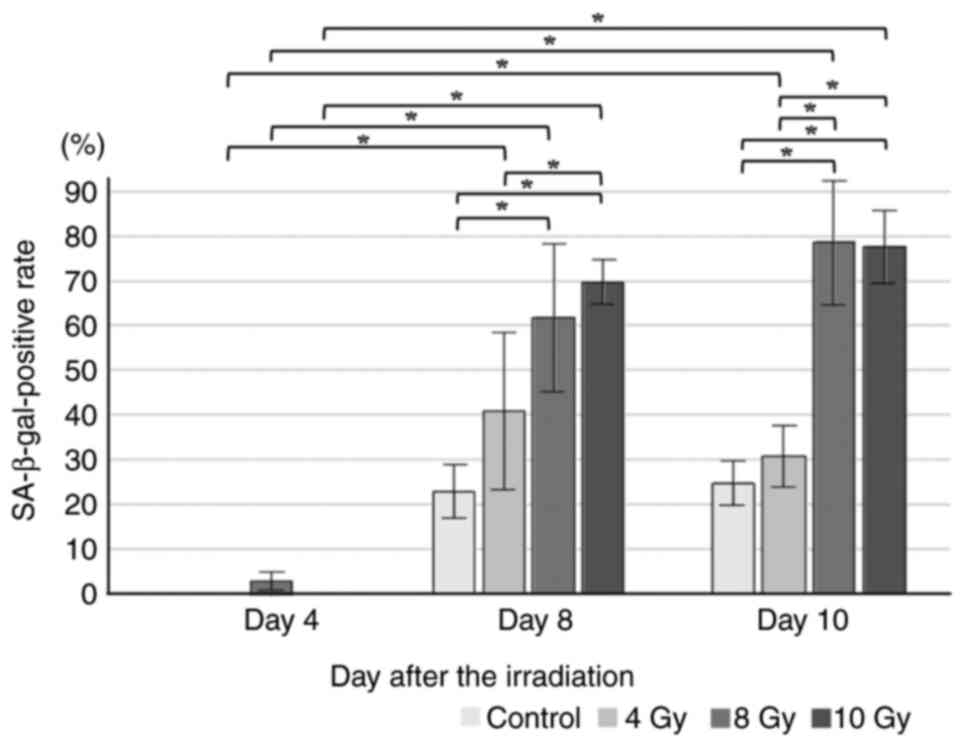
whether cellular senescence occurred (Fig. S1). With 4 Gy irradiation, 50% of
the cells were not stained in any time period. After 8 days of 8 Gy
irradiation, >50% of the cells exhibited positivity for SA-β-gal
staining (Fig. 1). In the
subsequent experiments, fibroblasts that had been irradiated with 8
Gy for 5–8 days were used as senescent cells.
Cytotoxicity of metformin to
fibroblasts
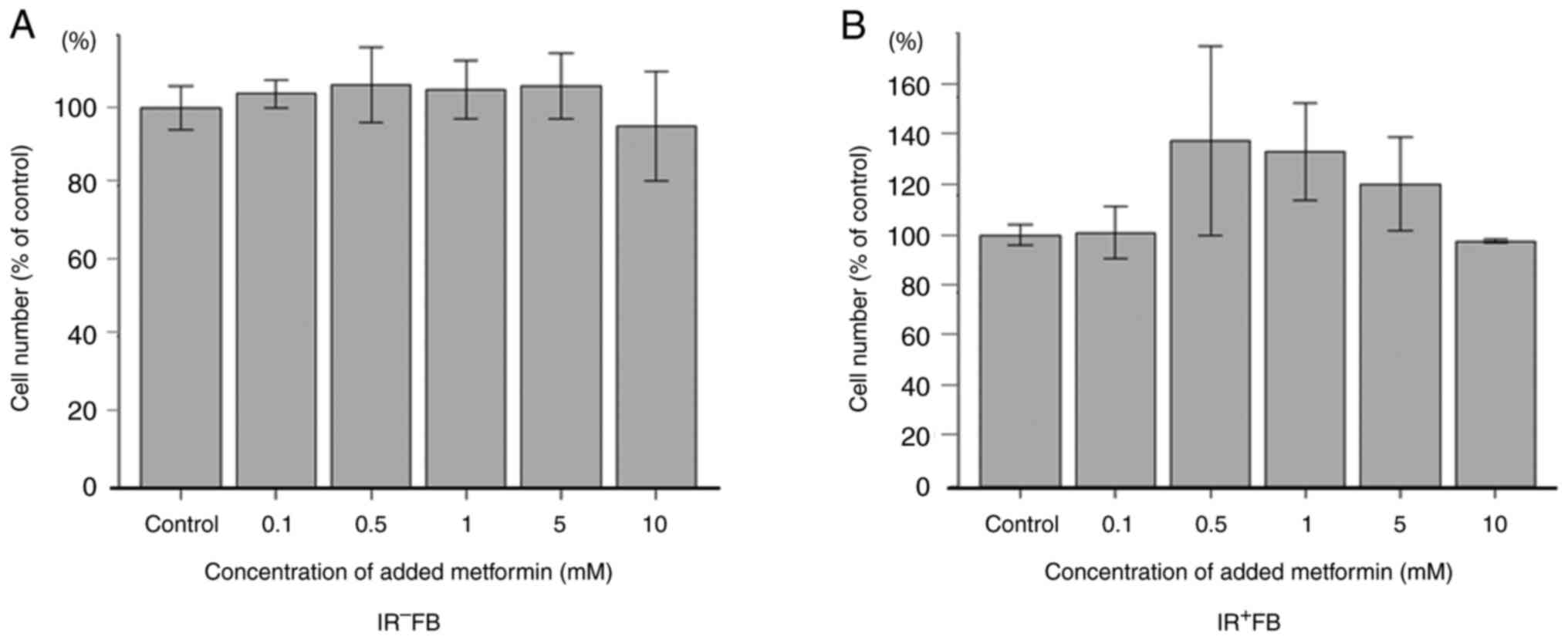
The present study examined the effects of metformin
on the proliferation of fibroblasts using MTT assay. Metformin
loading did not significantly inhibit cell proliferation at any
concentration used, with or without irradiation (Fig. 2). Subsequent experiments were
performed using a dilution of 5 mM metformin.
