Introduction
Cancer is one of the most prevalent diseases and the
leading cause of death worldwide. The global cancer incidence and
mortality were estimated at nearly 19 million new cases and 10
million deaths in 2020, with lung, colorectal, breast and prostate
cancer accounting for the majority of the diagnosed cancers
worldwide (1,2). Despite the increasing survival rates
of cancer patients observed in the past decades due to improved and
game-changing therapies, the global incidence of cancer keeps
increasing and is estimated to reach 28 million per year in 2040,
leading to a physical, emotional and financial burden to the
patients and their families, and a strain on resources of the
public health system (2,3). The cancer burden has been attributed
to the inequitable access to cancer prevention, early detection,
screening and treatment of the population, particularly in low- and
middle-income countries. In addition, an increase in the price of
cancer drugs has been recorded in recent years. Cancer is predicted
to cost $25.2 trillion to the world economy from 2020 to 2050,
bringing severe financial distress to the economy, affected
patients and families, and the public health system (4,5).
Furthermore, highly effective treatments, including radiotherapy,
chemotherapy, surgery, targeted therapy and hormonal treatments,
which significantly contribute to the overall reduction of cancer
mortality, recurrence and spread and increase in survival rate,
have limitations in curing cancer, particularly in patients with
delayed diagnoses and in patients with cancer therapy resistance,
resulting in a worse survival outcome (6,7).
Drug repositioning, an approach to using approved
drugs to treat diseases outside their original indication scope,
has gained significant attention in recent years (8). Repurposing existing drugs reduces the
exorbitant cost of developing new drugs, estimated to be between
$314 million and $2.8 billion per medication in 2018. It shortens
the time taken for the drug approval process, which can otherwise
take 10 to 15 years to reach the market (8,9).
Furthermore, repurposed drugs have a decreased likelihood of
clinical failure due to adverse effects, as their safety and dosing
have already received clinical approval (10). Consequently, repurposing drugs for
cancer treatment represents a cost-effective method for cancer
therapy.
Various antipsychotic drugs including penfluridol
(PF), phenothiazine, pimozide, chlorpromazine and thioridazine have
been discovered to have anticancer properties and have been
identified as a promising alternative cancer treatment (11). Several studies have reported that
patients taking antipsychotic drugs have a lower cancer incidence
compared to the general population (12). PF is a well-established
first-generation diphenylbutylpiperidine (DPBP) antipsychotic drug
used to treat chronic schizophrenia and other psychotic disorders,
and has been found to inhibit cancer (11,13).
Indeed, PF inhibits cancer cell proliferation and induces apoptosis
and autophagy in various cancer cell lines (14). PF has a half-life of 70 h, can cross
the blood-brain barrier (BBB) and block the dopamine receptor
(DRD2) binding sites, making it a potent drug for the treatment of
brain cancers, as well as cancers with high brain metastasis
ability (13,15,16).
This scoping review aimed to briefly investigate the anticancer
properties of PF, focusing on in vivo and in vitro
studies and providing a concise overview of PF's anticancer
mechanism of action.
Methodology
Protocol
This scoping review followed the Preferred Reporting
Items for Systematic Reviews and Meta-Analyses extension for
Scoping Reviews (PRISMA-ScR) checklist and PRISMA-ScR Tip sheets
(17). The review was conducted in
five stages according to Mak and Thomas's (18) steps for conducting a scoping review:
i) Identify the research question; ii) identify relevant studies;
iii) select the studies to be included in the review; iv) charting
data; and v) collate, summarize and report results.
Identification of the research
question
This review aimed to investigate the effect of PF on
cancer. Following the standard recommendation for a broader
research question (19), a primary
research question was developed: What are the effects of PF on
cancer? This research question provided sufficient literature to
warrant the scoping review and envelop the studies on PF as a
treatment for cancer. Hence, no further narrowing of the research
question was necessary.
Identification of relevant
studies
A literature search was conducted using the Scopus
(https://www-scopus-com.uitm.idm.oclc.org), PubMed
(https://pubmed.ncbi.nlm.nih.gov) and Web
of Science (WoS; http://www-webofscience-com.uitm.idm.oclc.org)
databases with the search string ‘penfluridol’ AND ‘cancer’. The
search, conducted in November 2023, included no additional
filters.
Study selection
Following the search, the identified records were
assessed in two phases. In the first phase, one author (AAIM)
evaluated the obtained articles by their titles and excluded
duplicate articles, review papers and retracted papers. The same
author screened the remaining journal papers based on their titles
and abstracts. Any articles with no relation to the roles of PF in
treating cancers according to their titles and abstracts and any
conference abstracts were excluded. In the second phase, one author
(AAIM) retrieved and assessed the remaining articles' full text. In
the end, original research articles on the roles, functions,
importance and mechanisms of PF in treating cancer written in
English were retained and included in the scoping review. The
second author (AAR) was consulted for their opinion on the obtained
data throughout the study selection stages.
Mendeley Desktop version 1.19.8 (Elsevier) and
Microsoft Excel version 16.89.1 (Microsoft Corporation) were used
to sort out the literature and identify the articles' duplication
by independently filtering out the articles' DOIs, titles and
abstracts.
Charting the data
Microsoft Excel version 16.89.1 (Microsoft
Corporation) was used to organize the data from the selected
articles. Data extraction was performed following the PRISMA
guidelines by (AAIM). The extracted data were categorised into
author names, publication year, type of cancer, dose of PF, study
design, significant findings, key findings and limitations.
Collating, summarizing and reporting
of the results
The extracted data were tabulated in Microsoft Excel
version 16.89.1 (Microsoft Corporation) with rows relating to the
articles and columns that classify the variables and contain the
relevant information. This table was used to classify and report
the information collected.
Results
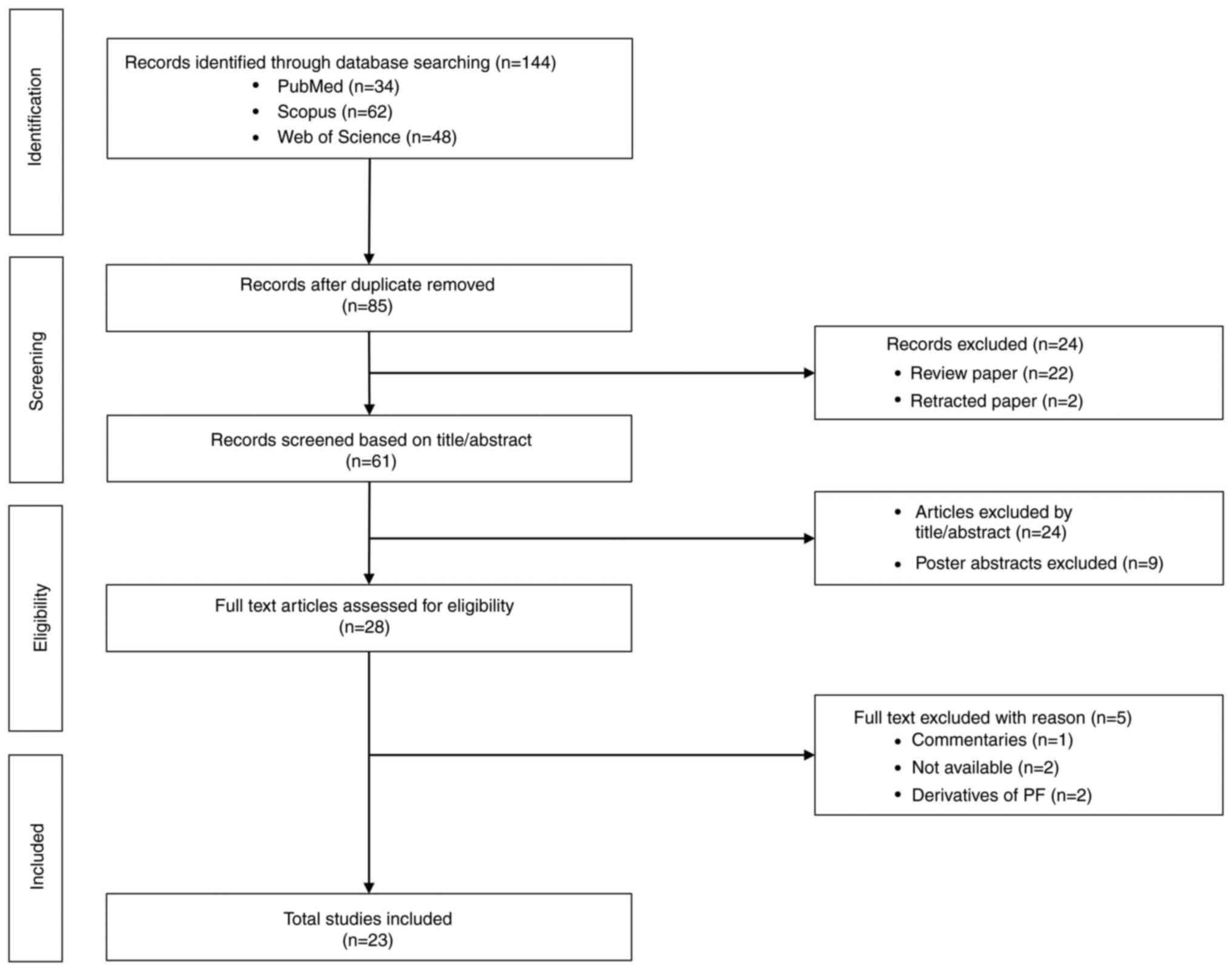
Article selection
The primary search yielded 144 articles, 34 from
PubMed, 62 from Scopus and 48 from WoS. A total of 59 duplicates
were removed, resulting in 85 records retained and screened
further. Subsequently, a total of 24 articles were eliminated due
to being reviews (n=22) and retracted papers (n=2). The remaining
61 articles were screened based on title and abstract and 24
articles with no relation to the roles of PF in treating cancers
and poster abstracts (n=9) were excluded. A total of 28 records
were identified as eligible for further full-text assessment. Of
the 28 articles, five were excluded due to lack of availability
(n=2), focusing on PF derivatives (n=2) and commentaries (n=1).
Finally, 23 original research studies in English were retained for
this scoping review. The PRISMA flowchart on the paper selection is
presented in Fig. 1.
