Introduction
Lung cancer is the leading cause of cancer-related
death, and was estimated to be responsible for >120,000 deaths
in the United States in 2023 (1).
Despite improvements in early detection and treatment leading to a
decline in lung cancer mortality rates, the 5-year relative
survival rate remains low, particularly in non-small cell lung
cancer (NSCLC) which accounts for ~82% of all reported lung cancer
cases (1,2). Due to the frequently asymptomatic
onset of NSCLC, most patients are diagnosed at an advanced stage,
during which only 21% of patients are suitable for surgical
resection, and most (61%) are treated with chemotherapy, radiation
and/or immunotherapy (3,4). However, radiotherapy is not suitable
for postoperative patients due to its severe side effects and
deleterious effects on survival (5,6). Due
to primary and acquired resistance to immunotherapy, most patients
with NSCLC treated with immunotherapy do not achieve durable
clinical responses (7,8). By contrast, chemotherapy exhibits a
greater benefit than immunotherapy for patients with NSCLC;
however, synergistic immunotherapy is also recommended as the
standard of care in most patients with advanced NSCLC (9,10).
Cisplatin, an adjuvant chemotherapeutic drug that
binds to DNA and induces cell death, has been recommended as a
first-line drug for postoperative patients with NSCLC (11). In addition, cisplatin combined with
immunotherapy has been approved for patients with inoperable
advanced NSCLC (12). A pooled
analysis of adjuvant cisplatin in lung cancer confirmed that
cisplatin-based adjuvant chemotherapy exhibited a disease-free
survival benefit of 5.8% and an overall survival benefit of 5.4%,
compared with in patients who did not receive cisplatin-based
adjuvant chemotherapy (13).
However, most patients with NSCLC exhibit intrinsic or acquired
cisplatin resistance (14,15), thus leading to ineffective
responses. Therefore, there is an urgent need to understand the
molecular mechanisms underlying spontaneous and acquired resistance
to cisplatin.
Golgi phosphoprotein 3 (GOLPH3) is a Golgi
oncoprotein that is upregulated in numerous tumors, including lung,
breast and prostate cancer, and melanoma (16). High levels of GOLPH3 have been shown
to be positively associated with poor survival, partly via
GOLPH3-associated mTOR and WNT signaling pathway activation, which
can mediate pro-tumorigenic and drug-resistant effects in patients
with cancer (17). In line with
this, high levels of GOLPH3 can promote the resistance to multiple
chemotherapeutic drugs, such as oxaliplatin (18), 5-fluorouracil (19) and sorafenib (20). However, to the best of our
knowledge, GOLPH3-mediated cisplatin resistance and the underlying
mechanisms in patients with NSCLC have not been clarified. The
present study aimed to investigate the role of GOLPH3 in cisplatin
resistance in human NSCLC cells, and to understand the mechanisms
by which GOLPH3 regulates chemotherapeutic resistance and promotes
tumor metastasis.
Materials and methods
Cell culture
The A549 human NSCLC cell line was purchased from
The Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences. A549-cisplatin resistant (A549-Cis) cells were obtained
from Otwo Biotech (Shenzhen) Inc., and were created by screening
cells after long-term treatment with 5 µg/ml cisplatin. Therefore,
the present study also used a cisplatin concentration of 5 µg/ml.
The A549 cells were cultured in RPMI 1640 medium (HyClone; Cytiva)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin.
The cells were maintained in a humidified incubator containing 5%
CO2 at 37°C. The A549 cells used in the present
experiments were passaged a maximum of 50 times.
Establishment of cell lines with
stable expression
GOLPH3 short hairpin (sh)RNAs (shGOLPH3) and GOLPH3
overexpression (GOLPH3-OE) plasmid vectors were designed and
obtained from VectorBuilder GmbH. Notably,
pLV[shRNA]-EGFP:T2A:Puro-U6 was used as the vector backbone for
knockdown and pLV[Exp]-EGFP:T2A:Puro-EF1A was used as the vector
backbone for overexpression. Briefly, 1×106/dish 293T
cells (The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences) were plated on 10-cm dishes to clone the
lentiviruses encoding shGOLPH3, noncoding negative control (NC)
shRNA (shNC), GOLPH3-OE or GOLPH3-NC (empty vector). The lentiviral
vectors were produced using a 3rd generation system. For each
transfection, 10 µg lentiviral plasmid was used along with a 3:2:1
ratio of lentiviral vector to packaging plasmids (psPAX2) and
envelope plasmids (pMD2.G). The cells were transfected at 37°C for
48 h using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.). Lentiviral particles were collected 48 h
post-transfection, concentrated by ultracentrifugation and stored
at −80°C. For stable expression, A549 cells were infected with the
lentiviruses encoding shGOLPH3, shNC, GOLPH3-OE and GOLPH3-NC at a
multiplicity of infection of 10 at 37°C for 24 h, followed by
selection with 2 µg/ml puromycin (cat. no. P8230; Beijing Solarbio
Science & Technology Co., Ltd.) at 37°C for 72 h. GOLPH3
expression levels were verified by reverse
transcription-quantitative PCR (RT-qPCR) and western blotting. The
oligonucleotide sequences for shRNAs were as follows: sh1,
5′-GCTTGTGGAATGAGACGTAAACTCGAGTTTACGTCTCATTCCACAAGC-3′ (target
sequence: GCTTGTGGAATGAGACGTAAA); sh2,
5′-GCTTGCTTCAATCATGGTTATCTCGAGATAACCATGATTGAAGCAAGC-3′ (target
sequence: GCTTGCTTCAATCATGGTTAT); shNC,
5′-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′ (target
sequence: CCTAAGGTTAAGTCGCCCTCG; this sequence has no corresponding
target was found in both humans and mice). Cells were allowed to
recover for 72 h post-transduction before any subsequent
experimentation.
RNA isolation and RT-qPCR
Total RNA was isolated from NSCLC cells using a
PureLink™ RNA isolation kit (cat. no. 12183018A; Invitrogen; Thermo
Fisher Scientific, Inc.). cDNA was generated from the RNA using a
PrimeScipt™ RT reagent kit (cat. no. RR037A; Takara Biotechnology
Co., Ltd.) according to the manufacturer's protocol. RT was
performed at 37°C for 15 min, followed by 85°C for 5 sec to
inactivate the reverse transcriptase. qPCR was performed using SYBR
Green (Bio-Rad Laboratories, Inc.) on the CFX96 touch real-time PCR
system (iQ5; Bio-Rad Laboratories, Inc.). The thermocycling
conditions were as follows: Initial denaturation at 95°C for 3 min,
followed by 40 cycles at 95°C for 10 sec and 60°C for 30 sec.