Effects of metformin on cellular
senescence
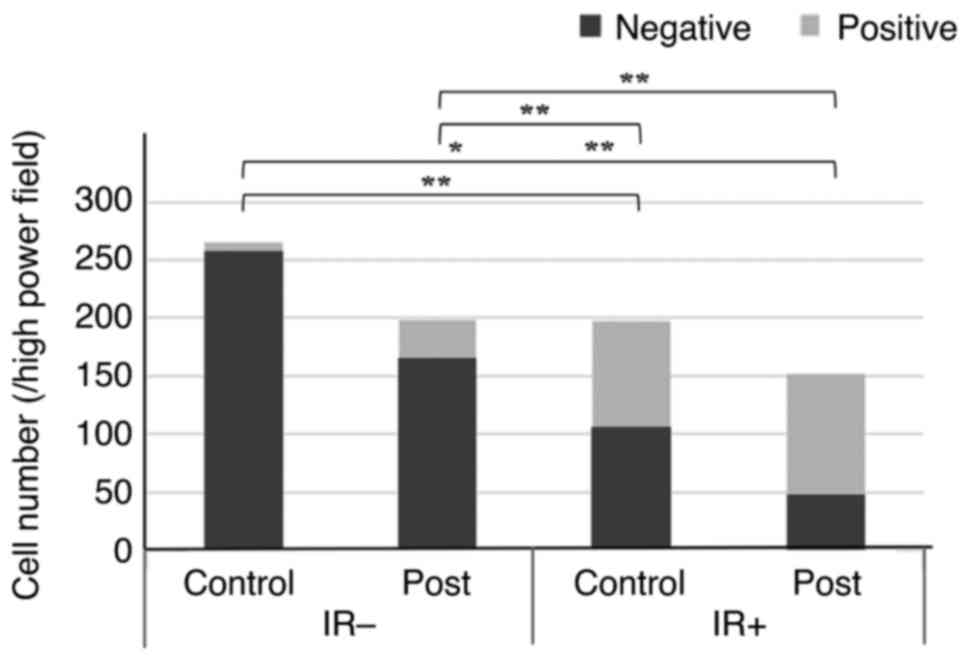
The irradiation of fibroblasts was found to induce a
decrease in the number of viable cells and cellular senescence. The
treatment of irradiated fibroblasts with metformin decreased the
number of viable cells to 57.3% (P=0.02) and increased the
proportion of senescent cells to 68.4% (P<0.001) (Fig. 3).
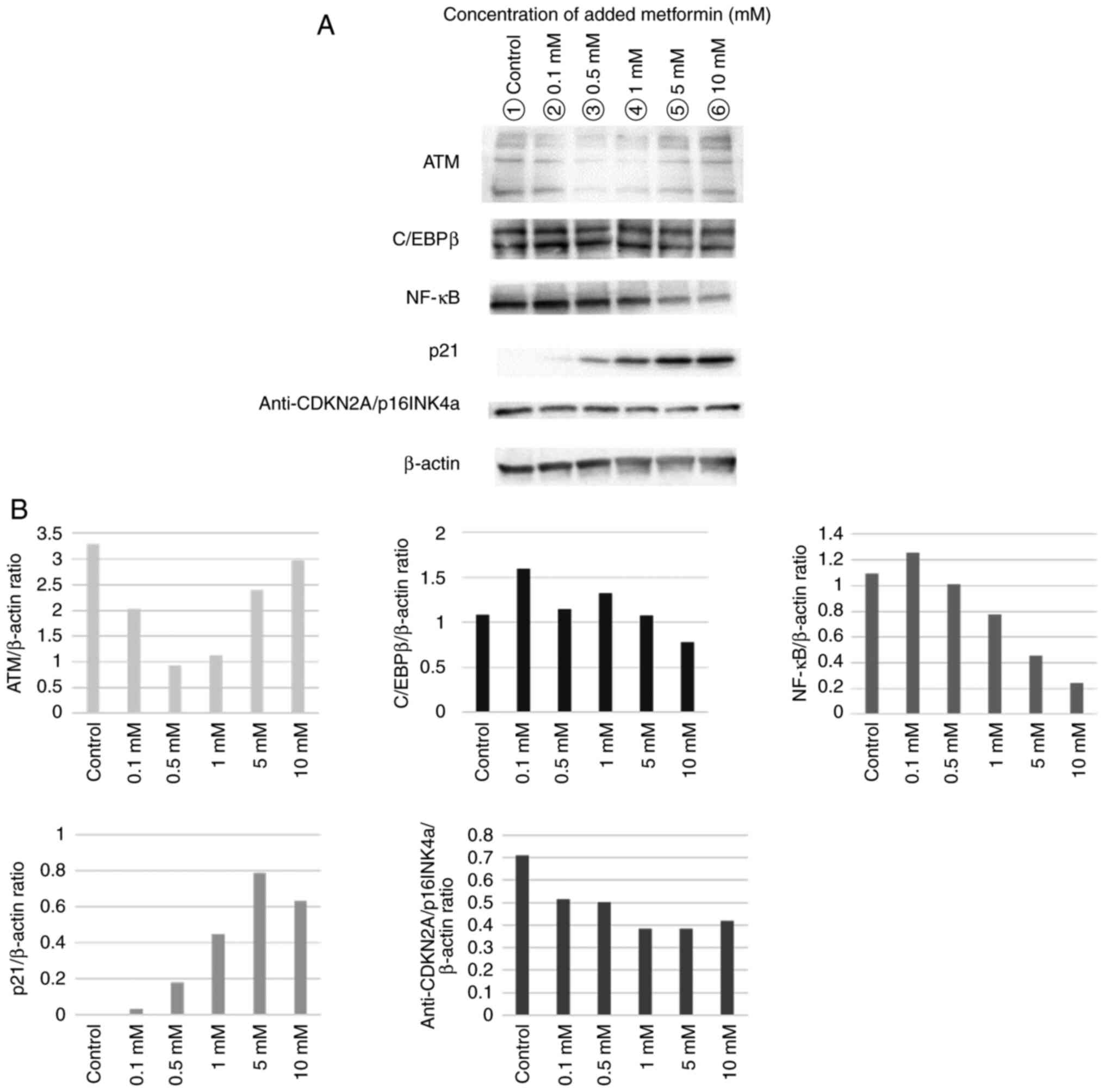
Various concentrations of metformin were added to
the irradiated fibroblasts and specific proteins were evaluated by
performing western blot analysis. Loading higher metformin
concentrations tended to suppress the expression of NF-κB, which is
responsible for major signaling that stimulates the expression of
SASP, whereas the expression of anti-p21, a senescence marker, was
enhanced (Fig. 4).
Metformin suppresses
irradiation-induced SASP secretion
The present study investigated the effects of SASP
factors secreted by irradiated fibroblasts on cancer. The present
study also investigated the effects of the addition of metformin to
fibroblasts on SASP secretion. As illustrated in Fig. 5, Fig.