Study characteristics
The main findings are summarised and presented in
Table I. Out of the 23 original
articles included in this review, 4 evaluated the anticancer effect
of PF on breast cancer, 4 on lung cancer, 4 on pancreatic cancer, 3
on glioblastoma, 1 on bladder cancer, 1 on gallbladder cancer, 1 on
oesophageal cancer, 1 on leukaemia cancer and 1 on renal cancer.
The remaining 3 articles included in this review studied the effect
of PF on various types of cancer. All of the studies in this review
included an in vitro model of a cell line supplemented with
0 to 40 µM PF, while most studies used an in vivo model
treated with PF at 1 to 10 mg/kg.
 | Table I.The effect of penfluridol on
cancer. |
Table I.
The effect of penfluridol on
cancer.
| Type of cancer | Author(s),
year | Study model | Doses | Treatment
duration | Findings | (Refs.) |
|---|
| Breast | Hedrick | SKBR3 & | 0.7–7.5 µM | 24 h | PF inhibited cell
growth and cell migration and | (20) |
|
| et al
2017 | MDA-MB- |
|
| induced cell
apoptosis. PF induced ROS in breast |
|
|
|
| 231 cells |
|
| cancer cells by
downregulating the expression of |
|
|
|
|
|
|
| Sp transcript
factor (Sp1, Sp2 and Sp4) and cMyc |
|
|
|
|
|
|
| (miR-27a and
miR-20/miR-17). PF repressed the |
|
|
|
|
|
|
| expression of α5-,
α6-, β1 and β4-integrin. |
|
|
|
| Female | 10 mg/kg/ | 19 days | PF decreased the
tumour volume, inhibited tumour |
|
|
|
| athymic | day |
| growth and induced
apoptosis. PF ROS-dependently |
|
|
|
| nude mice |
|
| reduced the
expression of Sp1, Sp3, Sp4 and Sp- |
|
|
|
|
|
|
| regulated genes
(α6-, α5-, β1- and β4-integrins). |
|
|
| Gupta | MCF-7, | 0-20 µM | 24-72 h | PF treatment
inhibited the cell growth and colony | (21) |
|
| et al
2019 | MCF-7HH, |
|
| formation of
PXT-sensitive and PXT-resistant cells |
|
|
|
| MCF-7PR, |
|
| in a concentration-
and time-dependent manner. PF |
|
|
|
| 4T1 &
4T1PR |
|
| downregulated the
expression of HER2, β-catenin, |
|
|
|
| cells |
|
| c-Myc, TCF-1,
TCF-4, p-GSK3β and cyclin D1. PF |
|
|
|
|
|
|
| induced apoptosis
in breast cancer cells. A |
|
|
|
|
|
|
| combination of PXT
and PF significantly reduced |
|
|
|
|
|
|
| the cancer cells'
viability, suppressing the |
|
|
|
|
|
|
| chemoresistance
markers while enhancing apoptosis. |
|
|
|
| Female | 10 mg/kg/ | 23 days | PF significantly
suppressed the growth of paclitaxel- |
|
|
|
| Balb/c mice | day |
| resistant cells
alone or in combination with 5 mg/kg/3 |
|
|
|
|
|
|
| days PXT. PF alone
and PF combined with PXT |
|
|
|
|
|
|
| inhibited HER2 and
β-catenin expression and |
|
|
|
|
|
|
| increased
apoptosis. |
|
|
| Ranjan | MDA-MB- | 0-20 µmol/l | 24-72 h | PF suppressed the
proliferation, migration and | (22) |
|
| et al
2016 | 231, 4T1 & |
|
| invasion of breast
cancer. PF treatment inhibited |
|
|
|
| HCC1806 |
|
| cells' survival and
motility by reducing the |
|
|
|
| cells |
|
| expression of
integrin α6, integrin β4, FAK, paxillin, |
|
|
|
|
|
|
| Rac1/2/3 and ROCK1
and inducing apoptosis. |
|
|
|
| Female Balb/ | 10 mg/kg | 27 days | PF suppressed
tumour growth by inhibiting |
|
|
|
| c mice |
|
| β4-integrin and
enhancing apoptosis in mice. |
|
|
|
|
|
|
| Intracardiac or
intracranial injection of breast cancer |
|
|
|
|
|
|
| cells revealed
similar results. |
|
|
| Srivastava | HUVECs & | 0-5 µM | 24-48 h | Low doses of PF
blocked the angiogenesis- | (23) |
|
| et al
2020 | MDA-MB-23 |
|
| suppressed
VEGF-induced primary endothelial cell |
|
|
|
| cells |
|
| migration and tube
formation of the cancer cells. PF |
|
|
|
|
|
|
| inhibited the Src
and Akt signalling pathways. |
|
|
|
| C57B/L6 | 1 mg/kg | 11 days | Low dose of PF
blocked VEGF- and FGF-induced |
|
|
|
| mice |
|
| angiogenesis. |
|
| Lung | Lai et
al | A549 & | 0-7.5 µM | 24 h | PF inhibited lung
tumour growth by reducing | (24) |
|
| 2022 | HCC827 |
|
| mitochondrial ATP
production and downregulating |
|
|
|
| cells |
|
| NADH levels. PF
mediated the suppression of |
|
|
|
|
|
|
| mitochondrial
biogenesis, impeded the level |
|
|
|
|
|
|
| of PGC-1α protein
and mRNA, SIRT1 and p53, and |
|
|
|
|
|
|
| induces AMPK
activation. |
|
|
|
| NOD-SCID | 5 m/kg | 28 days | PF inhibited the
growth and metastasis of lung |
|
|
|
| mice |
|
| cancer in mice.
These results were enhanced in |
|
|
|
|
|
|
| combination with
2DG. PF decreased the expression |
|
|
|
|
|
|
| of PGC-1α and
SIRT1. |
|
|
| Xue et
al | A549, H446, | 0-20 µM | 24-72 h | PF inhibited lung
cancer cell growth and viability | (26) |
|
| 2020 | H1993, |
|
| with G0/G1 cell
cycle phase arrest by enhancing the |
|
|
|
| SPC-A1 & |
|
| expression level of
p21/p27 and decreasing the |
|
|
|
| LL2 cells |
|
| expression levels
of the cyclin-CDK complex. PF |
|
|
|
|
|
|
| induced apoptosis
via the mitochondria-mediated |
|
|
|
|
|
|
| intrinsic apoptosis
pathway and inhibited the |
|
|
|
|
|
|
| migration and
invasion of lung cancer cells. |
|
|
|
| Female | 10 mg/kg | 26 days | PF inhibited tumour
growth in an A549 cell |
|
|
|
| Balb/c |
|
| xenograft mouse
model. PF blocked the proliferation |
|
|
|
| nude mice |
|
| and metastasis of
lung cancer in mice by |
|
|
|
|
|
|
| regulating the AKT
and MMP signalling pathways |
|
|
|
|
|
|
| while inducing
apoptosis. |
|
|
| Hung | NSCLC, | 7.5–10 µM | 24-72 h | PF suppressed cell
proliferation by inducing ER | (25) |
|
| et al
2019 | A549, |
|
| stress- mediated
autophagosome accumulation to |
|
|
|
| HCC827 & |
|
| deplete ATP energy.