β-actin was used as a normalization control and relative mRNA
expression levels were calculated using the 2−ΔΔCq
method (21). The primer sequences
for qPCR were as follows (5′-3′): GOLPH3, forward
GATGCTCCAACAGGGGATGT, reverse TGGTGAGGGGATGTGTTGTC; ATP7A forward
GCCAGCCTCTGACACAAGAA, reverse GCTCCTCTCAACGTTTCTGGA; ABCG2, forward
TGGCTGTCATGGCTTCAGTA, reverse GCCACGTGATTCTTCCACAA; MATE1, forward
CTTCAGGCAGGACCCAGAT, reverse CAGATAGTTGGCGAGGGCAT; MATE2K, forward
ATCCTAGCCACCAGGCACTA, reverse GTGTCCACCTGCACTAGACC; ALDH1A1,
forward TGGACCAGTGCAGCAAATCA, reverse ACGCCATAGCAATTCACCCA; C-myc,
forward TACTGCGACGAGGAGGAGAA, reverse CGAAGGGAGAAGGGTGTGAC;
β-actin, forward AACTGGGACGACATGGAGAAAA, reverse,
GGATAGCACAGCCTGGATAGCA.
Western blot analysis
Total proteins were extracted from NSCLC cells with
RIPA buffer (Roche Diagnostics). The protein concentration was
determined using the bicinchoninic acid (BCA) assay (Pierce BCA
Protein Assay Kit; Thermo Fisher Scientific, Inc.). Equal amounts
of protein (30 µg/lane) were separated by SDS-PAGE on a 10%
polyacrylamide gel and were transferred to polyvinylidene fluoride
(PVDF) membranes (Bio-Rad Laboratories, Inc.). After blocking in 5%
BSA at room temperature for 1 h, the PVDF membranes were probed
with primary anti-GOLPH3 (1:1,000; cat. no. A13121; ABclonal
Biotech Co., Ltd.) and anti-GAPDH (1:5,000; cat. no. AC026;
ABclonal) antibodies overnight at 4°C. Subsequently, the membranes
were incubated with horseradish peroxidase-linked secondary
antibodies (1:5,000; cat. no. A6154; Sigma-Aldrich; Merck KGaA) at
room temperature for 1 h. The ECL Kit (Wanleibio Co., Ltd.) was
used for detecting protein bands, and protein levels were
semi-quantified with Image Lab software (Bio-Rad Laboratories,
Inc.), normalized to GAPDH levels.
Cell viability assay
Cell viability was evaluated using the Cell Counting
Kit-8 (CCK-8) assay (Dojindo Laboratories, Inc.). Briefly,
0.5–1×104 cells/well of NSCLC cells (infected with shNC,
sh1, sh2, GOLPH3-OE and GOLPH3-NC) were seeded in 96-well plates.
After an overnight incubation, the cells were treated with various
concentrations of cisplatin (0, 1, 2.5, 5, 10, 25 and 50 µg/ml;
Shanghai Aladdin Biochemical Technology Co., Ltd.) at 37°C for 24
h. Subsequently, CCK-8 solution was added to the phenol red- and
serum-free cell medium for another 1 h. Light absorbance at 450 nm
was then recorded using an Infinite M200 Pro microplate reader
(Tecan Group, Ltd.). Each concentration was tested at least three
times.
EdU assay
A total of 5×104 cells/well of NSCLC
cells (infected with shNC, sh1, sh2, GOLPH3-OE and GOLPH3-NC) were
seeded in 24-well plates. After overnight incubation, the cells
were treated with 5 µg/ml cisplatin at 37°C for 24 h. Subsequently,
the cells were cultured with EdU reagent (1:1,000 dilution;
Beyotime Institute of Biotechnology) for another 2 h, and were
fixed with 4% paraformaldehyde at 37°C for 15 min, followed by
staining with DAPI (1 µg/ml) at 37°C for 10 min. All cells were
detected using fluorescence microscopy (Leica Microsystems,
Inc.).
Cell death assay
Cell death was evaluated using an Annexin
V-FITC/propidium iodide (PI) Staining Kit (cat. no. CA001;
Signalway Antibody LLC). Stably transduced NSCLC cells were plated
onto 24-well plates at a density of 5×104 cells/well.
After incubation for 24 h, the cells were treated with 5 µg/ml
cisplatin at 37°C for 24 h. Subsequently, the cells were washed
with phosphate-buffered saline (PBS), digested with EDTA-free
trypsin, resuspended in 500 µl binding buffer, and stained with 10
µl Annexin V-FITC and 10 µl PI at 37°C for 15 min in the dark.
After washing twice with PBS, the cells were immediately analyzed
by flow cytometry (FACSCelesta; BD Biosciences) and fluorescence
microscopy (Leica Microsystems, Inc.), the results were analyzed
using FlowJo-V10-CL software (FlowJo LLC).
Cell cycle assay
Cell cycle progression was examined using a Cell
Cycle Analysis Kit (cat. no. C1052; Beyotime Institute of
Biotechnology). Stably transduced NSCLC cells were plated onto
24-well plates at a density of 5×104 cells/well and were
incubated for 24 h, after which, the cells were treated with 5
µg/ml cisplatin at 37°C for 24 h. After washing twice with PBS to
remove excess cisplatin, the cells were fixed with 75% ethanol for
24 h at 4°C, were washed twice with PBS and were treated with RNAse
(100 µg/ml) and PI (50 µg/ml) solution at 37°C for 30 min in the
dark. After washing twice with PBS, the cells were assessed using
flow cytometry (FACSCelesta; BD Biosciences) and the results were
analyzed using FlowJo-V10-CL software.
Intracellular cisplatin concentration
detection
NSCLC cells were plated onto 24-well plates at a
density of 5×104 cells/well and were incubated at 37°C
for 24 h. The cells were treated with 5 µg/ml cisplatin-Cy5 (Xi'an
Qiyue Biotechnology Co., Ltd.) at 37°C for 24 h, and, after washing
twice with PBS, intracellular cisplatin-Cy5 concentration was
assessed under a fluorescence microscope (Leica Microsystems,
Inc.).
Intracellular GSH detection
Intracellular GSH levels were detected using a GSH
Assay Kit (cat. no. S0053; Beyotime Institute of Biotechnology).
NSCLC cells were plated onto 24-well plates at a density of
5×104 cells/well and were incubated at 37°C for 24 h.
The cells were treated with 5 µg/ml cisplatin at 37°C for 24 h,
after which, intracellular GSH levels were measured according to
the kit instructions.
Intracellular reactive oxygen species
(ROS) detection
Intracellular ROS were detected using a Reactive
Oxygen Species Assay Kit (cat. no. S0033S; Beyotime Institute of
Biotechnology). NSCLC cells were plated onto 24-well plates at a
density of 5×104 cells/well and were incubated for 24 h.