6, Fig. 7, Fig. 8, the esophageal cancer cells
cultured with the supernatant obtained from the irradiated
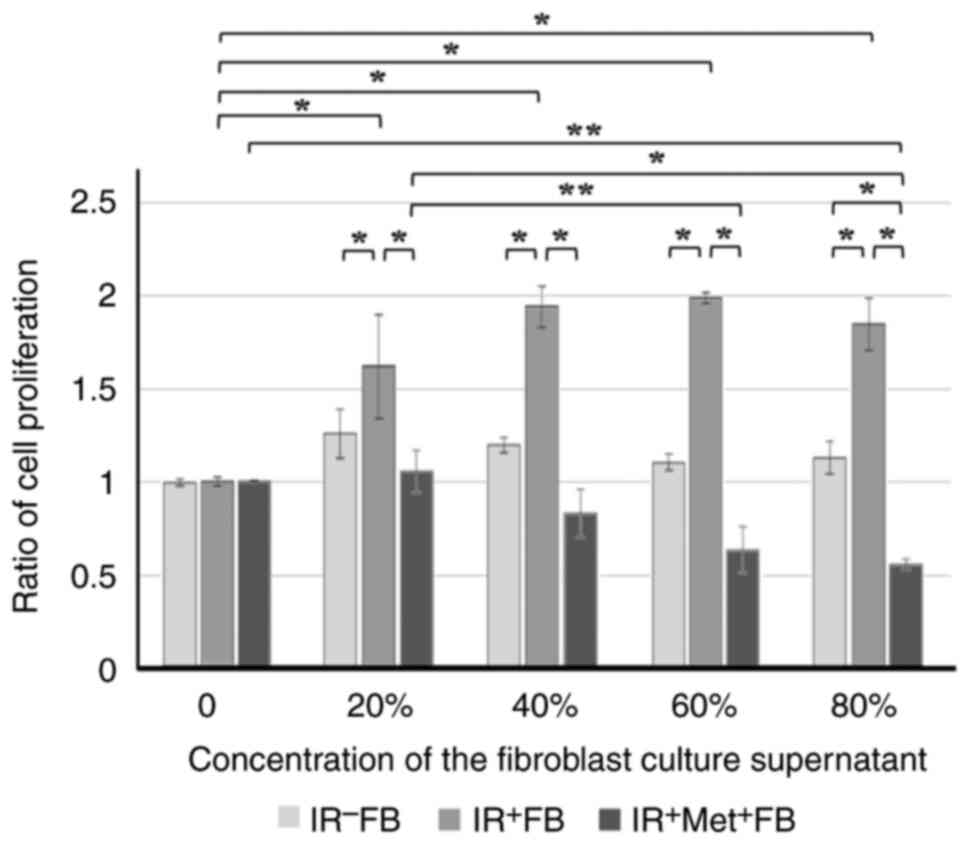
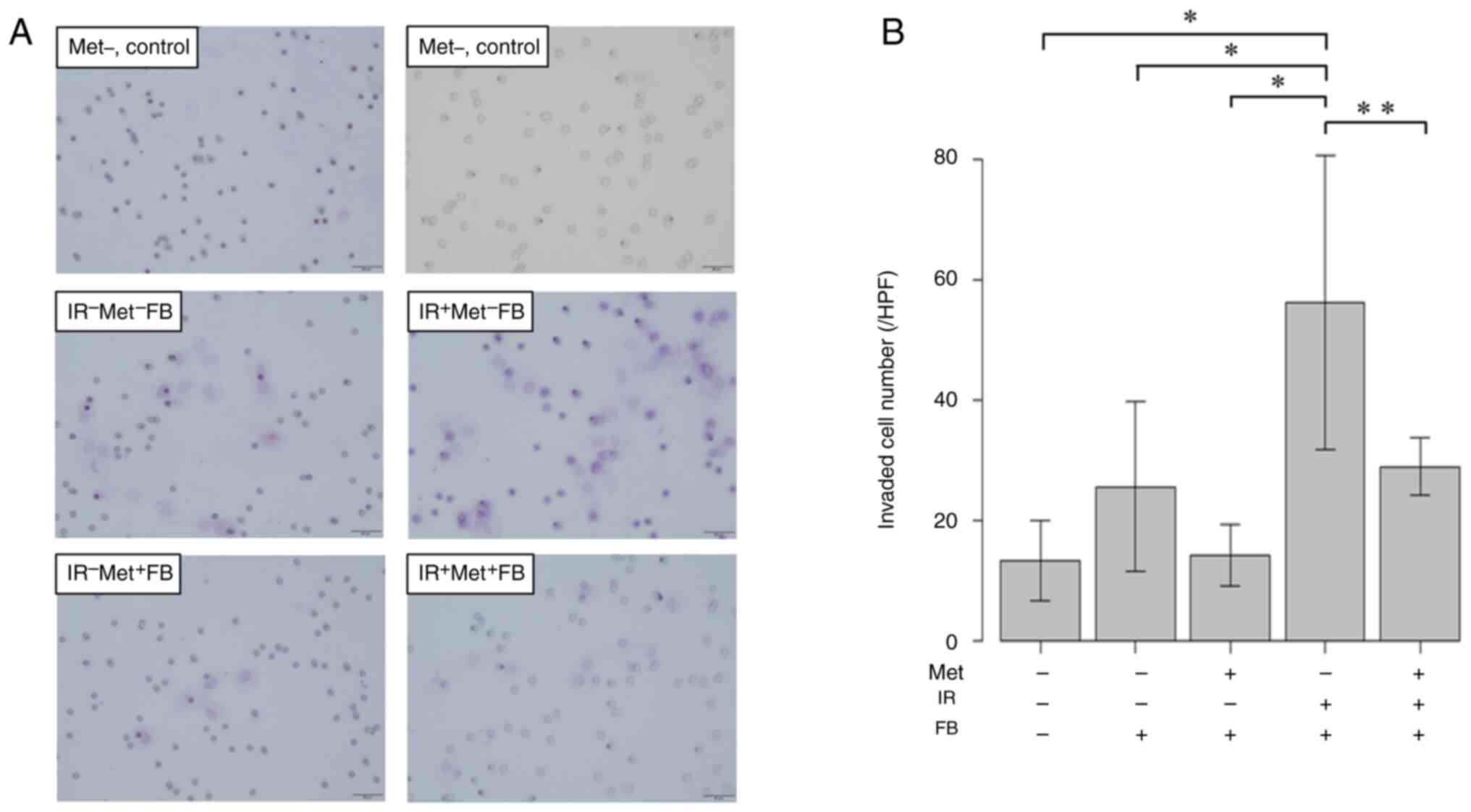
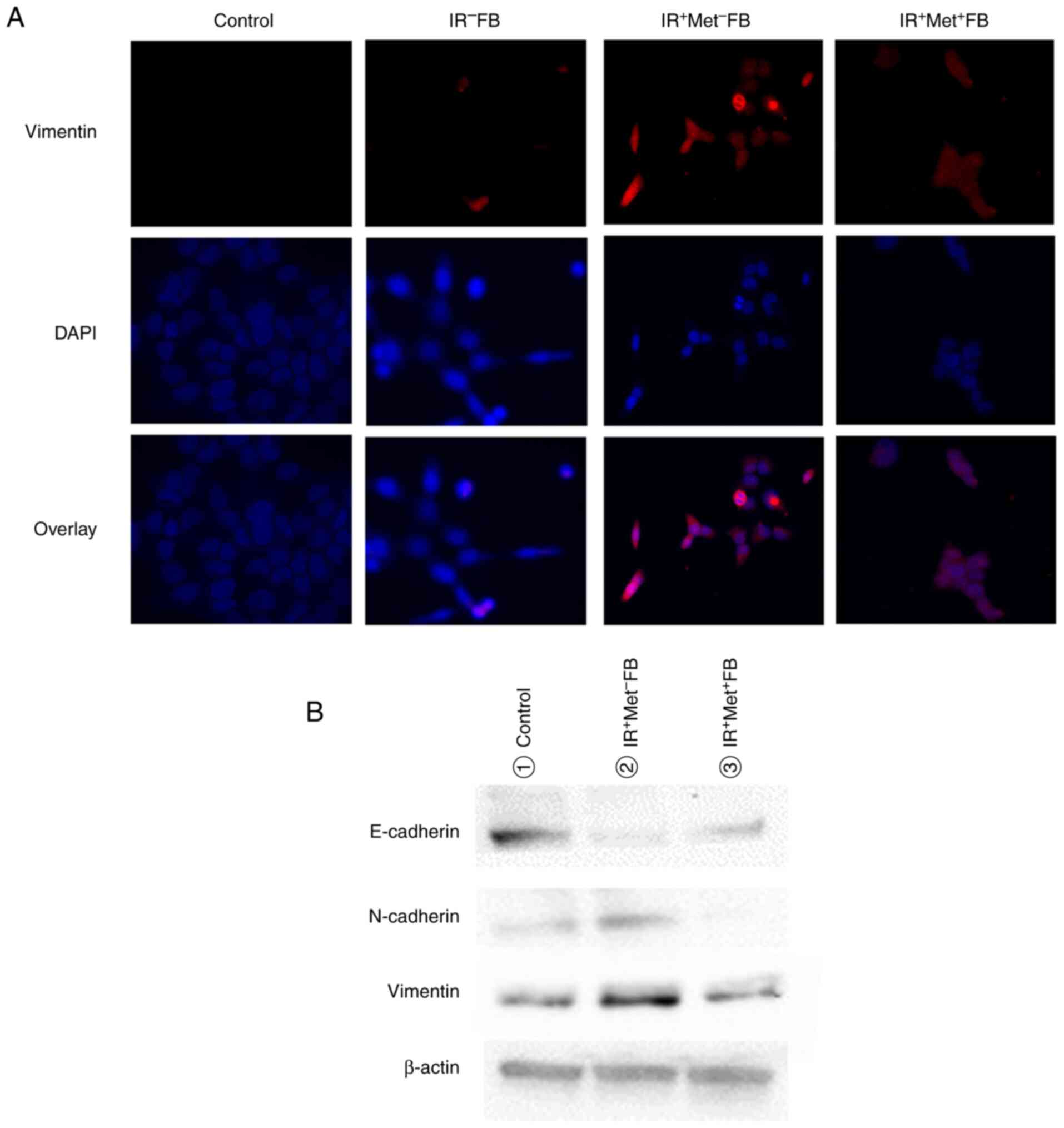
fibroblasts exhibited cell proliferation (Fig. 5), migratory ability (Fig. 6) and invasiveness (Fig. 7), and EMT was promoted (Fig. 8). When the fibroblasts were cultured
in the supernatant from the irradiated fibroblasts pre-treated with