PF inhibited cell migration. |
|
|
|
| BEAS-2B |
|
| PF induced
nonapoptotic cell death by blocking |
|
|
|
| cells |
|
| autophagic flux and
autophagosome formation |
|
|
|
|
|
|
| of LC3B-II protein
in lung cancer cells. |
|
|
|
| NOD-SCID | 5-10 mg/kg | 35 days | PF inhibited the
growth and the metastasis of lung |
|
|
|
| mice |
|
| cancer in mice by
inducing an accumulation of |
|
|
|
|
|
|
|
autophagosome-related protein. |
|
|
| Hung | A549, H23, | 0-20 µM | 24-72 h | PF reduced the
migration, invasion and adhesion | (27) |
|
| et al
2021 | HCC827, |
|
| of LADC cells. PF
inhibited MMP-12 by |
|
|
|
| PC9, H1975, |
|
| downregulating the
uPA/uPAR/TGF-β/Akt axis to |
|
|
|
| HMEC-1 & |
|
| modulate the
motility and adhesion of LADC cells. |
|
|
|
| BEAS-2B |
|
| PF inhibited EMT
byupregulating E-cadherin and |
|
|
|
|
|
|
| downregulating
N-cadherin. |
|
|
|
| Male NOD- | 2.5 µM | 5 weeks | PF suppressed lung
metastasis and colony formation. |
|
|
|
| SCID mice |
|
| PF increased the
expression of E-cadherin levels and |
|
|
|
|
|
|
| decreased
N-cadherin and MMP-12 levels in tumour |
|
|
|
|
|
|
| tissues. |
|
| Pancreatic | Ranjan | AsPC-1, | 0-10 µM | 24-72 h | PF induced ER
stress in pancreatic cancer cells | (28) |
|
| et al
2017 | BxPC-3 & |
|
| characterised by
the upregulation of ER stress |
|
|
|
| Panc-1 cells |
|
| markers of BIP,
CHOP and IRE1α. PF-induced ER |
|
|
|
|
|
|
| stress led to
autophagy. |
|
|
|
| Female | 10 mg/kg | 3 weeks | PF-treated mice had
a high level of BIP, CHOP |
|
|
|
| athymic |
|
| and IRE1α
expression in the tumour lysates and a |
|
|
|
| nude mice |
|
| reduction of tumour
mass. |
|
|
| Ranjan and | BxPC-3 & | 0-20 µM | 24-72 h | PF induced
apoptosis and inhibited the growth of | (29) |
|
| Srivastava | AsPC-1 |
|
| pancreatic cancer
cells. PF enhanced autophagy in |
|
|
| 2016 |
|
|
| pancreatic cancer
cells mediated by apoptosis. PF |
|
|
|
|
|
|
| impeded the
formation of lysosomes. |
|
|
|
| Female | 10 mg/kg | 27 & 59 | PF reduced the
growth of BxPC-3 tumour xenografts |
|
|
|
| athymic |
| days | and the development
of orthotopically implanted |
|
|
|
| nude mice |
|
| pancreatic tumours
by 80% by inducing autophagy- |
|
|
|
|
|
|
| mediated apoptosis
in the tumours. |
|
|
| Dandawate | AsPC-1, | 0-40 µM | 24-72 h | PF highly bound to
the JAK2 domain PRLR. PF | (30) |
|
| et al
2020 | BxPC-3, |
|
| suppresses
pancreatic cancer cells' proliferation and |
|
|
|
| Panc-1, |
|
| colony and spheroid
formation. PF enhanced PDAC |
|
|
|
| MiaPaCa-2,
& |
|
| autophagy. The PF
mechanism of inhibition of |
|
|
|
| UNKC-6141 |
|
| PDAC cell growth
did not proceed through DRD2. |
|
|
|
| Male C57BL/6 | 5 mg/kg | 28 & 35 | PF decreased the
weight of the orthotopic tumours. |
|
|
|
|
|
| days | PF significantly
inhibited the tumour weight and |
|
|
|
| mice &
PDX- |
|
| volume in xenograft
tumours. PF reduced PDAC |
|
|
|
| carrying |
|
| tumour growth by
inducing autophagy. |
|
|
|
| NGS mice |
|
|
|
|
|
| Chien | Panc0403, | 0-10 µM | 0-36 h | PF inhibited
pancreatic cancer cell proliferation and | (31) |
|
| et al
2015 | SU8686, |
|
| growth and induced
apoptosis. PF decreased the |
|
|
|
| MiaPaCa2, |
|
| phosphorylation
levels of SRC, AKT and p70S6K. |
|
|
|
| Panc1, |
|
| PF induced the
activation of PP2A protein |
|
|
|
| Panc0504, |
|
| phosphatase leading
to pancreatic cancer cell death. |
|
|
|
| AsPc1, |
|
|
|
|
|
|
| Panc1005, |
|
|
|
|
|
|
| Panc0203, |
|
|
|
|
|
|
| Panc0327, |
|
|
|
|
|
|
| BxPc3 & |
|
|
|
|
|
|
| HPDE |
|
|
|
|
| Glioblastoma | Ranjan and | GBM43, | 0-20 µM | 24-72 h | PF reduced the
survival rate and induced apoptosis | (32) |
|
| Srivastava | GBM10, |
|
| in glioblastoma
cell lines. PF treatment suppressed |
|
|
| 2017 | GBM44, |
|
| the phosphorylation
of Akt at Ser473 and decreased |
|
|
|
| GBM28, |
|
| the expression of
GLI1, OCT4, Nanog and Sox2. |
|
|
|
| GBM14 & |
|
|
|
|
|
|
| U251MG |
|
|
|
|
|
|
| Athymic nude | 10 mg/ | 39-54 | PF treatment
inhibited the growth and reduced the |
|
|
|
| mice | kg/day |
| volume of days
in vivo glioblastoma tumour models. |
|
|
|
|
|
|
| PF reduced pAkt,
GL1 and OCT4 and enhanced |
|
|
|
|
|
|
| apoptosis. |
|
|
| Kim et
al | CSC2, X01, | 0-20 µM | 24-72 h | PF suppressed the
growth of GSCs in a dose- and | (33) |
|
| 2019 | 0315, 528NS, |
|
| time- dependent
manner. PF suppressed the stemness |
|
|
|
| 83NS, U87MG |
|
| of GSCs and
inhibited their sphere-forming ability |
|
|
|
| & T98G
cells |
|
| and invasiveness.
PF inhibited the EMT potential |
|
|
|
|
|
|
| in human GSCs. |
|
|
|
| Female Balb/c | 0.8 | 60 days | PF decreased
invasion and tumour volume in |
|
|
|
| nude mice | mg/kg/ |
| orthotopic
xenografts by reducing GLI1, uPAR, |
|
|
|
|
| week |
| SOX2 and vimentin
expression. With a |
|
|
|
|
|
|
| combination of PF
and TMZ, the tumour |
|
|
|
|
|
|
| disappeared in the
mice. |
|
|
| Ranjan | Female | 10 mg/kg | 40-48 | PF treatment
suppressed glioblastoma tumour | (34) |
|
| et al
2017 | athymic |
| days | growth by reducing
murine myeloid-derived |
|
|
|
| nude mice
& |
|
| suppressor cells
(MDSC). PF enhanced splenic cell |
|
|
|
| female SCID- |
|
| proliferation,
increased M1 macrophages and |
|
|
|
| NOD mice |
|
| suppressed
T-regulatory cells and inflammation in |
|
|
|
|
|
|
| the tumour. |
|
| Gallbladder | Hu et
al | EH-GB1, | 0-10 µM | 24-72 h | PF inhibited the
proliferation and migration of | (35) |
|
| 2022 | GBC-SD & |
|
| gallbladder cancer
(GBC) cells and enhanced |
|
|
|
| SGC-996 |
|
| apoptosis. PF
treatment caused the activation |
|
|
|
| cells |
|
| of
AMPK/PFKB3-mediated glycolysis in GBCs, |
|
|
|
|
|
|
| and a combination
of PF and 2-DG or AMPK |
|
|
|
|
|
|
| inhibitor CC
improved the anticancer effect of PF. |
|
|
|
| Old female | 10 mg/kg | 15 days | PF suppressed lung
tumour growth. PF promoted |
|
|
|
| nude mice |
|
| apoptosis and the
activation of AMPK/PFKFB3 |
|
|
|
|
|
|
| signaling. |
|
| Bladder | van der | UCB & | 0-100 µM | 0-40 h | PF inhibited the
cell viability and clonogenicity | (36) |
|
| Horst et
al | UM-UC- |
|
| of the UCB cell
line. PF induced lysosomal |
|
|
| 2020 | 3luc2 cells |
|
| leakage and
destabilized lysosomal structures in |
|
|
|
|
|
|
| human UCB cells,
resulting in the redistribution |
|
|
|
|
|
|
| of
phosphatidylserine from the internal to the |
|
|
|
|
|
|
| external membrane
surface, an early indicator |
|
|
|
|
|
|
| of apoptosis. |
|
|
|
| Female Balb/c | 130 µg/kg | 29 days | PF significantly
decreased tumour growth, |
|
|
|
| nude mice | per week |
| invasion and
metastasis in mice. |
|
| Oesophageal | Zheng | KYSE30, | 0-10 µmol/l | 24-72 h | PF inhibited
proliferation and colony formation | (37) |
|
| et al
2020 | KYSE150 & |
|
| and enhanced the
apoptosis of the esophageal |
|
|
|
| KYDEE270 |
|
| squamous cell
carcinoma (ESCC) cells. The |
|
|
|
|
|
|
| inhibition of the
cells was mediated by |
|
|
|
|
|
|
| AMPK/FOXO3a/BIM
signalling. PF |
|
|
|
|
|
|
| inhibited
glycolysis. |
|
|
|
| Female Balb/c | 9-18 mg/kg | 42 days | PF decreased tumour
volume and cell |
|
|
|
| nude mice |
|
| proliferation and
induced apoptosis in tumour |
|
|
|
|
|
|
| xenograft mice. PF
inhibited glycolysis. |
|
| Leukaemia | Wu et
al | HL-60, U937 | 7.5–20 µM | 24-72 h | PF inhibited the
proliferation of AML cells. | (38) |
|
| 2019 | & MV4-11 |
|
| PF induced
apoptosis by activating PP2A to |
|
|
|
| cell lines |
|
| suppress Akt and
MAPK activities. PF |
|
|
|
| with FLT3-WT |
|
| triggered
autophagic responses by inducing |
|
|
|
| or FLT3-ITD |
|
| the elevation of
intracellular ROS levels. |
|
| Renal | Tung | 786-O, | 0-25 µM | 24-72 h | PF decreased the
proliferation and colony | (39) |
|
| et al
2022 | Caki-1, A498 |
|
| formation of RCC
cells. PF induced autophagy |
|
|
|
| & ACHN
cells |
|
| through the ER
stress-mediated UPR, leading |
|
|
|
|
|
|
| to apoptosis. PF
reduced stemness and sphere |
|
|
|
|
|
|
| formation in RCC
cells. |
|
|
|
| NOD-scid | 5 µM | 5 weeks | PF reduced the
tumour volume and growth by |
|
|
|
| IL2Rγ |
|
| suppressing the
GLI1 and proliferation |
|
|
|
| (NSG) mice |
|
| index Ki-67. |
|
| Diverse | Wu et
al | B16, LL/2, | 0-9 µmol/l | 24-72 h | PF inhibited the
proliferation of melanoma, | (40) |
|
| 2014 | CT26 & |
|
| lung carcinoma,
colon carcinoma and breast |
|
|
|
| 4T1 cells |
|
| cancer cells in a
time-dependent manner. PF |
|
|
|
|
|
|
| increased the
accumulation of unesterified |
|
|
|
|
|
|
| cholesterol in the
cancer cells. |
|
|
|
| Female | 0.06–0.12 mg/ | 45 days | PF suppressed lung,
colon, melanoma and |
|
|
|
| C57BL/6 | week |
| breast tumour
growth, and tumour weight of |
|
|
|
| mice & |
|
| the mice. PF
decreased the total cholesterol |
|
|
|
| Balb/c mice |
|
| in the tumour
tissue of the mice. |
|
|
| Varalda | HCT116, | 10-160 µmol/l | 24-72 h | PF inhibited the
proliferation of colorectal | (14) |
|
| et al
2020 | SW620, |
|
| and breast cancer
and glioblastoma cells. |
|
|
|
| MCF7, MDA- |
|
| PF reduced MCF7 and
HCT116 cell motility |
|
|
|
| MB-231, |
|
| and migration. PF
induced mitochondrial |
|
|
|
| U87 & U251 |
|
| and lysosomal
membrane alteration and |
|
|
|
| cells |
|
| phospholipid
aggregation in cancer cells. PF |
|
|
|
|
|
|
| suppressed the mTOR
pathway, induced |
|
|
|
|
|
|
| by AMPK activation
and autophagy. |
|
|
| Du et
al | HeLa | 5 µmol/l | 24 h | PF has high
radiosensitivity activity. PF | (41) |
|
| 2018 |
|
|
| inhibited the
repair of DSBs post-IR in HeLa |
|
|
|
|
|
|
| cells by reducing
NHEJ repair and preventing |
|
|
|
|
|
|
| the activation of
DNA-PKcs. |
|
Synthesis of the results
Breast cancer
PF was found to exert its anticancer properties on
breast cancer through multiple pathways. PF inhibits cell
proliferation in breast cancer cells (20–23).