The cells were treated with 5 µg/ml cisplatin at 37°C for 24 h,
and, after washing twice with PBS, the DCFH-DA probe was added to
the culture medium and co-incubated 37°C for another 2 h. After
washing twice with PBS to remove excess DCFH-DA, the cells were
resuspended in PBS and were detected using fluorescence microscopy
(Leica Microsystems, Inc.).
Sphere formation assay
NSCLC cells were seeded into ultra-low cluster
6-well microplates at a density of 500 cells/well and were cultured
in serum-free medium (containing 20 ng/ml basic fibroblast growth
factor and epidermal growth factor, 5 µg/ml insulin, and 0.4% BSA;
Dalian Meilun Biology Technology Co., Ltd.) supplemented with 5
µg/ml cisplatin. Cells were cultured for ~10 days before 3D tumor
spheres (tight, spherical, nonadherent masses >50 µm in
diameter) were observed and images were captured under a
phase-contrast fluorescence microscope (Leica Microsystems,
Inc.).
Statistical analysis
Data are presented as the mean ± SD (n=3). The
experimental data were statistically analyzed by one-way analysis
of variance using GraphPad Prism 7.0 statistical software
(Dotmatics). For post hoc comparisons, the Fisher's least
significant difference method was used, or the Tukey honestly
significant difference method was used when >3 groups were
compared. P<0.05 was considered to indicate a statistically
significant difference.
Results
GOLPH3 knockdown is associated with
cisplatin sensitivity
To evaluate the effects of GOLPH3 on cisplatin
resistance, knockdown and overexpression of GOLPH3 using
lentiviruses encoding shGOLPH3 or GOLPH3-OE was performed. The
protein and mRNA expression levels of GOLPH3 were increased in
GOLPH3-OE cells, and were decreased in shGOLPH3 cells (Figs. 1A and S1). Subsequently, the inhibitory effects
of cisplatin on the viability of these cells were assessed. As
shown in Fig. 1B, GOLPH3 knockdown
significantly increased the inhibitory effects of 5 µg/ml cisplatin
on cell viability by ~2 fold. By contrast, 2.5 µg/ml cisplatin
reduced cell viability by ~25% in GOLPH3-knockdown cells, but had
minimal impact on control cells. Moreover, the inhibitory effect of
cisplatin on A549-Cis cells was significantly lower than that on
A549 cells (Fig. S2). By contrast,
overexpression of GOLPH3 resulted in minimal impact on the
inhibitory effects of cisplatin on cell viability (Fig. 1C), indicating that GOLPH3
overexpression does not affect cisplatin resistance. Since EdU is
commonly used to detect cell proliferation, cells with GOLPH3
overexpression or knockdown were treated with EdU in the presence
of 5 µg/ml cisplatin. Consistent with the cell viability assay, the
numbers of EdU-positive cells were markedly decreased in the GOLPH3
knockdown groups compared with those in the control group (Fig. 1D). By contrast, the numbers of
EdU-positive cells were comparatively higher in the GOLPH3
overexpression group than in the control group. These findings
indicated that GOLPH3 knockdown may increase cisplatin sensitivity
in NSCLC cells.
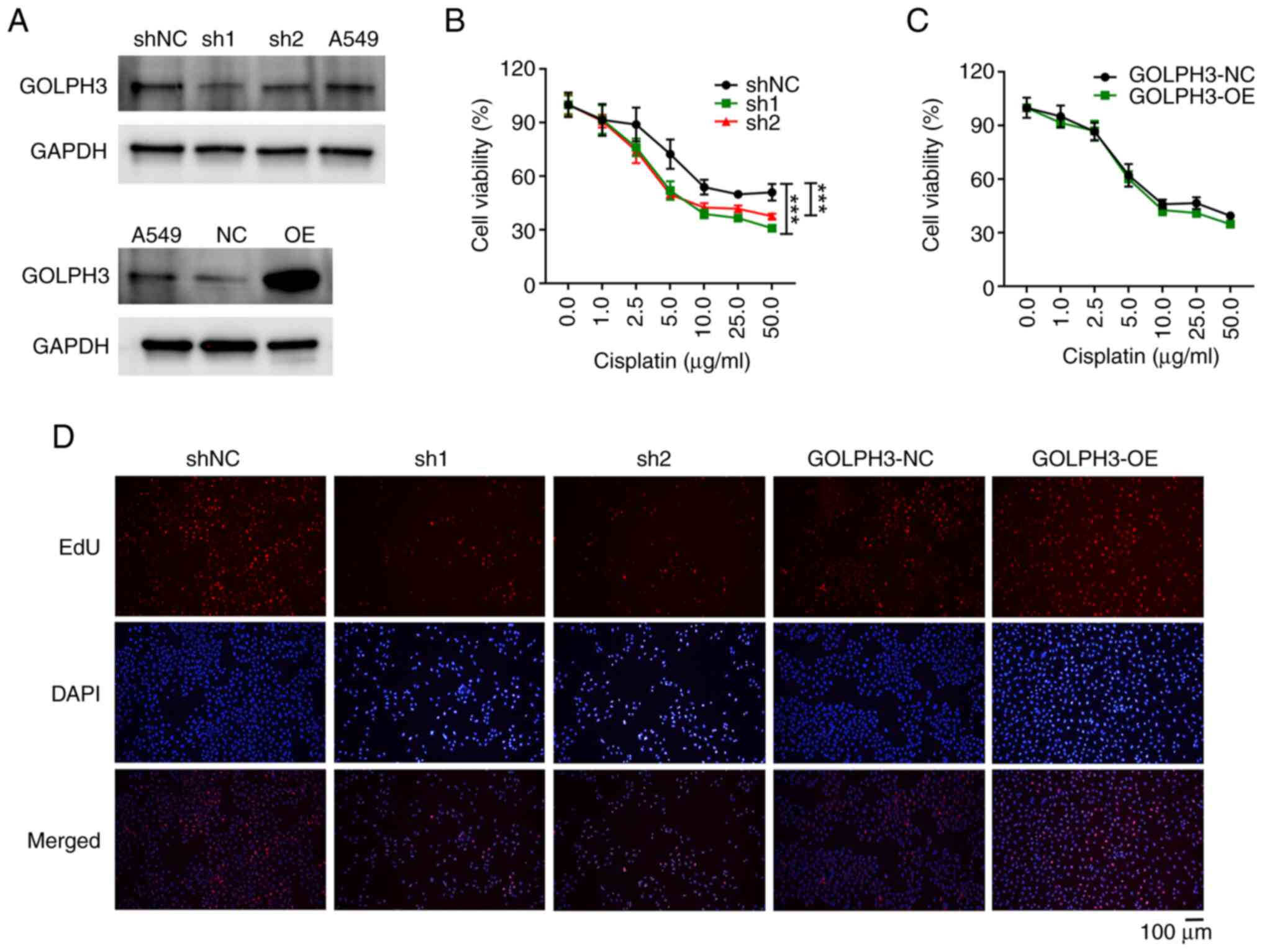 | Figure 1.GOLPH3 knockdown inhibits the
viability and proliferation of A549 cells. (A) GOLPH3 knockdown or
OE was examined by western blot analysis. Following treatment with
the indicated concentrations of cisplatin for 24 h, the viability
of A549 cells infected with (B) GOLPH3 shNC, sh1 and sh2, or (C)
GOLPH3-NC and GOLPH3-OE was evaluted by Cell Counting Kit-8 assay.