metformin, there was no effect on the migratory ability of the
cancer cells; however, the proliferative ability, invasiveness and
EMT were confirmed to be suppressed.
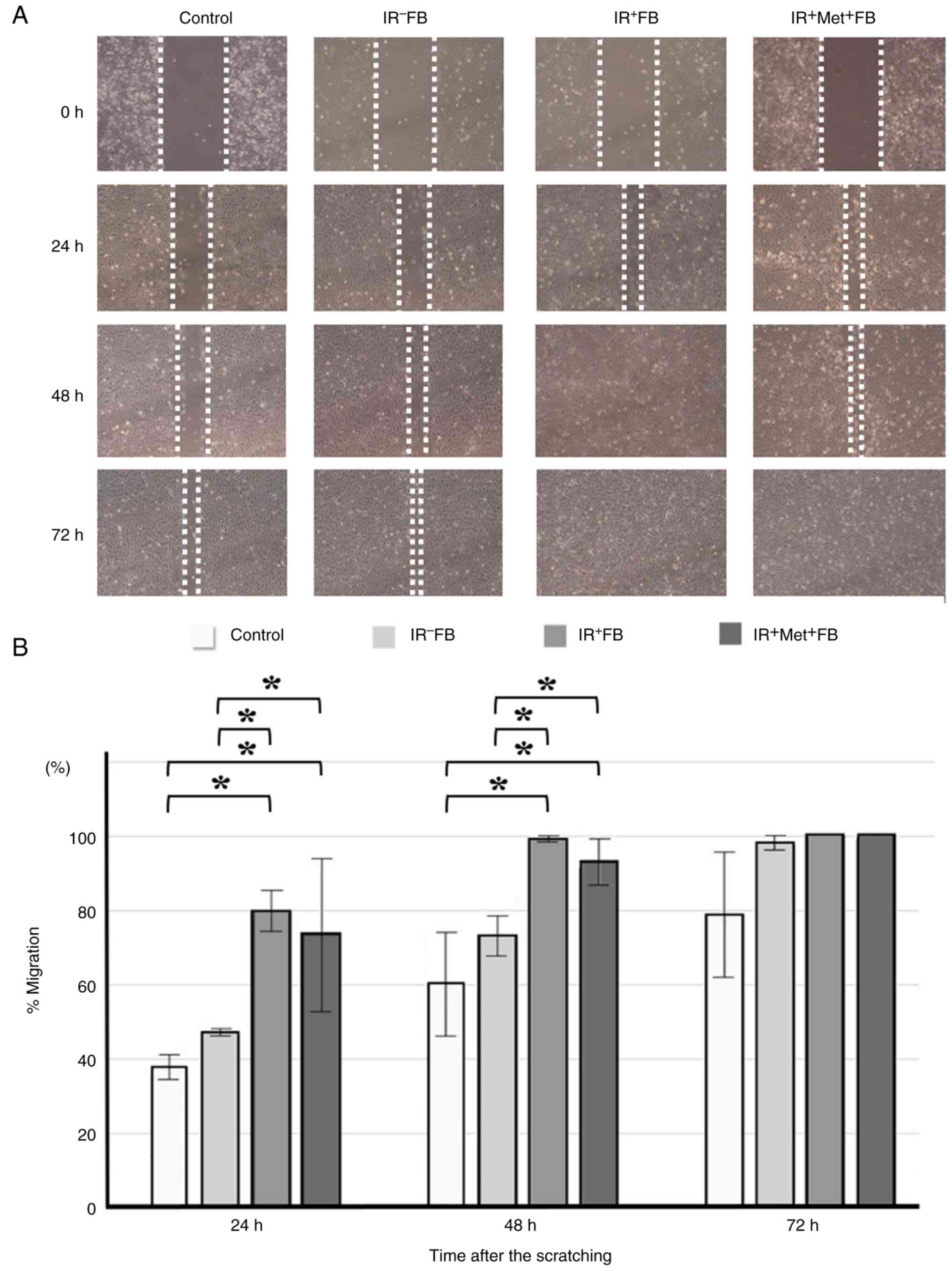 | Figure 6.The effect of fibroblast culture
supernatant and metformin on the migration ability of KES. (A)
Representative image of migration assay (scratch wound assay). Cell
monolayers were scratched with a 1,000-µl pipette tip and after
washing, the medium was replaced with the respective supernatant.