For instance, in the relevant studies, PF successfully reduced the
tumour volume and weight in the in vivo model of breast
cancer. Furthermore, PF was found to reduce cell migration through
reactive oxygen species (ROS) (20), the integrin pathway (22) and the VEGF pathway (23). In addition, PF was capable of
inducing apoptosis in breast cancer cells (20–22).
While Hedrick et al (20)
imparted that the anticancer properties of PF in breast cancer are
ROS-dependent, Gupta et al (21) revealed that PF downregulates the
expression of chemoresistance markers in paclitaxel (PXT)-resistant
breast cancer, leading to apoptosis. In fact, cotreatment of PF
with glutathione attenuated the effect of PF on breast cancer
(20), and the combination of PF
and PXT decreased the expression of human EGFR 2 (HER2), β-catenin
and cyclin D, simultaneously enhancing apoptosis (21). It is worth mentioning that chronic
administration of PF failed to elicit any significant toxic or
behavioural side effects in mice (22) and that low concentrations of PF can
block angiogenesis in vitro and in vivo (23).
Lung cancer
Studies on PF in the treatment of lung cancer
revealed that PF inhibits lung tumour growth by inducing
mitochondrial ATP energy loss in vivo and in vitro
(24). Similar results were
obtained pertaining to autophagy (25). PF inhibited lung cancer
proliferation, migration and metastasis in vivo and in
vitro by regulating the AKT and MMP signalling pathway
(26,27). PF suppressed MMPs and
epithelial-mesenchymal transition (EMT) required for cancer cell
motility and adhesion (27).
Induced G0/G1 phase arrest, ROS and endoplasmic reticulum (ER)
stress, as well as loss of the mitochondrial membrane potential
(ΔΨm), were noted in PF-treated lung cancer (25,26).
On the other hand, PF in combination with glycolysis inhibitor
2-deoxy-D-glucose synergistically enhanced the inhibitory effect of
PF on lung cancer cell growth and the total amount of mitochondria
(24). No side effects were noted
in the mice used in these studies.
Pancreatic cancer
In pancreatic cancer, PF was reported to induce
autophagy (28–30) and apoptosis (29,31) in
in vitro and in vivo models. PF inhibited the
proliferation and colony formation of pancreatic cancer cells
(28–31) through the suppression of prolactin
receptor (PRLR) signalling (30)
and the activation of protein phosphatase 2A (PP2A) protein
phosphatase (31). A study by
Ranjan & Srivastava (29)
revealed that PF induced autophagy-mediated apoptosis in pancreatic
cancer. However, while PF exerts its antipsychotic activity by
blocking the DRD2 receptors, the antiproliferative activity of PF
in pancreatic ductal adenocarcinoma is not related to DRD2 and does
not trigger protein degradation (30).
Glioblastoma
In glioblastoma, PF appeared to reduce the growth of
glioblastoma cells and a tumour model through the apoptosis pathway
(32). In addition, GLA1 was
downregulated in a glioblastoma cancer model treated with PF
(32,33). On the other hand, Ranjan et
al (34) showed that PF
treatment suppresses glioblastoma growth through immune regulation,
while Kim et al (33)
denoted that it is by reducing the sphere formation, stemness and
invasiveness of the cells. Of note, a combination of PF and
temozolomide (TMZ) produced maximal antiproliferative and antitumor
effects in in vivo and in vitro models (33).
Other cancers
PF inhibited the cell proliferation and colony
formation of esophageal squamous cell carcinoma (ESCC) cells
(37), renal cell carcinoma (RCC)
cell lines (39), acute myeloid
leukaemia (AML) cells (38),
gallbladder cancer (35), bladder
cancer (36), colorectal and breast
cancer, glioblastomas, lung cancer and melanoma cells (14,40).
For instance, PF reduces glycolysis and promotes cell apoptosis
(35,36). Apoptosis was induced in cancer cells
through repressed glycolysis (37)
and activation of PP2A (38) in
ESCC and AML cells, respectively. Combining 2-deoxy-D-glucose
(2-DG) or AMP-activated protein kinase (AMPK) inhibitor Compound C
(CC) significantly improved PF's anticancer effect in gallbladder
cancer (35). Furthermore, PF
enhanced autophagy in cancer cells by upregulating ER stress and
ROS (38,39). It is worth highlighting that the
anticancer activities of PF on RCC partially occurred through DRD2
(39) and that PF suppresses the
mTOR pathway in various cell lines (14). Besides that, in vivo studies
uncovered that PF effectively prolongs the survival rate of mice
bearing lung tumours and enhances the decrease of total cholesterol
in tumour tissues with no significant difference in the serum
cholesterol levels of the PF-treated mice (40). No side effects to the vital organs
of the PF-treated mice were noted despite the decrease in tumour
growth and volume in vivo (37,39).
In addition, PF-treated urothelial carcinoma of the bladder (UCB)
orthotopic xenograft mice exhibited normal murine urothelium,
epithelial tissue integrity, proliferation index (proliferating
cell nuclear antigen), viable epithelial cell numbers and
fragmented keratin (36). In the
meantime, extensive research by Du et al (41) unveiled that PF has high
radiosensitizing activity and can suppress the activity of
non-homologous end joining (NHEJ).
Discussion
PF
PF
{4-(4-chloro-α,α,α,-trifluoro-m-tolyl)-1-[4,4-bis-(p-fluorophenyl)-butyl]-4-piperidinol}
is a DPBP, first-generation antipsychotic drug used to treat
chronic schizophrenia, Tourette's syndrome, acute psychosis and
other psychotic disorders discovered by Janssen Pharmaceutical in
1968 (15,42,43).
PF is a long-lasting neuroleptics drug orally administered once a
week with a half-life of 66 h and a terminal plasma half-life of
199 h (44). Indeed, PF is highly
lipophilic and is deposited in the adipose tissue after absorption
in the gastrointestinal tract, which heightens its prolonged
duration of action, since PF is gradually released from these
reservoirs (15,44). PF is excreted via urine and faeces
(45). The maximal weekly dose of
PF treatment for patients with chronic schizophrenia varies between
60 and 140 mg (46,47). According to Andrade (48), long half-life antipsychotic drugs
significantly reduce the withdrawal or discontinuation syndrome of
the drug but can be disadvantageous in case of toxicity or side
effects. For instance, PF treatment fosters various adverse
effects, including depression, agitation, drowsiness, tachycardia,
hypotension, insomnia and neuroleptic malignant syndrome (49). Therefore, a low dose of PF treatment
is recommended.
PF is capable of penetrating the BBB, making it a
promising option for the treatment of brain-metastasized cancers
(33). Airoldi et al
(43) revealed that despite the
distribution of PF in the adipose tissue being higher, PF remains
concentrated in the brain with the highest concentration recorded
in the hemisphere part of the brain 1 h after the injection and the
mesencephalon 24 h after the injection of 0.5 mg/kg PF in rats. PF
acts by blocking the dopamine receptor, particularly DRD2, where it
binds with a high affinity, leading to the inhibition of
dopaminergic neurotransmission (13,15,16). A
former study by Enyeart et al (50) revealed that the PF also blocks the
T-type and L-type calcium channels at low concentrations.
Additionally, Vlachos et al (12) have reported that patients taking
antipsychotic drugs have a lower cancer incidence compared to the
general population (12).
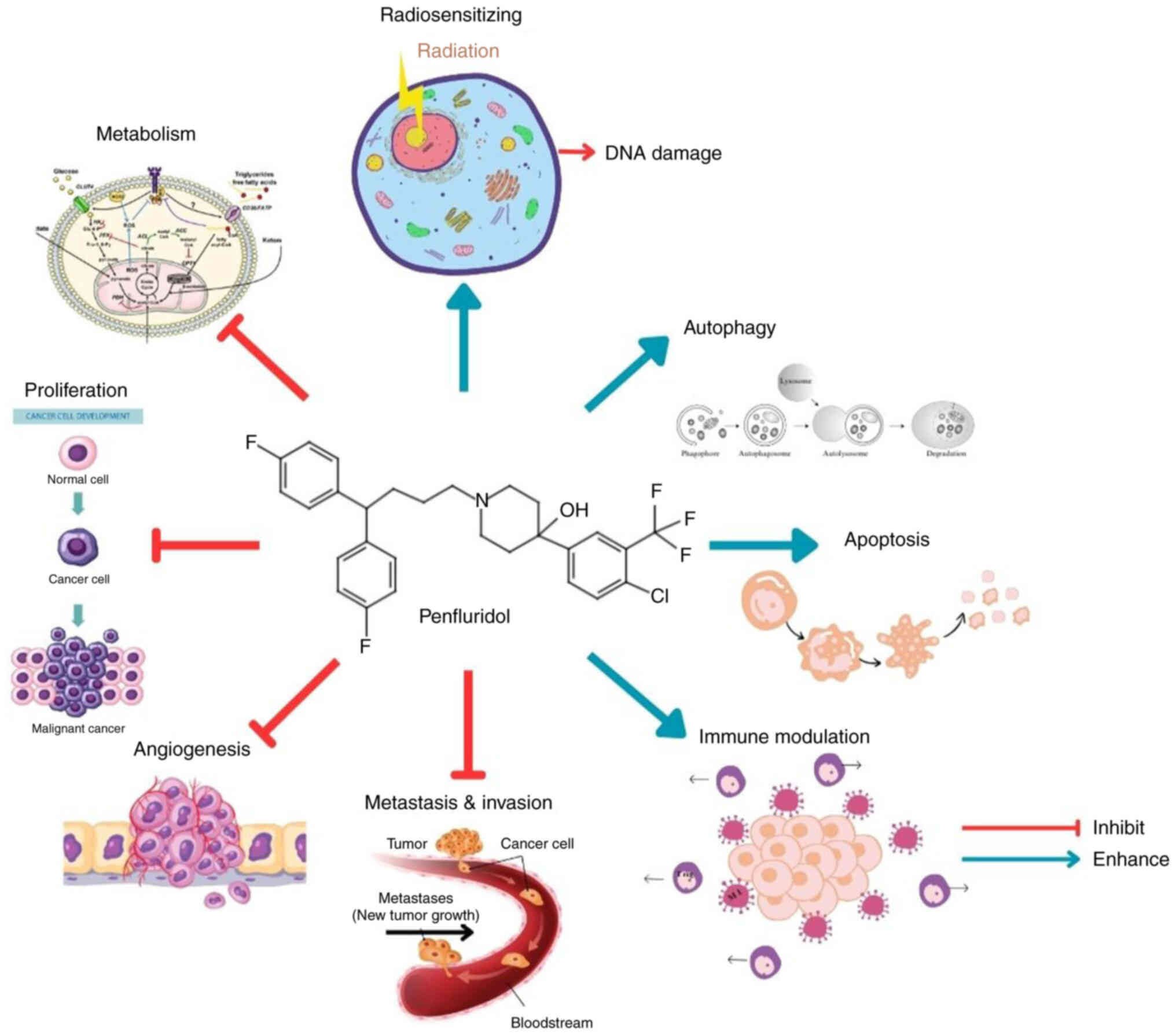
Henceforth, examining the anticancer properties of PF may offer
valuable insight. Fig. 2 summarises
the anticancer activities of PF.