Cell viability was suppressed in sh1 and sh2 cells, but no
difference was detected in GOLPH3-OE cells. Data are presented as
the mean ± SD (n=3). ***P<0.001. (D) Proliferation of A549 cells
infected with GOLPH3 shNC, sh1, sh2, GOLPH3-NC and GOLPH3-OE
treated with 5 µg/ml cisplatin for 24 h were examined by EdU assay.
Cell proliferation was suppressed in sh1 and sh2 cells, but
increased in GOLPH3-OE cells. GOLPH3, Golgi phosphoprotein 3; NC,
negative control; OE, overexpression; sh, short hairpin. |
The present study further investigated whether
GOLPH3 knockdown could enhance the apoptosis of cells in the
presence of 5 µg/ml cisplatin. A low proportion of apoptotic cells
was observed in the control group, whereas the proportion was
increased to 10–15% in the GOLPH3 knockdown groups (Fig. 2A and B). Consistent with the results
of flow cytometry, immunofluorescence analysis showed that GOLPH3
knockdown markedly increased the proportion of Annexin
V+ and/or PI+ cells (Fig. 2C). Moreover, cisplatin-mediated DNA
toxicity was elevated in cells with GOLPH3 knockdown. As shown in
Fig. 2D, GOLPH3 knockdown in A549
cells resulted in the prolongation of cisplatin-mediated cell cycle
arrest at G2 phase compared with that in the shNC group.
Without cisplatin treatment, the cell cycle distribution was
consistent for all cells (Fig.
S3). By contrast, GOLPH3 overexpression partially abrogated
cisplatin-mediated cell cycle block at G2 phase compared
with that in the GOLPH3-NC group (Fig.
2E), suggesting that GOLPH3 may impair cisplatin-DNA adducts
and cytotoxicity. These data indicated that GOLPH3 knockdown not
only enhances the pro-apoptotic effect of cisplatin, but may also
elevate its DNA toxicity.
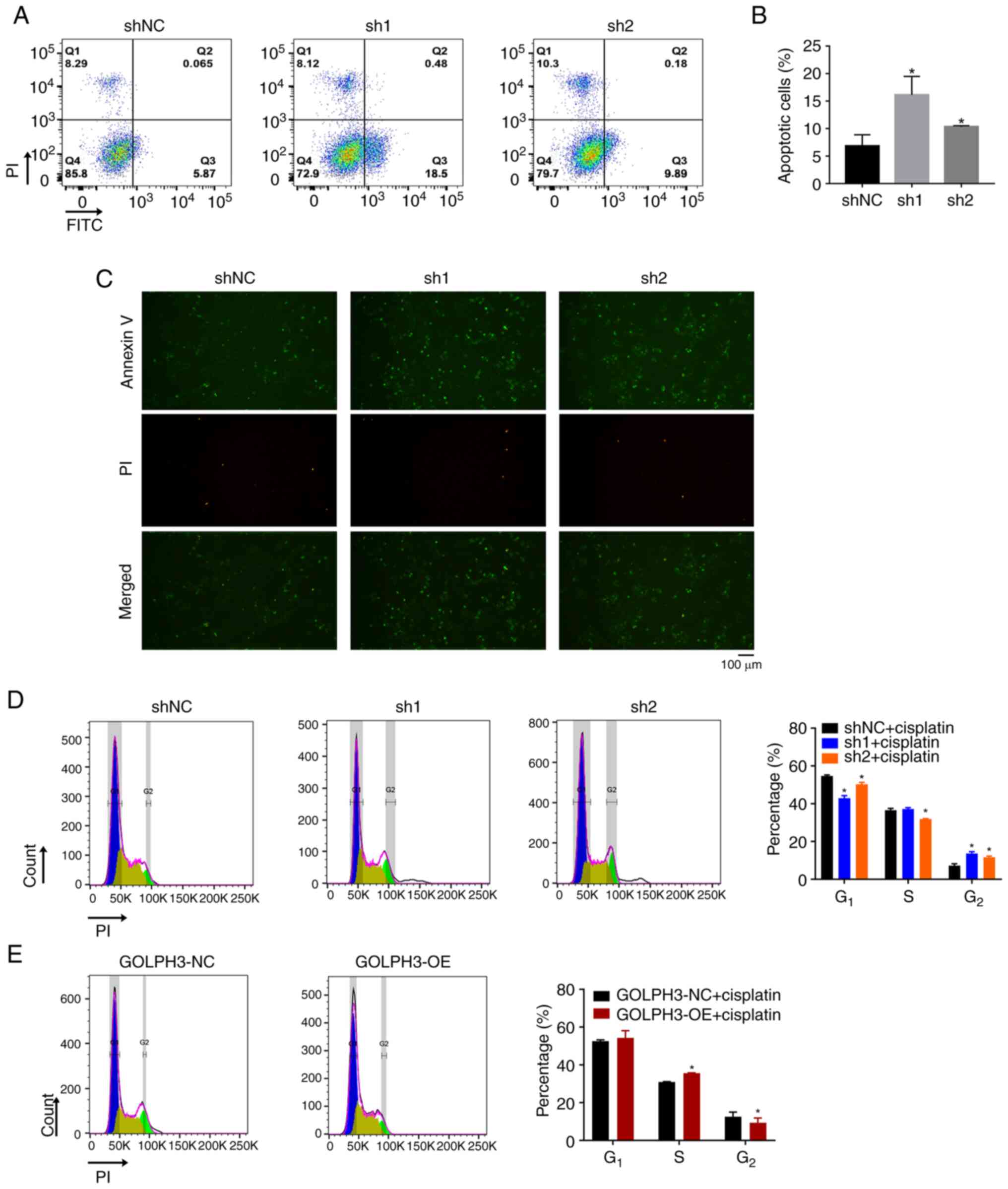 | Figure 2.GOLPH3 knockdown enhances apoptosis
and cell cycle arrest in A549 cells. Following treatment with 5
µg/ml cisplatin for 24 h, A549 cells infected with GOLPH3 shNC,
sh1, sh2, GOLPH3-NC and GOLPH3-OE were stained with Annexin V/PI or
PI for cell apoptosis or cell cycle assays, respectively. GOLPH3
knockdown increased the proportion of apoptotic cells. (A)
Representative plots of flow cytometric analysis of cell apoptosis
and (B) statistical analysis of the proportion of apoptotic cells.