Images were obtained after 24, 48 and 72 h. (B) In the scratch
wound assay, significant differences (*P<0.001) were observed
between the control vs. the IR+FB, control vs.
IR+Met+FB, IR−FB vs.
IR+FB, and IR−FB vs.
IR+Met+FB groups. The addition of supernatant
significantly enhanced the migratory activity of cancer cells.
However, there was no difference between the IR+FB and
IR+Met+FB groups, and the addition of
metformin had a minimal effect on migration. Data were analyzed by
repeated measures two-way ANOVA followed by the Bonferroni post hoc
test. IR+FB, irradiated fibroblasts; IR−FB,
non-irradiated fibroblasts; IR+Met+FB,
irradiated fibroblasts treated with metformin. |
Discussion
The aim of the present study was to clarify the role
of SASP-mediated interactions between the ESCC cells and
radiation-induced senescent CAFs and to examine the effects of
preventing cancer progression through the regulation of SASP by
metformin. In the present study, the irradiation of fibroblasts
induced cellular senescence and subsequent SASP secretion.
Moreover, metformin inhibited the radiation-induced cellular
senescence and subsequent SASP in the cell lines and ESCC
progression via the activation of EMT.
It is clinically important to perform chemotherapy
or radiotherapy to control the primary lesions, metastasis, and
circulating tumor cells in the blood to treat cancer and improve
disease prognosis. Contrarily, in recent years, cellular senescence
has attracted attention as a reaction induced by DNA damage that
causes carcinogenesis, including the shortening of telomeres and
activation of oncogenes, in contrast to apoptosis, which has long
been known as a cancer-suppressing mechanism (7–9). In
the SASP state, senescent cells secrete inflammatory cytokines,
such as IL-6 and IL-1, chemokines, such as IL-8 and CXCL, and
proteins, such as MMPs and various growth factors (1–3). Some
fundamental studies have reported that SASP is deeply involved in
cancer progression through the paracrine action in cancer tissues
caused by SASP (1,2,18).
Additionally, the administration of anticancer drugs or
irradiation, which are major means of treatment for esophageal
cancer, also causes cellular senescence in normal cells including
fibroblasts in the host. Hernandez-Segura et al (19) reported that cellular senescence
occurred in benign cells due to irradiation. This is considered to
play a critical role in further cancer progression and the
acquisition of treatment resistance.
The present study investigated whether cellular
senescence occurs in fibroblasts isolated and cultured from
resected human esophageal cancer tissues. A time-dependent increase
was observed in the positive counts in SA-β gal staining following
a single dose of radiation. Additionally, an expression of
increased p16 and p21 was observed, which comprehensively supports
the occurrence of cellular senescence in fibroblasts. In the
experimental results presented in Fig.
4, p21 expression was not observed in fibroblasts at day 8
(data not shown) following irradiation; thus, those at day 5
following irradiation were used. The reason for this is that once
activated, p16 and p21 are inactivated during the aging process
(9), and the measurement on day 8
may have been made at a time when they were inactivated. Another
possibility is that there may have been a procedural error.
The expression of Lys53 (Ac-p382) and acetylated p21
at p53 has been shown to be increased in hepatocellular carcinoma
following treatment with metformin (20). Metformin also enhanced the
expression of p21, a senescence marker in a concentration-dependent
manner. Moreover, irradiation increased the expression of NF-κB in
fibroblasts and induced the transcriptional enhancement of IL-1,
IL-2, IL-6, IL-8, IL-12 and TNF-α, among others. The irradiation
also promoted SASP-induced cytokine secretion (21). Contrarily, p53 functions
suppressively in the induction of SASP and the upregulation of p16,
and it has been reported that the expression of the F-box protein
gene, known as FBXO22, is specifically induced among the genes
specifically expressed in senescent cells (22).
Low-dose metformin also induces p53-dependent
senescence in hepatoma cells by activating AMPK signaling (20). The data of the present study also
suggested metformin-induced fibroblast senescence. Therefore, it
was expected that the administration of metformin in addition to
irradiation would further induce cellular senescence. However, the
percentage of senescent cells increased, although the number of
viable cells decreased. The present study was not able to examine
in detail how metformin acts on the target cells. Studies to date
have revealed a variety of effects of metformin. Metformin
activates AMPK by increasing intracellular AMP concentrations
(23). This acts on energy
metabolism and inhibits cell proliferation. It has also been shown
to inhibit the progression of the cell cycle from G1 to S phase by
decreasing the level of cyclin D1 (24). It is considered that these factors
contribute to the decreased survival rate.
Whether DNA damage induces apoptosis or cellular
senescence is dependent on the amount of stress present. Excessive
stress was considered to induce increased apoptosis beyond cellular
senescence. Recently, several genes controlling these factors have
been reported (25).