The anticancer properties of PF
PF was found to inhibit cell proliferation in a
dose- and time-dependent manner in all the cancer cells reported in
this review (14,20–41).
For instance, <10 µmol/l of PF induced 50% cell death in various
cancer cell lines (14), while
<2.5 µM of PF inhibited the proliferation and colony formation
of lung and renal cancer cells (25,39).
Meanwhile, 0.12 mg/week of PF remarkably prolonged the survival
time in the LL/2 in vivo cancer model (40), indicating that a low concentration
of PF can provide anticancer properties. The antiproliferative
effects of PF are not mediated by major neuroreceptor systems
(14), but instead by interfering
with the mechanism involved in cancer growth. Indeed, PF induced
cell death by downregulating the cell growth proteins (cyclin D and
MYC) and expression of the G2-M phase of cell cycle arrest protein
cyclin B1 and p21 in pancreatic cells (31). The regulatory protein of the
transition between the G2 to M phase has a significant role in the
proliferation of the tumour and inhibition of these proteins led to
cell-cycle arrest and eventually apoptosis (51,52).
In addition, PF activates PP2A, mediating apoptosis and cell death
(31,38). PP2A is a positive regulator of
apoptosis and activation of PP2A dephosphosphorylates and
inactivates anti-apoptotic Bcl-2, inducing apoptosis (53).
On the other hand, PF increased ROS levels in lung
cancer cells, leading to cell death (26). ROS functions as a double-edged sword
in cancer cells and can act as a tumour-promoting or suppressing
agent (54,55). Cancer cells possess elevated levels
of ROS in comparison to normal cells due to their fast
proliferation and alteration in cellular metabolism, particularly
the reprogrammed metabolism to aerobic glycolysis (56,57).
Moderate levels of ROS are necessary for cancer cell proliferation,
invasion, migration and angiogenesis, while excessively elevated
ROS increase cancer cell damage and death (58,59).
Indeed, PF induced ROS production triggering autophagy in AML cells
(38). Furthermore, Hedrick et
al (20) revealed that PF
enhances ROS production, leading to the downregulation of cMyc
expression and decreased expression of Sp1, Sp3, Sp4 and
Sp-regulated genes in vitro and in an in vivo model
of breast cancer, hence inhibiting cell viability and inducing
apoptosis. The specificity proteins Sp and the multifaceted
oncogene c-Myc are essential regulators of cellular proliferation,
metabolism, metastasis and apoptosis, which, when upregulated,
conduce to the progression of cancer (60–63).
Furthermore, Sp regulates the activation or repression of
integrins, transmembrane receptors which partake in cancer
development, progression, invasion, metastasis and resistance to
therapy (64–67). Ranjan et al (22) showed that PF reduced the expression
of α6, α4, αv, β4, β1 and β3 integrins, as well as the downstream
effector molecules of integrin signalling, including focal adhesion
kinase (FAK), phosphorylated paxillin and paxillin in breast cancer
(22). Desgrosellier & Cheresh
(68) revealed that integrin
signalling plays a significant role in cancer migration,
proliferation and invasion and inhibition of integrins lead to the
repression of tumour invasion and growth.
Apart from that, PF enhances energy loss. For
instance, according to Lai et al (24), PF inhibits non-small cell lung
cancer (NSCLC) cell growth by increasing ATP energy loss through
the suppression of mitochondrial ATP production activity and
biogenesis. Similarly, suppression of ATP increased the formation
and accumulation of autophagosomes in NSCLC, leading to death
(25). Indeed, cancer cells require
a considerable energy supply for their metabolism (69). This phenomenon known as the Warburg
effect is marked by an elevated glycolysis in the cancer cells,
including aerobic glycolysis to meet the energy needs of the cells
to grow (70). Several studies have
shown that targeting the metabolism of cancer cells impacts the
growth of the cancer and inhibition of the glycolysis reduced the
biosynthesis of ATP, a constituent of DNA, RNA and phospholipids
(71), thus suppressing cancer cell
growth. PF inhibited ESCC cell proliferation by repressing
glycolysis through AMPK signalling (37). The activation of AMPK/forkhead box
(FOX)O3a/BCL-2-interacting mediator (BIM) signalling in an
oesophageal cell line induced apoptosis (37). In addition, Hu et al
(35) reported that the combination
of glycolytic inhibitor 2-DG or AMPK inhibitor CC with PF treatment
significantly inhibited the proliferation of gallbladder cancer.
Furthermore, PF dysregulated cholesterol homeostasis by enhancing
the accumulation of unesterified cholesterol in LL/2, 4T1, B16 and
CT26 cancer cells and decreased the total cholesterol in tumour
tissues of PF-treated mice (40).
Cholesterol plays a significant role in tumour cell growth
(72). Henceforth, PF has potential
anti-metabolic properties for the treatment of cancer.
Apoptosis and autophagy, programmed cell death
pathways crucial in maintaining cellular and organismal
homeostasis, are the main mechanisms researched in cancer (73,74).
PF was found to enhance apoptosis by inducing nuclear
fragmentation, poly-ADP ribose polymerase cleavage, caspase-3
activation and condensed nuclear chromatin (21,29,38).
Similarly, PF upregulated the expression levels of the proapoptotic
proteins BIM, BAX and p53-upregulated modulator of apoptosis (PUMA)
and downregulated the antiapoptotic gene Bcl-2 in pancreatic cancer
(31). Then again, PF induced G0/G1
phase arrest and, vehemently, cell apoptosis in lung cancer via the
mitochondrial apoptotic pathway (26). For instance, Xue et al
(26) reported that PF increased
the loss of ΔΨm. Thus, PF induces tumour suppression through
apoptosis. Despite that, Hung et al (25) remarked that PF's antiproliferative
effect on NSCLC cells is independent of apoptosis. In fact, instead
of the apoptotic characteristics, formation of autophagosome
protein light chain (LC)3B-II and expression of autophagosome
markers autophagy-related protein 5, Beclin-1 and p62 were recorded
in PF-treated NSCLC cells, implying that PF enhances cell death
through autophagy (25,73,75,76).
Indeed, PF promoted an increase in LC3-II and p62 protein
expression, phospholipid aggregates and lysosomal membrane damage
and consequently cell death in various cell lines (14,29,30).
In addition, in in vivo and in vitro studies of
PF-treated lung and pancreatic cancer, an increase in ER stress was
observed, which stimulated autophagy, noticeable by the
overexpression of binding protein, C/EBP homologous protein (CHOP)
and inositol-requiring 1α, increased unfolded protein response
(UPR) signals and activation of p38/MAPK (25,28).
Pre-treatment of the pancreatic cells with pharmacological
inhibitors such as sodium phenylbutyrate and mithramycin or
silencing of CHOP, followed by PF treatment, considerably
suppressed autophagy, revealing that PF-induced enhancement of ER
stress leads to autophagy in pancreatic cancer (28). Of note, blocking autophagy with
pharmacological inhibitors significantly increases PF-induced
apoptosis in AML cells (38).
However, treatment of BxPC-3 and AsPC-1 with autophagy inhibitors
and LC3B silencing followed by PF treatment resulted in a
significant reduction of apoptosis, suggesting that PF induces
autophagy-mediated apoptosis in pancreatic cancer (29). Similar results were obtained in RCC
cell lines, where PF induced autophagy-mediated apoptosis through
the upregulation of ER stress-mediated UPR (39). Therefore, PF enhances autophagy and
autophagy-mediated apoptosis in cancer.
In addition, PF constrained migration, adhesion,
invasion and metastasis in cancer cells (27,36).
Metastasis, a hallmark of cancer-cell migration from their initial
site, is a crucial factor of cancer therapy failure associated with
an unfavourable prognosis and cancer-associated death (77). PF treatment significantly reduced
cell migration and invasion by reducing the expression of
phosphorylated Rac family small GTPase 1 (Rac1), Rac1/2/3 and
Rho-associated coiled-coil containing protein kinase 1 proteins in
triple-negative breast cancer cells (22). Furthermore, Kim et al
(33) revealed that PF inhibited
stemness by reducing the expression of SOX2, Nestin and optical
coherence tomography angiography and invasiveness by decreasing the
expression of integrin α6 and urokinase plasminogen activator
receptor in glioma-sphere forming cells. In addition, PF reduced
the potential for EMT, associated with cancer migration, invasion,
stemness and metastasis, of glioblastoma cancer models by
downregulating the expression of EMT markers (vimentin, Zeb 1,
N-cadherin, Snail and Slug) (33,78,79).
Besides that, PF reduced the expression of glioma-associated
oncogene homolog 1 and OCT4 in several glioblastoma cell lines
(32) and reduced lung cancer
migration in vivo and in vitro by blocking the
FAK-related migration signalling pathway and regulating the AKT and
MMP signalling pathway (26).