*P<0.05 vs. shNC. (C) Representative immunofluorescence images
of Annexin V+ and/or PI+ cells. GOLPH3
knockdown prolonged the cell cycle arrest in G2 phase. (D)
Representative plots of flow cytometric analysis of cell cycle
progression in cells infected with shNC, sh1 and sh2 (left panel).
Statistical analysis of the proportion of cells in each stage of
the cell cycle (right panel). (E) Representative plots of flow
cytometric analysis of cell cycle progression in cells infected
with GOLPH3-NC and GOLPH3-OE cells (left panel). Statistical
analysis of the proportion of cells in each stage of the cell cycle
(right panel). Data are presented as the mean ± SD (n=3). GOLPH3,
Golgi phosphoprotein 3; NC, negative control; OE, overexpression;
PI, propidium iodide; sh, short hairpin. |
GOLPH3 knockdown elevates
intracellular cisplatin level
Reduced drug accumulation is a main factor involved
in cisplatin resistance (14,15).
To verify the hypothesis that GOLPH3 decreased intracellular
concentrations of cisplatin, the present study assessed the
intracellular cisplatin concentration using cisplatin-Cy5
fluorescence. As expected, GOLPH3 knockdown in A549 cells led to
enhanced cisplatin-fluorescence signal accumulation (Fig. 3A). By contrast, GOLPH3
overexpression notably decreased intracellular cisplatin
concentration (Fig. 3B).
Intracellular drug concentrations depend on cellular uptake or
efflux, and passive drug diffusion is the main form of drug
accumulation. The present study aimed to uncover the biological
functions of GOLPH3 on intracellular drug efflux by examining the
expression levels of drug efflux transporters. RT-qPCR showed that
GOLPH3 knockdown significantly downregulated ATP7A (Fig. 3C), ABCG2 (Fig. 3D), MATE1 (Fig. 3E) and MATE2K (Fig. 3F) mRNA expression levels compared
with those in the shNC group, indicating that GOLPH3 knockdown may
increase cisplatin sensitivity by downregulating the expression of
ABC transporters and decreasing drug efflux.
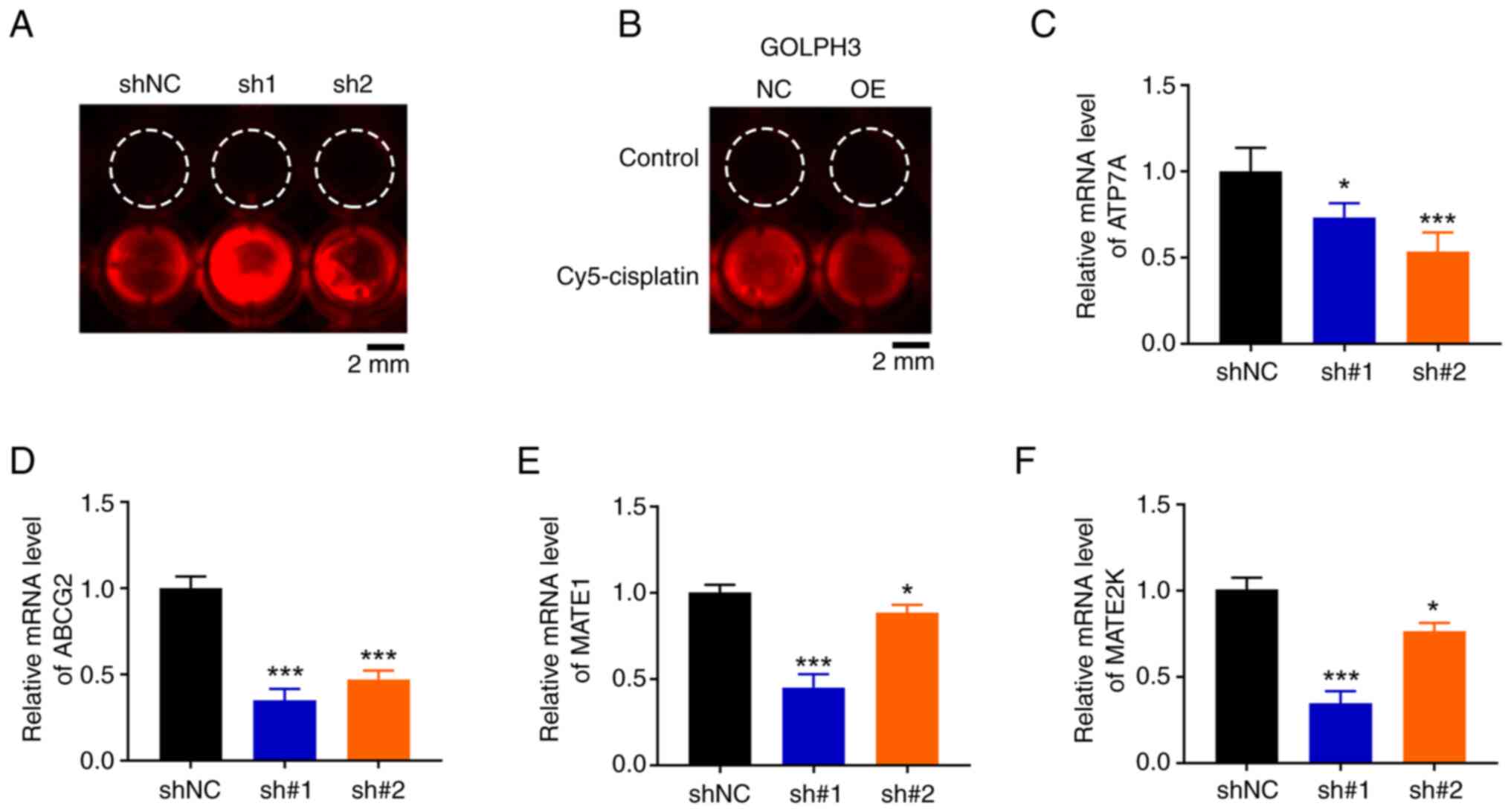 | Figure 3.GOLPH3 knockdown increases cisplatin
accumulation in A549 cells. A549 cells infected with (A) GOLPH3
shNC, sh1 or sh2, and (B) GOLPH3-NC or GOLPH3-OE were treated with
5 µg/ml cisplatin-Cy5 for 24 h and intracellular cisplatin-Cy5
concentration was assessed by fluorescence microscopy.