In the experimental results presented in Figs. 2 and 3, fibroblasts were used 5–8 days following
irradiation and 72 h following treatment with metformin in the MTT
assay as the cause of the difference in the number of viable cells.
In the cell count experiment, metformin was administered for 8 days
immediately following irradiation. Thus, the decrease in cell
number may be due to spontaneous cell dropout over time or
increased cell death due to excessive DNA stress from radiation and
metformin. As a limitation, however, this hypothesis was not
experimentally proven in the present study and is an issue for the
future.
For controlling cancer, not only the proliferative
activity of cancer cells themselves, but also the metastatic
ability, invasiveness, fibrogenesis of cancer stroma, and
overcoming treatment resistance are considered critical. The
fibroblasts of the host are induced into the tumor tissue by
microenvironment changes caused by growth factors and cytokines
secreted by autocrine or paracrine manners from tumor cells and
CAFs in and around the cancer stroma. Induced CAFs generate
extracellular matrices, such as hyaluronic acid and collagen, which
accelerate tumor growth and stromal fibrosis (26). SASP factors do not only promote
tumor cell proliferation, but also have synergistic effects on the
induction of EMT and stromal fibrosis. Okamoto et al
(13) previously reported that the
interaction between CAFs, including hepatic stellate cells and
myofibroblasts, and cholangiocarcinoma cells promotes cancer cell
proliferation and EMT through the function of angiotensin II,
stromal derived factor-1 and TGF-β derived from activated CAFs,
which is deeply involved in cancer progression, metastasis and the
invasiveness of cholangiocarcinoma. Additionally, abnormally high
levels of membrane type 1-MMP are often expressed in the tumor
microenvironment. MMP has been reported to be highly expressed in
senescent fibroblasts (27).
Membrane type 1-MMP promotes the EMT of cancer cells by degrading
the extracellular matrix, invading the surrounding tissue, and
gaining the ability to form new blood vessels (28). Takino et al (29) demonstrated that MT1-MMP inhibited
nascent fibronectin assembly by not only cleaving, it but also
controlling the cell-matrix adhesions in the close proximity of
cell-cell contacts, resulting in the reduction of N-cadherin
adhesions and fibronectin matrix assembly. Nakayama et al
(30) conducted fundamental
research on radiation-induced EMT in ESCC cells and the EMT
inhibitory effect of metformin through TGF-β regulation (30). The present study demonstrated that
the addition of supernatant from irradiated fibroblasts to KES
cells significantly increased the cancer cell proliferation,
invasive and migratory abilities, and EMT marker activation. This
result supports the findings that CAF-derived inflammatory
cytokines and chemokines in the SASP state-induced EMT and the
CAF-mediated paracrine action synergistically have a significant
effect on cancer cell activation (14). In the experimental results presented
in Fig. 7, the results of Matrigel
assay revealed the promotion of cancer cell invasion by irradiated
fibroblasts and its suppression by metformin. However, there was a
large variation in the number of invading cells in the
IR+Met−FB group. The possible reasons for
this event were that the cancer cells did not adhere uniformly or
were not distributed uniformly due to a partial lack of
Matrigel.
In solid malignant tumors, the fibrous tissue
development in the cancer stroma inhibits the development of
peripheral blood vessels and the distribution of anticancer drugs
to the cancer cells, which can be among the causes of anticancer
drug resistance. To the best of our knowledge, there has been no
basic research on the mechanisms of cancer progression, fibrosis
and treatment resistance through cellular senescence in fibroblasts
coexisting in cancer tissues, and their influence on the cancer
microenvironment via SASP in esophageal cancer, which is among the
refractory solid tumors.
Metformin exerts antitumor effects by inhibiting
mTORC1 through AMPK activation (31). Metformin also inhibits NF-κB via
mTOR (32). Additionally, metformin
inhibits ROS production in senescent cells (33), and mitochondrial reactive oxygen
species are involved in NF-κB activation (34,35).
Metformin inhibits IKKα/β kinases, but not p38 MAPK. Since these
pathways are downstream of TGF-β-activated kinase 1 (TAK1), they
are considered to interfere only with the IKK activation pathway.
In addition, in a previous study, qPCR analysis confirmed that the
expression of multiple cytokines and chemokines, such as CXCL5,
IL-1b, IL-6 and IL-8, was inhibited (16).
Several factors have been reported to alter the
expression of genes encoding SASP factors in the molecular
mechanisms involved in induction. The present study focused on
NF-κB, which is considered to be a major factor, with the aim of
regulating SASP in senescent fibroblasts. NF-κB is a transcription
factor that regulates the gene expression of chemokines that bind
to CXCR2 (18). Cellular senescence
signals phosphorylate p65 and translocate the p65/p50 complex
(NF-κB) into the nucleus. As a result, the p65 subunit of NF-κB
functions as a transcription factor by binding to the
transcriptional regulatory region of the SASP gene (36). mTOR is involved in regulating NF-kB.
mTOR positively regulates SASP gene expression through the NF-KB
pathway by promoting IL-1a translation (37). IL-1a is a master regulator that acts
upstream of many SASP factors, including IL-6 and IL-8.
Furthermore, SASP factors induced by IL-1a expression exhibit
paracrine effects on the surrounding cells and induce cellular
senescence (38,39). In the present study, mTORC1 was
hypothesized to inhibit metformin-suppressed SASP, resulting in an
antitumor effect. However, the present study did not examine the
secretion of SASP, such as IL-6. Further studies are thus required
to elucidate this matter.
Clinical studies have reported several beneficial
effects of metformin in the treatment of cancer. A previous
retrospective study demonstrated that patients with type 2 diabetes
taking metformin for >4 years had a significantly lower
incidence of cancer than patients receiving other diabetes
treatments (40). Additionally,
metformin increases the pathological response rate following
neoadjuvant chemotherapy for breast cancer in diabetic patients
(41). In another study, the
administration of metformin to patients who had undergone surgery
for early-stage colorectal cancer reduced the cancer recurrence by
37% and increased the survival rate by 31% (42). Metformin use also reduced the risk
of mortality in patients with pancreatic cancer by 12%. The effect
was particularly large in patients with stage I or II cancer
detected at a relatively early stage, and the risk of mortality
decreased by 26% (43). All these
effects may have been the result of antitumor effects, as the
secondary effects of metformin may have reflected the effect of the
drug's rearrangement on the cancer cells themselves and their
microenvironment.
The present study has some limitations. First, the
majority of in vitro studies investigating the anticancer
effects of metformin have used metformin at concentrations of 1–10
mM (165–1,650 mg/l), which are well below therapeutic plasma levels
(0.465–2.5 mg/l or 2.8–15 µM) (44). The present study utilized a high
dose of metformin relative to the therapeutic plasma level of 5 mM.