Despite PF's ability to constrain the mobility of bladder cancer
cells, no notable effects were registered in the urothelium and
epithelial tissue of UCB orthotopic xenograft mice (36). Therefore, PF exhibits anti-migration
and anti-metastasis properties without any side effects.
PF significantly inhibits angiogenesis, a mechanism
by which tumours acquire new nutrients for cell metabolism, leading
to cancer growth (80). Research by
Srivastava et al (23)
revealed that PF inhibits angiogenesis in in vitro and in
vivo models of triple-negative breast cancer by suppressing
VEGF-induced Src and Akt activation to prevent VEGF activity
(23). VEGF is a major angiogenic
agent in tumours that can initiate the growth and metastasis of
tumours (81). Furthermore, PF
increased the immune surveillance in glioblastoma cancer (33). Indeed, PF reduced the number of
regulatory T cells (Treg), suppressed the expression of FoxP3 and
CD4 by Tregs and increased M1 macrophages (increase of CD86 and
IL-12 expression), but also inhibited tumour inflammation markers
[C-C motif chemokine ligand 4 (CCL4) and IFNγ] responsible for
tumour progression, as observed in PF-treated mouse glioblastoma
cancer model (33,34). CCL4 has been identified as a marker
of tumour proliferation in RCC and is associated with immune
checkpoint genes, including lymphocyte activating 3, cytotoxic
T-lymphocyte-associated protein 4 and programmed cell death protein
1 in RCC (82). Hence, the
reduction of CCL4 in glioblastoma by PF indicates that PF
suppresses the progression and immunity of the cancer cells.
Therefore, PF can impede angiogenesis in tumours and modulate the
immune system.
Lastly, PF considerably downregulated the expression
of the chemoresistance markers HER2, β-catenin, cyclin D1 and c-Myc
in PXT-resistant breast cancer (21). Indeed, chemoresistance promoted
cancer relapses and metastasis, thus leading to a more aggressive
form of cancer (83,84). Gupta et al (21) revealed that PF reduced the tumour
volume in PXT-resistant breast cancer female Balb/c mice by 40%,
but the combination of PF and PXT induced apoptosis and decreased
the expression of HER2, β-catenin and cyclin D1. Moreover, Du et
al (41) unveiled that PF has
radiosensitizing activity and can suppress the NHEJ activity/repair
of the ionizing radiation (IR)-induced DNA double-strand breaks
(DSBs). Radiosensitizers heighten the lethal effect of radiotherapy
on tumours by boosting DNA damage and free radical production
without affecting normal tissue (85) and PF impaired the IR-induced DNA
damage repair, enhanced the formation of DSB markers (γ-H2AX and
tumour protein 53 binding protein 1 foci) and inhibited the repair
of DSB post-IR in HeLa cells, thus promoting the cytotoxicity of IR
in HeLa cells (41). Taken
together, it can be suggested that PF is likely an excellent
chemo-radiotherapeutic agent.
Overall, PF exhibits several anticancer effects in
various cancer cell lines. PF affects the proliferation of cancer
cells, provides radiosensitizing activity, induces apoptosis and
autophagy, inhibits the invasion, metastasis and angiogenesis of
cancer, and reduces the immunosuppressive effect of cancer
cells.
PF treatment induced apoptosis via the
mitochondrial apoptotic pathway in lung cancer and via the
suppression of Akt in glioblastoma. In addition, PF instigated
apoptosis through nuclear fragmentation, PARP cleavage and caspase
3 activation in leukaemia and through the activation of the BIM,
BAX and PUMA proapoptotic proteins in pancreatic cancer. PF
inhibited cell proliferation in lung cancer via mitochondrial ATP
energy loss and the inhibition of glycolysis, which consequently
led to autophagy and cell death. Similarly, in gallbladder, bladder
and oesophageal cancer, PF promoted apoptosis via inhibition of
glycolysis. However, in pancreatic cancer, PF induced autophagy via
induction of ER stress but suppressed the mTOR pathway in numerous
cancer cell lines. Upregulation of ROS by PF has been associated
with the ability of PF to enhance autophagy in renal cancer and
leukaemia. However, ROS led to the downregulation of cMyc and Sp
resulting in apoptosis in breast cancer. PF inhibited cell
migration via integrin and the VEGF pathway in breast cancer. PF
induced cell death via the activation of PP2A and PRLR signalling
in pancreatic cancer but PP2A in leukaemia cells. PF modulated the
immune system in glioblastoma cells by reducing the number of Tregs
and increasing M1 macrophages. In HeLa cells, PF was able to reduce
DNA repair by inhibiting the activity of NHEJ.
Limitation of PF and future direction
The dosage of PF as an anticancer drug exceeds the
clinical therapeutic range of PF as an antipsychotic. Indeed, the
10 mg/kg dose treatment of PF regularly used in in vivo
studies, including in chronic treatment, has successfully reduced
the tumour size and weight without triggering any major side
effects, as no significant changes in the body weight of the mice
and clinical chemical blood analysis parameters were observed
(29), and this 10 mg/kg dose of PF
is equivalent to the dose of 0.83 mg/kg in humans. Thus, for a
person weighing 60 kg, the human equivalent dose of PF would be ~50
mg (21). However, most of the
available studies indicate that mice are given PF treatment daily,
which exceeds the maximum weekly dose of PF for chronic
schizophrenia patients, which varies between 60 and 140 mg
(46,47). Of note, this high dose may cause
various adverse effects (49). In
fact, since PF interacts extensively with most G-protein coupled
receptors at levels consistent with the recommended anticancer
dosage, which is 50 mg per day for humans, the off-target central
nervous system (CNS) activity of PF as an anticancer drug will
exacerbate the drug's neurological side effects (86). Therefore, studying other mechanisms
of PF delivery to targeted sites, including the use of
nanotechnology, could potentially reduce the CNS side effects of
the drug.
Nonetheless, a study by Kim et al (33) on an orthotopic mouse glioblastoma
model revealed that a lower dose of 0.8 mg/week of PF, which is
equivalent to 4 mg/week in an adult patient with a body weight of
60 kg, showed effective antitumor activity. In this study, PF
significantly reduced the proliferation, invasion, migration and
stemness of glioblastoma cells, as well as the tumour size and
invasion in vivo, indicating that a lower dose of PF has a
potent anticancer effect (33).
Similarly, 0.12 mg/week of PF resulted in significant inhibition of
the tumour and prolonged the survival time in an LL/2 lung cancer
in vivo model (40).
Furthermore, Kim et al (33)
demonstrated that a combination of 0.8 mg/kg of PF with TMZ
provides better anticancer properties and prolonged survival in the
mouse model. Hence, a lower dose of PF alone or in combination with
other chemotherapeutic drugs may be considered for future clinical
and in vivo studies.
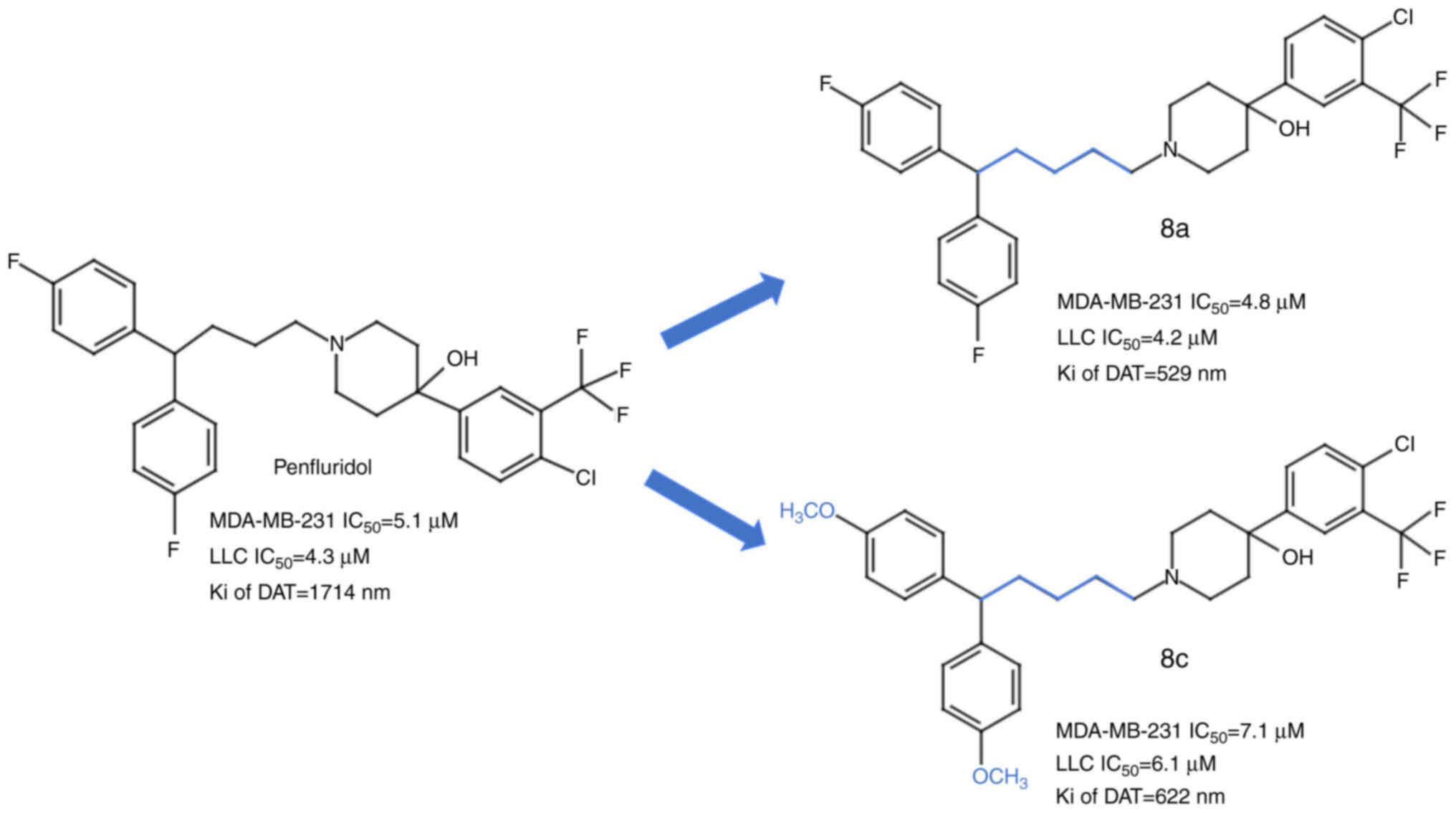
In addition, Ashraf-Uz-Zaman et al (86,87)
suggested the usage of PF derivatives with enhanced anticancer
activity and a weakened antipsychotic effect. Their study
identified 2 PF analogue compounds with less affinity for the CNS
and greater anticancer properties than PF (87) (Fig.