Representative fluorescence images of intracellular cisplatin-Cy5
accumulation are shown. mRNA expression levels of ABC transporters,
including (C) ATP7A, (D) ABCG2, (E) MATE1 and (F) MATE2K were
examined relative to levels in GOLPH3-shNC cells by reverse
transcription-quantitative PCR. β-actin was used as an endogenous
control. Data are presented as the mean ± SD (n=3). *P<0.05,
***P<0.001 vs. shNC. GOLPH3, Golgi phosphoprotein 3; NC,
negative control; OE, overexpression; PI, propidium iodide; sh,
short hairpin. |
GOLPH3 impairs glutathione (GSH)/ROS
balance
Antioxidant ability assists in the survival of tumor
cells treated with chemotherapy (22); therefore, the present study assessed
whether GOLPH3 was associated with redox homeostasis. The levels of
GSH were examined in cells with GOLPH3 knockdown or overexpression
under 5 µg/ml cisplatin treatment. As shown in Fig. 4A, cells with GOLPH3 knockdown
exhibited a reduction in GSH levels compared with in the control
cells. By contrast, GOLPH3-overexpressing cells displayed higher
levels of GSH compared with in the control cells (Fig. 4B), indicating that GOLPH3 could
impair redox homeostasis by maintaining a reduced state. Notably,
A549-Cis cells exhibited higher levels of GSH compared with A549
cells, which is consistent with the results of
GOLPH3-overexpressing cells (Fig.
S4). The present study further assessed the dependence of ROS
accumulation on GOLPH3 expression. Enhanced ROS accumulation was
detected in cells with GOLPH3 knockdown compared with in the
control cells, whereas no notable ROS signal was detected in
GOLPH3-overexpressing cells, following treatment with 5 µg/ml
cisplatin (Fig. 4C). These data
strongly indicated that cisplatin-mediated oxidative stress depends
on cellular GOLPH3 expression, and that GOLPH3 overexpression may
disrupt the GSH/ROS balance and result in cisplatin resistance.
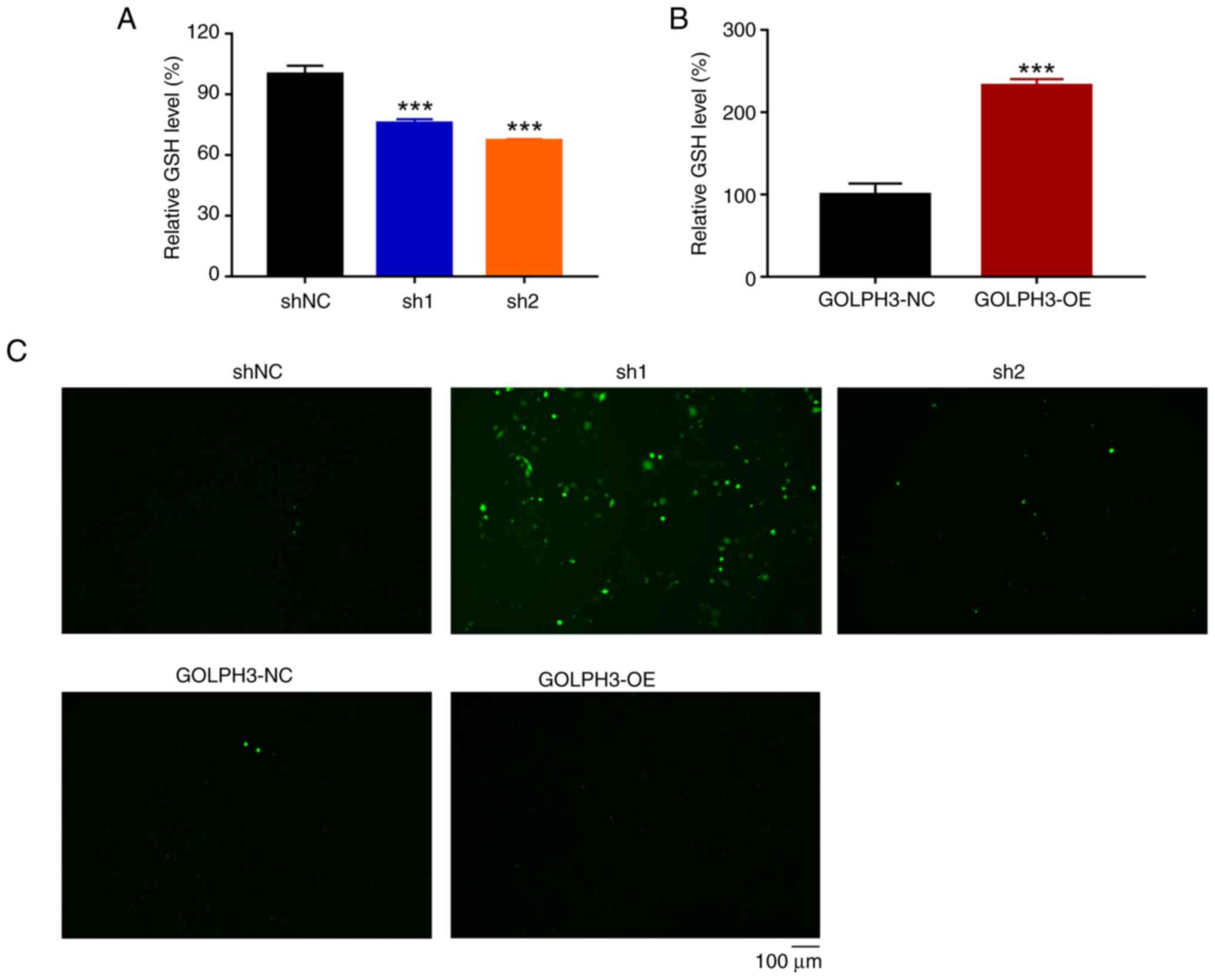 | Figure 4.GOLPH3 knockdown impairs the
antioxidant capacity of A549 cells. A549 cells infected with GOLPH3
shNC, sh1, sh2, GOLPH3-NC and GOLPH3-OE were treated with 5 µg/ml
cisplatin for 24 h and intracellular GSH and ROS level were
examined. Relative GSH level in cells infected with (A) shNC, sh1
or sh2, and (B) GOLPH3-NC or GOLPH3-OE. Data are presented as the
mean ± SD (n=3). ***P<0.001 vs. shNC. (C) Cisplatin-induced ROS
levels were elevated in cells with GOLPH3 knockdown, as examined
using a DCFH-DA probe. Green fluorescence indicates ROS levels.