Second, the present study ensured the prevention of the direct
effects of metformin on cancer cells by avoiding the exposure of
ESCC cells to metformin. However, the possibility that metformin
remained in the supernatant and directly affected cancer cells
cannot be denied and the serum concentration of metformin will
affect both fibroblasts and cancer cells in vivo. The
authors deemed that this experimental environment was close to the
actual condition in vivo. Third, no in vivo
experiments were performed herein. This is necessary for clinical
application and is an issue to be addressed in the future. Finally,
the present study demonstrated only the radiation-induced cellular
senescence of fibroblasts and events resulting in the
paracrine-style activation of SASP factors secreted from them
against cancer cells. The detailed composition of the SASP factors
and the role of each factor were shown in the present study, and
these are worthy of investigation in the future. To date, reports
on this mechanism of cancer cell activation in esophageal cancer
are limited (45,46), and the present study is a
preliminary experiment for further research and the results may
prove useful as a mechanism of cancer progression.
In conclusion, the present study demonstrated that
the radiation-induced cellular senescence of CAFs and subsequent
SASP secretion induced an increase in cell proliferation, migratory
and invasive activities, and EMT in cancer cells. Moreover,
metformin is expected to be effective as a chemical modulator and a
therapeutic agent of esophageal cancer that targets cellular
senescence and SASP through a novel mechanism. The antitumor effect
by suppressing radiation-induced SASP is expected to be very
promising in future anticancer therapy as a drug repositioning.
Supplementary Material
Supporting Data
Acknowledgements
The authors are grateful to Futakuchi and Yoshida,
the Medical Technologist of Gastrointestinal Surgery, Kanazawa
University, who provided technical support for the experiments and
made significant contributions.
Funding
The present study was supported by Grants-in-Aid for Scientific
Research from the Japan Society for the Promotion of Science (Grant
no. 18K15627).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS, KO, IN, TO and NI contributed to the conception
and design of the study. KO and NI were supervised the work. YS
collected data. YS and KO drafted the manuscript. KO, HS, TY, JK,
KN, IN and NI surgically resected the specimens, which were used in
the separation and culture of the fibroblasts. TT and YE
contributed to establishing the fibroblasts and the analysis of the
data. TT, TO and NI contributed to the editing and reviewing of the
manuscript. YS and KO confirm the authenticity of all raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Kanazawa University (study no. 2016-463). Written
informed consent was obtained from all patients for the use of
surgically resected specimens.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SASP
|
senescence-associated secretory
phenotype
|
|
ESCC
|
esophageal squamous cell carcinoma
|
|
CAFs
|
cancer-associated fibroblasts
|
|
IL
|
interleukin
|
|
GRO
|
growth-related oncogene
|
|
CXCL
|
C-X-C motif chemokine ligand
|
|
MMPs
|
matrix metalloproteases
|
|
EMT
|
epithelial-mesenchymal transition
|
|
TGF-β
|
transforming growth factor-β
|
|
TOR
|
target of rapamycin
|
|
FBS
|
fetal bovine serum
|
|
SDS
|
sodium dodecyl sulfate
|
|
PVDF
|
polyvinylidene difluoride
|
References
|
1
|
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz
DP, Goldstein J, Nelson PS, Desprez PY and Campisi J:
Senescence-associated secretory phenotypes reveal
cell-nonautonomous functions of oncogenic RAS and the p53 tumor
suppressor. PLoS Biol. 6:2853–2868. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuilman T, Michaloglou C, Vredeveld LC,
Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ and Peeper DS:
Oncogene-induced senescence relayed by an interleukin-dependent
inflammatory network. Cell. 133:1019–1031. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coppé JP, Desprez PY, Krtolica A and
Campisi J: The senescence-associated secretory phenotype: The dark
side of tumor suppression. Ann Rev Pathol. 5:99–118. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang G, Rosen DG, Zhang Z, Bast RC Jr,
Mills GB, Colacino JA, Mercado-Uribe I and Liu J: The chemokine
growth-regulated oncogene 1 (Gro-1) links RAS signaling to the
senescence of stromal fibroblasts and ovarian tumorigenesis. Proc
Natl Acad Sci USA. 103:16472–16477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Serrano M and Blasco MA: Putting the
stress on senescence. Curr Opin Cell Biol. 13:748–753. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inomata K, Aoto T, Binh NT, Okamoto N,
Tanimura S, Wakayama T, Iseki S, Hara E, Masunaga T, Shimizu H and
Nishimura EK: Genotoxic stress abrogates renewal of melanocyte stem
cells by triggering their differentiation. Cell. 137:1088–1099.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Collado M, Blasco MA and Serrano M:
Cellular senescence in cancer and aging. Cell. 130:223–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Childs BG, Durik M, Baker DJ and van
Deursen JM: Cellular senescence in aging and age-related disease:
From mechanisms to therapy. Nat Med. 21:1424–1435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Isohata N, Aoyagi K, Mabuchi T, Daiko H,
Fukaya M, Ohta H, Ogawa K, Yoshida T and Sasaki H: Hedgehog and
epithelial-mesenchymal transition signaling in normal and malignant
epithelial cells of the esophagus. Int J Cancer. 125:1212–1221.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thiery JP, Acloque H, Huang RYJ and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okamoto K, Tajima H, Nakanuma S, Sakai S,
Makino I, Kinoshita J, Hayashi H, Nakamura K, Oyama K, Nakagawara
H, et al: Angiotensin II enhances epithelial-to-mesenchymal
transition through the interaction between activated hepatic
stellate cells and the stromal cell-derived factor-1/CXCR4 axis in
intrahepatic cholangiocarcinoma. Int J Oncol. 41:573–582. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Laberge RM, Awad P, Campisi J and Desprez
PY: Epithelial-mesenchymal transition induced by senescent
fibroblasts. Cancer Microenviron. 5:39–44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Velarde MC, Demaria M and Campisi J:
Senescent cells and their secretory phenotype as targets for cancer
therapy. Interdiscip Top Gerontol. 38:17–27. 2013.PubMed/NCBI
|
|
16
|
Moiseeva O, Deschênes-Simard X, St-Germain
E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN and Ferbeyre
G: Metformin inhibits the senescence-associated secretory phenotype
by interfering with IKK/NF-κB activation. Aging Cell. 12:489–498.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Makita N, Ninomiya I, Tsukada T, Okamoto
K, Harada S, Nakanuma S, Sakai S, Makino I, Kinoshita J, Hayashi H,
et al: Inhibitory effects of valproic acid in DNA double-strand
break repair after irradiation in esophageal squamous carcinoma
cells. Oncol Rep. 34:1185–1192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Acosta JC, O'Loghlen A, Banito A, Guijarro
MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N,
et al: Chemokine signaling via the CXCR2 receptor reinforces
senescence. Cell. 133:1006–1018. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hernandez-Segura A, Brandenburg S and
Demaria M: Induction and validation of cellular senescence in
primary human cells. J Vis Exp. 2018:e577822018.PubMed/NCBI
|
|
20
|
Yi G, He Z, Zhou X, Xian L, Yuan T, Jia X,
Hong J, He L and Liu J: Low concentration of metformin induces a
p53-dependent senescence in hepatoma cells via activation of the
AMPK pathway. Int J Oncol. 43:1503–1510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hellweg CE, Spitta LF, Koch K, Chishti AA,
Henschenmacher B, Diegeler S, Konda B, Feles S, Schmitz C, Berger T
and Baumstark-Khan C: The role of the nuclear factor κB pathway in
the cellular response to low and high linear energy transfer
radiation. Int J Mol Sci. 19:22202018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johmura Y, Sun J, Kitagawa K, Nakanishi K,
Kuno T, Naiki-Ito A, Sawada Y, Miyamoto T, Okabe A, Aburatani H, et
al: SCF(Fbxo22)-KDM4A targets methylated p53 for degradation and
regulates senescence. Nat Commun. 7:105742016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nagano T, Nakano M, Nakashima A, Onishi K,
Yamao S, Enari M, Kikkawa U and Kamada S: Identification of
cellular senescence-specific genes by comparative transcriptomics.