3). These compounds, which harbour modifications of PF in the
spacer linker by the addition of one carbon in the chain and the
introduction of a functional methoxy group (8c), displayed no
toxicity in mice and successfully inhibited the cell proliferation
of the metastatic triple-negative breast cancer cell line
MDA-MB-231 and the lung carcinoma cell line LLC (87). A further study by Ashraf-Uz-Zaman
et al (86) identified other
PF derivatives with the ability to induce apoptosis in the
MDA-MB-231 cell line without affecting the cell cycle. Nonetheless,
these compounds have yet to be tested and compared with PF in
various cancers. Therefore, more thorough research needs to be done
on the PF and PF derivatives to validate them as a clinical
therapeutic option for cancer.
Conclusion
This scoping review provides an overview of the
anticancer properties of PF in various cancers. It is crucial to
investigate potential cost- and time-effective drugs for managing
cancer. Based on the available studies, repurposing PF for cancer
treatment has promising effects, particularly in cancer with low
survival rates and high resistance to the current treatment. PF
inhibited the growth, metastasis and migration of various cancers,
including glioblastoma, as well as breast, lung, pancreatic, renal,
bladder and oesophageal cancer in vitro and in vivo
through distinctive mechanisms. Less than 10 µM of PF can provide
anticancer activity in vitro. Nonetheless, the in
vivo dose of PF exceeds the dose required for antipsychotic
treatment; therefore, the subsequent development of PF derivatives
with elevated anticancer and reduced antipsychotic activities was
deemed necessary. In addition, despite the approval of PF as a
valuable anticancer agent, dose-related side effects in the
clinical setting and in other types of cancer have yet to be
studied. Henceforth, extensive research on the effect of PF on
other cancers and in patients needs to be performed. In addition,
this scoping review focused on original articles published and
indexed in PubMed, Scopus and WoS before November 2023 in English.
Therefore, non-indexed, grey literature, studies in other languages
and review papers were overlooked. Furthermore, only articles
focusing on PF were considered and no clinical studies were
included in this review. Thus, future scoping reviews on the
anticancer properties of PF should include PF derivatives and
clinical studies when available.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the Ministry of
Higher Education Malaysia (grant no.
FRGS/1/2023/SKK10/UITM/02/1).
Availability of data and materials
Not applicable.
Authors' contributions
AAIM contributed to the conceptualization,
methodology, validation, formal analysis, data collection,
visualisation, writing of the original draft and reviewing and
editing. AAR contributed to the conceptualization of the study,
reviewed and edited the manuscript as well as supervised the
preparation of and validated the manuscript. Data authentication is
not applicable. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Asma Ali Ibrahim Mze (ORCID no.
0009-0002-1848-6160); Amirah Abdul Rahman (ORCID no.
0000-0002-3566-1787).
References
|
1
|
Ferlay J, Colombet M, Soerjomataram I,
Parkin DM, Piñeros M, Znaor A and Bray F: Cancer statistics for the
year 2020: An overview. Int J Cancer. Apr 5–2021.(Epub ahead of
print). View Article : Google Scholar
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
World Health Organization (WHO), . Cancer.
WHO; Geneva: 2022
|
|
4
|
Chen S, Cao Z, Prettner K, Kuhn M, Yang J,
Jiao L, Wang Z, Li W, Geldsetzer P, Bärnighausen T, et al:
Estimates and projections of the global economic cost of 29 cancers
in 204 countries and territories from 2020 to 2050. JAMA Oncol.
9:465–472. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gordon N, Stemmer SM, Greenberg D and
Goldstein DA: Trajectories of injectable cancer drug costs after
launch in the United States. J Clin Oncol. 36:319–325. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hendouei N, Saghafi F, Shadfar F and
Hosseinimehr SJ: Molecular mechanisms of anti-psychotic drugs for
improvement of cancer treatment. Eur J Pharmacol. 856:1724022019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qu LG, Brand NR, Chao A and Ilbawi AM:
Interventions addressing barriers to delayed cancer diagnosis in
low- and middle-income countries: A systematic review. Oncologist.
25:e1382–e1395. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Masuda T, Tsuruda Y, Matsumoto Y, Uchida
H, Nakayama KI and Mimori K: Drug repositioning in cancer: The
current situation in Japan. Cancer Sci. 111:1039–1046. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wouters OJ, McKee M and Luyten J:
Estimated Research and Development Investment Needed to Bring a New
Medicine to Market, 2009–2018. JAMA. 323:844–853. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Low ZY, Farouk IA and Lal SK: Drug
Repositioning: New approaches and future prospects for
life-debilitating diseases and the COVID-19 pandemic outbreak.
Viruses. 12:10582020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brown JS: Treatment of cancer with
antipsychotic medications: Pushing the boundaries of schizophrenia
and cancer. Neurosci Biobehav Rev. 141:1048092022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vlachos N, Lampros M, Voulgaris S and
Alexiou GA: Repurposing antipsychotics for cancer treatment.
Biomedicines. 9:17852021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaw V, Srivastava S and Srivastava SK:
Repurposing antipsychotics of the diphenylbutylpiperidine class for
cancer therapy. Semin Cancer Biol. 68:75–83. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Varalda M, Antona A, Bettio V, Vachamaram
A, Yellenki V, Massarotti A, Baldanzi G and Capello D: Psychotropic
drugs show anticancer activity by disrupting mitochondrial and
lysosomal function. Front Oncol. 10:5621962020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Soares BG and Lima MS: Penfluridol for
schizophrenia. Cochrane Database Syst Rev.
2006:CD0029232006.PubMed/NCBI
|
|
16
|
Chokhawala K and Lee S: Antipsychotic
medications. StatPearls [Internet]. StatPearls Publishing; Treasure
Island, FL: 2023
|
|
17
|
Tricco AC, Lillie E, Zarin W, O'Brien KK,
Colquhoun H, Levac D, Moher D, Peters MDJ, Horsley T, Weeks L, et
al: PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist
and Explanation. Ann Intern Med. 169:467–473. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mak S and Thomas A: Steps for conducting a
scoping review. J Grad Med Educ. 14:565–567. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arksey H and O'Malley L: Scoping studies:
Towards a methodological framework. Int J Soc Res Methodol.
8:19–32. 2005. View Article : Google Scholar
|
|
20
|
Hedrick E, Li XX and Safe S: Penfluridol
represses integrin expression in breast cancer through induction of
reactive oxygen species and downregulation of Sp transcription
factors. Mol Cancer Ther. 16:205–216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gupta N, Gupta P and Srivastava S:
Penfluridol overcomes paclitaxel resistance in metastatic breast
cancer. Sci Rep. 9:50662019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ranjan A, Gupta P and Srivastava SK:
Penfluridol: An antipsychotic agent suppresses metastatic tumor
growth in triple-negative breast cancer by inhibiting integrin
signaling axis. Cancer Res. 76:877–890. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Srivastava S, Zahra FT, Gupta N, Tullar
PE, Srivastava SK and Mikelis CM: Low Dose of Penfluridol Inhibits
VEGF-Induced Angiogenesis. Int J Mol Sci. 21:7552020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lai TC, Lee YL, Lee WJ, Hung WY, Cheng GZ,
Chen JQ, Hsiao M, Chien MH and Chang JH: Synergistic tumor
inhibition via energy elimination by repurposing penfluridol and
2-Deoxy-D-Glucose in lung cancer. Cancers (Basel). 14:27502022.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hung WY, Chang JH, Cheng Y, Cheng GZ,
Huang HC, Hsiao M, Chung CL, Lee WJ and Chien MH: Autophagosome
accumulation-mediated ATP energy deprivation induced by penfluridol
triggers nonapoptotic cell death of lung cancer via activating
unfolded protein response. Cell Death Dis. 10:5382019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xue Q, Liu Z, Feng Z, Xu Y, Zuo W, Wang Q,
Gao T, Zeng J, Hu X, Jia F, et al: Penfluridol: An antipsychotic
agent suppresses lung cancer cell growth and metastasis by inducing
G0/G1 arrest and apoptosis. Biomed Pharmacother. 121:1095982020.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hung WY, Lee WJ, Cheng GZ, Tsai CH, Yang
YC, Lai TC, Chen JQ, Chung CL, Chang JH and Chien MH: Blocking
MMP-12-modulated epithelial-mesenchymal transition by repurposing
penfluridol restrains lung adenocarcinoma metastasis via
uPA/uPAR/TGF-β/Akt pathway. Cell Oncol (Dordr). 44:1087–1103. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ranjan A, German N, Mikelis C,
Srivenugopal K and Srivastava SK: Penfluridol induces endoplasmic
reticulum stress leading to autophagy in pancreatic cancer. Tumour
Biol. 39:10104283177055172017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ranjan A and Srivastava SK: Penfluridol
suppresses pancreatic tumor growth by autophagy-mediated apoptosis.