GOLPH3, Golgi phosphoprotein 3; GSH, glutathione; NC, negative
control; OE, overexpression; PI, propidium iodide; ROS, reactive
oxygen species; sh, short hairpin. |
Cisplatin resistance is associated
with GOLPH3-mediated stem cell-like phenotype
Tumor stem cell subpopulations are closely
associated with chemotherapeutic resistance. Therefore, the present
study explored the possibility that intrinsic cisplatin resistance
in NSCLC cells is due to the existence of a GOLPH3-mediated stem
cell subpopulation. Cancer stem cells (CSCs) are a subpopulation of
cancer cells with the ability to self-renew and drive tumorigenesis
(23). As expected, the mRNA
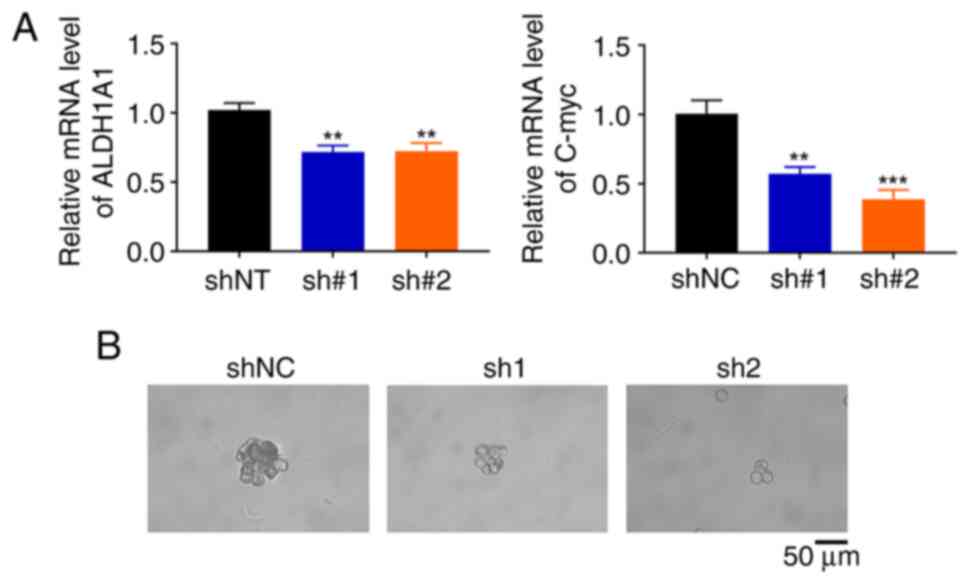
expression levels of CSC, markers (ALDH1A1, C-myc) were
significantly inhibited in NSCLC cells with GOLPH3 knockdown
(Fig. 5A). Tumor sphere formation
assay further verified this phenomenon. The size and number of
sphere colonies were markedly inhibited in cells with GOLPH3
knockdown compared with in the control group (Fig. 5B). These data suggested that GOLPH3
knockdown could suppress the stem cell-like phenotype of NSCLC
cells and further elevate cisplatin sensitivity.
Discussion
The heterogeneity of NSCLC results in the
short-lived potential of targeted therapy and immunotherapy.
Cisplatin-based chemotherapy is of benefit for postoperative
patients with NSCLC (13); however,
intrinsic and acquired therapeutic resistance severely limits its
clinical use (22). In addition,
the molecular mechanism of cisplatin resistance in NSCLC remains
unknown. The present study demonstrated the relevance of GOLPH3
protein in cisplatin resistance; knockdown of GOLPH3 markedly
augmented cisplatin sensitivity. This may be attributed to the fact
that low levels of GOLPH3 could inhibit the expression of ABC
transporters and further mitigate cisplatin efflux. Furthermore,
knockdown of GOLPH3 impaired the antioxidant ability of NSCLC cells
and elevated cisplatin-associated oxidative stress. Consequently,
cisplatin resistance may be attenuated by suppressing GOLPH3
protein expression.
High levels of ROS can impair tumor survival and
growth; however, most tumors possess marked antioxidant capacity
and thus exhibit therapeutic resistance (24,25).
In NSCLC, high levels of antioxidant enzymes, such as superoxide
dismutase 1 and GSH S-transferase, are commonly present in tumor
tissues, and inhibition of these enzymes can increase
chemotherapy-associated cell death, even in the presence of KRAS
mutations that promote continued proliferation of tumor cells
(26). Therefore, elevated
oxidative stress may be a novel avenue for cancer therapy. The
present study demonstrated a novel biological function of GOLPH3
that is relevant to cellular redox balance. Knockdown of GOLPH3
significantly reduced intracellular GSH levels, which is a key
antioxidant that could bind to cisplatin to form a deactivated
complex readily excreted by a GSH S-conjugated export pump,
resulting in enhanced drug accumulation and increased drug
sensitivity. By contrast, knockdown of GOLPH3 elevated
intracellular ROS levels. As high levels of GSH are often
associated with cisplatin resistance and tumor malignancy,
elucidating the relevance of GOLPH3 and GSH/ROS balance may expand
clinical application and overcome cisplatin resistance.
CSCs are a small heterogeneous subpopulation of
tumor cells that not only maintain the renewal capability of tumor
cells, but are also related to the resistance of tumor cells to
chemotherapy or radiotherapy (27).
Since increasing studies have reported that the PI3K-Akt-mTOR
signaling pathway is the major regulator of CSCs, which is involved
in the maintenance of stemness, proliferation, differentiation and
survival, inhibition of mTOR pathway activity may be a promising
therapeutic strategy for CSC-associated drug resistance (28,29).
Notably, GOLPH3, a Golgi oncoprotein, is an inherent initiator of
the activation of mTOR and its downstream signaling (17). The present study revealed that
knockdown of GOLPH3 markedly inhibited the expression levels of the
CSC markers ALDH1A and C-myc in NSCLC cells. Consistent with these
results, fewer tumor spheres were formed in cells with GOLPH3
knockdown than in control cells.
It is important to acknowledge that the absence of
another NSCLC cell line, primary NSCLC cells or an animal model in
the present study represents a potential limitation. The lack of
additional models restricts the generalizability and clinical
relevance of the findings; therefore, future investigations should
consider incorporating these supplementary models to provide a more
comprehensive evaluation of the therapeutic potential of GOLPH3
inhibition.
In summary, the present study revealed the relevance
of GOLPH3 in cisplatin resistance in the A549 NSCLC cell line.
Knockdown of GOLPH3 not only increased cisplatin accumulation but
also elevated oxidative stress in these cells. In addition,
knockdown of GOLPH3 diminished the CSC-like properties of NSCLC
cells and restored cisplatin sensitivity. Thus, inhibition of
GOLPH3 may be considered a promising therapeutic strategy for
cisplatin resistance in NSCLC. These findings should be further
investigated in vivo and in the clinic to determine whether
inhibition of GOLPH3 could be translated from experimental research
to clinical application.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the Education and Research Project
for Middle and Young Teachers in Fujian Province (Science and
Technology), China (grant no. JAT210124); the Startup Fund for
Scientific Research, Fujian Medical University, China (grant no.
2021QH1024); and Fujian Provincial Natural Science Foundation of
China (grant no. 2023J01635).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
DX conceived and designed the experiments. QW, ZL
and JL performed the experiments. QW, ZL, NY, YW, XT and JL
performed the data analyses. QW, ZL and JL drafted and revised the
manuscript. DX finalized the manuscript. QW and DX confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Siegel RL and Jemal A: Lung
cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miller KD, Nogueira L, Devasia T, Mariotto
AB, Yabroff KR, Jemal A, Kramer J and Siegel RL: Cancer treatment
and survivorship statistics, 2022. CA Cancer J Clin. 72:409–436.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang M, Herbst RS and Boshoff C: Toward
personalized treatment approaches for non-small-cell lung cancer.