Sci Rep. 6:317582016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kalluri R: The biology and function of
fibroblasts in cancer. Nat Rev Cancer. 16:582–598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ezure T, Sugahara M and Amano S: Senescent
dermal fibroblasts negatively influence fibroblast extracellular
matrix-related gene expression partly via secretion of complement
factor D. Biofactors. 45:556–562. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chun TH, Sabeh F, Ota I, Murphy H,
McDonagh KT, Holmbeck K, Birkedal-Hansen H, Allen ED and Weiss SJ:
MT1-MMP-dependent neovessel formation within the confines of the
three-dimensional extracellular matrix. J Cell Biol. 167:757–767.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takino T, Yoshimoto T, Nakada M, Li Z,
Domoto T, Kawashiri S and Sato H: Membrane-type 1 matrix
metalloproteinase regulates fibronectin assembly and N-cadherin
adhesion. Biochem Biophys Res Commun. 450:1016–1020. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakayama A, Ninomiya I, Harada S, Tsukada
T, Okamoto K, Nakanuma S, Sakai S, Makino I, Kinoshita J, Hayashi
H, et al: Metformin inhibits the radiation-induced invasive
phenotype of esophageal squamous cell carcinoma. Int J Oncol.
49:1890–1898. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gonzalez-Angulo AM and Meric-Bernstam F:
Metformin: A therapeutic opportunity in breast cancer. Clin Cancer
Res. 16:1695–1700. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee SY, Moon SJ, Kim EK, Seo HB, Yang EJ,
Son HJ, Kim JK, Min JK, Park SH and Cho ML: Metformin suppresses
systemic autoimmunity in roquinsan/san mice through
inhibiting b cell differentiation into plasma cells via regulation
of AMPK/mTOR/STAT3. J Immunol. 198:2661–2670. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Algire C, Moiseeva O, Deschênes-Simard X,
Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G and Pollak
MN: Metformin reduces endogenous reactive oxygen species and
associated DNA damage. Cancer Prev Res (Phila). 5:536–543. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Formentini L, Sánchez-Aragó M,
Sánchez-Cenizo L and Cuezva JM: The mitochondrial ATPase inhibitory
factor 1 triggers a ROS-mediated retrograde prosurvival and
proliferative response. Mol Cell. 45:731–742. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schreck R, Rieber P and Baeuerle PA:
Reactive oxygen intermediates as apparently widely used messengers
in the activation of the NF-kappa B transcription factor and HIV-1.
EMBO J. 10:2247–2258. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chien Y, Scuoppo C, Wang X, Fang X,
Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, et al:
Control of the senescence-associated secretory phenotype by NF-κB
promotes senescence and enhances chemosensitivity. Genes Dev.
25:2125–2136. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Laberge RM, Sun Y, Orjalo AV, Patil CK,
Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et
al: MTOR regulates the pro-tumorigenic senescence-associated
secretory phenotype by promoting IL1A translation. Nat Cell Biol.
17:1049–1061. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Orjalo AV, Bhaumik D, Gengler BK, Scott GK
and Campisi J: Cell surface-bound IL-1alpha is an upstream
regulator of the senescence-associated IL-6/IL-8 cytokine network.
Proc Natl Acad Sci USA. 106:17031–17036. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Acosta JC, Banito A, Wuestefeld T,
Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka
F, Andrulis M, et al: A complex secretory program orchestrated by
the inflammasome controls paracrine senescence. Nat Cell Biol.
15:978–990. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Evans JMM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiralerspong S, Palla SL, Giordano SH,
Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi
GN and Gonzalez-Angulo AM: Metformin and pathologic complete
responses to neoadjuvant chemotherapy in diabetic patients with
breast cancer. J Clin Oncol. 27:3297–3302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Coyle C, Cafferty FH, Vale C and Langley
RE: Metformin as an adjuvant treatment for cancer: A systematic
review and meta-analysis. Ann Oncol. 27:2184–2195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wan G, Sun X, Li F, Wang X, Li C, Li H, Yu
X and Cao F: Survival benefit of metformin adjuvant treatment for
pancreatic cancer patients: A systematic review and meta-analysis.
Cell Physiol Biochem. 49:837–847. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dowling RJ, Niraula S, Stambolic V and
Goodwin PJ: Metformin in cancer: Translational challenges. J Mol
Endocrinol. 48:R31–R43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang JW, Zhang D, Yin HS, Zhang H, Hong
KQ, Yuan JP and Yu BP: Fusobacterium nucleatum promotes esophageal
squamous cell carcinoma progression and chemoresistance by
enhancing the secretion of chemotherapy-induced
senescence-associated secretory phenotype via activation of DNA
damage response pathway. Gut Microbes. 15:21978362023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang P, Lv C, Zhang T, Liu J, Yang J, Guan
F and Hong T: FOXQ1 regulates senescence-associated inflammation
via activation of SIRT1 expression. Cell Death Dis. 8:e29462017.
View Article : Google Scholar : PubMed/NCBI
|