Nat Med. 27:1345–1356. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Banfill K, Giuliani M, Aznar M, Franks K,
McWilliam A, Schmitt M, Sun F, Vozenin MC and Faivre Finn C; IASLC
Advanced Radiation Technology committee, : Cardiac toxicity of
thoracic radiotherapy: Existing evidence and future directions. J
Thorac Oncol. 16:216–227. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bentzen SM: Preventing or reducing late
side effects of radiation therapy: Radiobiology meets molecular
pathology. Nat Rev Cancer. 6:702–713. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wennerberg E, Mukherjee S, Spada S, Hung
C, Agrusa CJ, Chen C, Valeta-Magara A, Rudqvist NP, Van Nest SJ,
Kamel MK, et al: Expression of the mono-ADP-ribosyltransferase ART1
by tumor cells mediates immune resistance in non-small cell lung
cancer. Sci Transl Med. 14:eabe81952022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Horvath L, Thienpont B, Zhao L, Wolf D and
Pircher A: Overcoming immunotherapy resistance in non-small cell
lung cancer (NSCLC)-novel approaches and future outlook. Mol
Cancer. 19:1412020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Ruysscher D, Faivre-Finn C, Nackaerts
K, Jordan K, Arends J, Douillard JY, Ricardi U and Peters S:
Recommendation for supportive care in patients receiving concurrent
chemotherapy and radiotherapy for lung cancer. Ann Oncol. 31:41–49.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rinaldi M, Cauchi C and Gridelli C: First
line chemotherapy in advanced or metastatic NSCLC. Ann Oncol. 17
(Suppl 5):v64–v67. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tsan DL, Lin CY, Kang CJ, Huang SF, Fan
KH, Liao CT, Chen IH, Lee LY, Wang HM and Chang JT: The comparison
between weekly and three-weekly cisplatin delivered concurrently
with radiotherapy for patients with postoperative high-risk
squamous cell carcinoma of the oral cavity. Radiat Oncol.
7:2152012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chaft JE, Rimner A, Weder W, Azzoli CG,
Kris MG and Cascone T: Evolution of systemic therapy for stages
I–III non-metastatic non-small-cell lung cancer. Nat Rev Clin
Oncol. 18:547–557. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pignon JP, Tribodet H, Scagliotti GV,
Douillard JY, Shepherd FA, Stephens RJ, Dunant A, Torri V, Rosell
R, Seymour L, et al: Lung adjuvant cisplatin evaluation: A pooled
analysis by the LACE collaborative group. J Clin Oncol.
26:3552–3559. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun Y, Shen W, Hu S, Lyu Q, Wang Q, Wei T,
Zhu W and Zhang J: METTL3 promotes chemoresistance in small cell
lung cancer by inducing mitophagy. J Exp Clin Cancer Res.
42:652023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang D, Zhao C, Xu F, Zhang A, Jin M,
Zhang K, Liu L, Hua Q, Zhao J, Liu J, et al: Cisplatin-resistant
NSCLC cells induced by hypoxia transmit resistance to sensitive
cells through exosomal PKM2. Theranostics. 11:2860–2875. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song JW, Zhu J, Wu XX, Tu T, Huang JQ,
Chen GZ, Liang LY, Zhou CH, Xu X and Gong LY: GOLPH3/CKAP4 promotes
metastasis and tumorigenicity by enhancing the secretion of
exosomal WNT3A in non-small-cell lung cancer. Cell Death Dis.
12:9762021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Scott KL, Kabbarah O, Liang MC, Ivanova E,
Anagnostou V, Wu J, Dhakal S, Wu M, Chen S, Feinberg T, et al:
GOLPH3 modulates mTOR signalling and rapamycin sensitivity in
cancer. Nature. 459:1085–1090. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu T, An Q, Cao XL, Yang H, Cui J, Li ZJ
and Xiao G: GOLPH3 inhibition reverses oxaliplatin resistance of
colon cancer cells via suppression of PI3K/AKT/mTOR pathway. Life
Sci. 260:1182942020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang MZ, Qiu CZ, Yu WS, Guo YT, Wang CX
and Chen ZX: GOLPH3 expression promotes the resistance of HT29
cells to 5-fluorouracil by activating multiple signaling pathways.
Mol Med Rep. 17:542–548. 2018.PubMed/NCBI
|
|
20
|
Gao Y, Yin Z, Qi Y, Peng H, Ma W, Wang R
and Li W: Golgi phosphoprotein 3 promotes angiogenesis and
sorafenib resistance in hepatocellular carcinoma via upregulating
exosomal miR-494-3p. Cancer Cell Int. 22:352022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meng F, Li Y, Liu Q, Sun L, Wang H, Li X,
Li G and Chen F: Experimental study of camptothecin combined with
drug-eluting bead transarterial chemoembolization in the rabbit VX2
liver tumor model. Front Oncol. 12:9069712022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qi Y, Wei J and Zhang X: Requirement of
transcription factor NME2 for the maintenance of the stemness of
gastric cancer stem-like cells. Cell Death Dis. 12:9242021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fuertes MA, Alonso C and Pérez JM:
Biochemical modulation of Cisplatin mechanisms of action:
Enhancement of antitumor activity and circumvention of drug
resistance. Chem Rev. 103:645–662. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zalewska-Ziob M, Adamek B, Kasperczyk J,
Romuk E, Hudziec E, Chwalińska E, Dobija-Kubica K, Rogoziński P and
Bruliński K: Activity of antioxidant enzymes in the tumor and
adjacent noncancerous tissues of non-small-cell lung cancer. Oxid
Med Cell Longev. 2019:29018402019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu C, Liu Z, Chen Z, Xu D, Chen L, Lin H
and Shi J: A nonferrous ferroptosis-like strategy for antioxidant
inhibition-synergized nanocatalytic tumor therapeutics. Sci Adv.
7:eabj88332021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Glasauer A, Sena LA, Diebold LP, Mazar AP
and Chandel NS: Targeting SOD1 reduces experimental non-small-cell
lung cancer. J Clin Invest. 124:117–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karami Fath M, Ebrahimi M, Nourbakhsh E,
Zia Hazara A, Mirzaei A, Shafieyari S, Salehi A, Hoseinzadeh M,
Payandeh Z and Barati G: PI3K/Akt/mTOR signaling pathway in cancer
stem cells. Pathol Res Pract. 237:1540102022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang
J, Zhang G, Wang X, Dong Z, Chen F and Cui H: Targeting cancer stem
cell pathways for cancer therapy. Signal Transduct Target Ther.
5:82020. View Article : Google Scholar : PubMed/NCBI
|