Sci Rep. 6:261652016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dandawate P, Kaushik G, Ghosh C, Standing
D, Ali Sayed AA, Choudhury S, Subramaniam D, Manzardo A, Banerjee
T, Santra S, et al: Diphenylbutylpiperidine Antipsychotic Drugs
Inhibit Prolactin Receptor Signaling to Reduce Growth of Pancreatic
Ductal Adenocarcinoma in Mice. Gastroenterology. 158:1433–1449.e27.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chien W, Sun QY, Lee KL, Ding LW, Wuensche
P, Torres-Fernandez LA, Tan SZ, Tokatly I, Zaiden N, Poellinger L,
et al: Activation of protein phosphatase 2A tumor suppressor as
potential treatment of pancreatic cancer. Mol Oncol. 9:889–905.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ranjan A and Srivastava SK: Penfluridol
suppresses glioblastoma tumor growth by Akt-mediated inhibition of
GLI1. Oncotarget. 8:32960–32976. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim H, Chong K, Ryu BK, Park KJ, Yu MO,
Lee J, Chung S, Choi S, Park MJ, Chung YG and Kang SH: Repurposing
penfluridol in combination with temozolomide for the treatment of
glioblastoma. Cancers (Basel). 11:13102019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ranjan A, Wright S and Srivastava SK:
Immune consequences of penfluridol treatment associated with
inhibition of glioblastoma tumor growth. Oncotarget. 8:47632–47641.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu J, Cao J, Jin R, Zhang B, Topatana W,
Juengpanich S, Li S, Chen T, Lu Z, Cai X and Chen M: Inhibition of
AMPK/PFKFB3 mediated glycolysis synergizes with penfluridol to
suppress gallbladder cancer growth. Cell Commun Signal. 20:1052022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van der Horst G, van de Merbel AF, Ruigrok
E, van der Mark MH, Ploeg E, Appelman L, Tvingsholm S, Jäätelä M,
van Uhm J, Kruithof-de Julio M, et al: Cationic amphiphilic drugs
as potential anticancer therapy for bladder cancer. Mol Oncol.
14:3121–3134. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng C, Yu X, Liang Y, Zhu Y, He Y, Liao
L, Wang D, Yang Y, Yin X, Li A, et al: Targeting PFKL with
penfluridol inhibits glycolysis and suppresses esophageal cancer
tumorigenesis in an AMPK/FOXO3a/BIM-dependent manner. Acta Pharm
Sin B. 12:1271–1287. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu SY, Wen YC, Ku CC, Yang YC, Chow JM,
Yang SF, Lee WJ and Chien MH: Penfluridol triggers cytoprotective
autophagy and cellular apoptosis through ROS induction and
activation of the PP2A-modulated MAPK pathway in acute myeloid
leukemia with different FLT3 statuses. J Biomed Sci. 26:632019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tung MC, Lin YW, Lee WJ, Wen YC, Liu YC,
Chen JQ, Hsiao M, Yang YC and Chien MH: Targeting DRD2 by the
antipsychotic drug, penfluridol, retards growth of renal cell
carcinoma via inducing stemness inhibition and autophagy-mediated
apoptosis. Cell Death Dis. 13:4002022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu LL, Liu YY, Li ZX, Zhao Q, Wang X, Yu
Y, Wang YY, Wang YQ and Luo F: Anti-tumor effects of penfluridol
through dysregulation of cholesterol homeostasis. Asian Pac J
Cancer Prev. 15:489–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Du J, Shang J, Chen F, Zhang Y, Yin N, Xie
T, Zhang H, Yu J and Liu F: A CRISPR/Cas9-Based screening for
non-homologous end joining inhibitors reveals ouabain and
penfluridol as radiosensitizers. Mol Cancer Ther. 17:419–431. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Janssen PA, Niemegeers CJ, Schellekens KH,
Lenaerts FM, Verbruggen FJ, Van Nueten JM and Schaper WK: The
pharmacology of penfluridol (R 16341) a new potent and orally
long-acting neuroleptic drug. Eur J Pharmacol. 11:139–154. 1970.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Airoldi L, Marcucci F, Mussini E and
Garattini S: Distribution of penfluridol in rats and mice. Eur J
Pharmacol. 25:291–295. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Andrade C: Psychotropic drugs with long
half-lives: Implications for drug discontinuation, occasional
missed doses, dosing interval, and pregnancy planning. J Clin
Psychiatry. 83:22f145932022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bhattacharyya R, Bhadra R, Roy U,
Bhattacharyya S, Pal J and Saha SS: Resurgence of penfluridol:
Merits and demerits. East J Psychiatry. 18:23–29. 2015. View Article : Google Scholar
|
|
46
|
Nikvarz N, Vahedian M and Khalili N:
Chlorpromazine versus penfluridol for schizophrenia. Cochrane
database Syst Rev. 9:CD0118312017.PubMed/NCBI
|
|
47
|
Wang RI, Larson C and Treul SJ: Study of
penfluridol and chlorpromazine in the treatment of chronic
schizophrenia. J Clin Pharmacol. 22:236–242. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Andrade C: The practical importance of
half-life in psychopharmacology. J Clin Psychiatry.
83:22f145842022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Clarke Z: Penfluridol. Elsevier; New York,
NY: pp. 1–4. 2007
|
|
50
|
Enyeart JJ, Biagi BA, Day RN, Sheu SS and
Maurer RA: Blockade of low and high threshold Ca2+ channels by
diphenylbutylpiperidine antipsychotics linked to inhibition of
prolactin gene expression. J Biol Chem. 265:16373–16379. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cabrera M, Gomez N, Remes Lenicov F,
Echeverría E, Shayo C, Moglioni A, Fernández N and Davio C: G2/M
cell cycle arrest and tumor selective apoptosis of acute leukemia
cells by a promising benzophenone thiosemicarbazone compound. PLoS
One. 10:e01368782015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Boudreau RT, Conrad DM and Hoskin DW:
Apoptosis induced by protein phosphatase 2A (PP2A) inhibition in T
leukemia cells is negatively regulated by PP2A-associated p38
mitogen-activated protein kinase. Cell Signal. 19:139–151. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nakamura H and Takada K: Reactive oxygen
species in cancer: Current findings and future directions. Cancer
Sci. 112:3945–3952. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yang H, Villani RM, Wang H, Simpson MJ,
Roberts MS, Tang M and Liang X: The role of cellular reactive
oxygen species in cancer chemotherapy. J Exp Clin Cancer Res.
37:2662018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shah MA and Rogoff HA: Implications of
reactive oxygen species on cancer formation and its treatment.
Semin Oncol. 48:238–245. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Singh R and Manna PP: Reactive oxygen
species in cancer progression and its role in therapeutics. Explor
Med. 3:43–57. 2022. View Article : Google Scholar
|
|
58
|
Perillo B, Di Donato M, Pezone A, Di Zazzo
E, Giovannelli P, Galasso G, Castoria G and Migliaccio A: ROS in
cancer therapy: The bright side of the moon. Exp Mol Med.
52:192–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kim SJ, Kim HS and Seo YR: Understanding
of ROS-Inducing strategy in anticancer therapy. Oxid Med Cell
Longev. 2019:53816922019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Miller DM, Thomas SD, Islam A, Muench D
and Sedoris K: c-Myc and cancer metabolism. Clin Cancer Res.
18:5546–5553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gao FY, Li XT, Xu K, Wang RT and Guan XX:
c-MYC mediates the crosstalk between breast cancer cells and tumor
microenvironment. Cell Commun Signal. 21:282023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Safe S: Specificity Proteins (Sp) and
Cancer. Int J Mol Sci. 24:51642023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vellingiri B, Iyer M, Devi Subramaniam M,
Jayaramayya K, Siama Z, Giridharan B, Narayanasamy A, Abdal Dayem A
and Cho SG: Understanding the role of the transcription factor sp1
in ovarian cancer: From theory to practice. Int J Mol Sci.
21:11532020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Dufour S, Broders-Bondon F and Bondurand
N: Chapter 13 - β1-Integrin Function and Interplay during Enteric
Nervous System Development. Academic Press; Boston, MA: pp.
153–166. 2015
|
|
65
|
Bergonzini C, Kroese K, Zweemer AJM and
Danen EHJ: Targeting integrins for cancer therapy-disappointments
and opportunities. Front cell Dev Biol. 10:8638502022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Valdembri D and Serini G: The roles of
integrins in cancer. Fac Rev. 10:452021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yousefi H, Vatanmakanian M, Mahdiannasser
M, Mashouri L, Alahari NV, Monjezi MR, Ilbeigi S and Alahari SK:
Understanding the role of integrins in breast cancer invasion,
metastasis, angiogenesis, and drug resistance. Oncogene.
40:1043–1063. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Desgrosellier JS and Cheresh DA: Integrins
in cancer: Biological implications and therapeutic opportunities.
Nat Rev Cancer. 10:9–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kim SY: Cancer energy metabolism: Shutting
power off cancer factory. Biomol Ther (Seoul). 26:39–44. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chelakkot C, Chelakkot VS, Shin Y and Song
K: Modulating glycolysis to improve cancer therapy. Int J Mol Sci.
24:26062023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Fadaka A, Ajiboye B, Ojo O, Adewale O,
Olayide I and Emuowhochere R: Biology of glucose metabolization in
cancer cells. J Oncol Sci. 3:45–51. 2017. View Article : Google Scholar
|
|
72
|
Lu J, Chen S, Bai X, Liao M, Qiu Y, Zheng
LL and Yu H: Targeting cholesterol metabolism in Cancer: From
molecular mechanisms to therapeutic implications. Biochem
Pharmacol. 218:1159072023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fan YJ and Zong WX: The cellular decision
between apoptosis and autophagy. Chin J Cancer. 32:121–129.
2013.PubMed/NCBI
|
|
74
|
Das S, Shukla N, Singh SS, Kushwaha S and
Shrivastava R: Mechanism of interaction between autophagy and
apoptosis in cancer. Apoptosis. 26:512–533. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mulcahy Levy JM and Thorburn A: Autophagy
in cancer: Moving from understanding mechanism to improving therapy
responses in patients. Cell Death Differ. 27:843–857. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Koukourakis MI, Kalamida D, Giatromanolaki
A, Zois CE, Sivridis E, Pouliliou S, Mitrakas A, Gatter KC and
Harris AL: Autophagosome Proteins LC3A, LC3B and LC3C have distinct
subcellular distribution kinetics and expression in cancer cell
lines. PLoS One. 10:e01376752015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Signal Transduct Target Ther. 5:282020.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ribatti D, Tamma R and Annese T:
Epithelial-Mesenchymal transition in cancer: A historical overview.
Transl Oncol. 13:1007732020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Huang Y, Hong W and Wei X: The molecular
mechanisms and therapeutic strategies of EMT in tumor progression
and metastasis. J Hematol Oncol. 15:1292022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Liu ZL, Chen HH, Zheng LL, Sun LP and Shi
L: Angiogenic signaling pathways and anti-angiogenic therapy for
cancer. Signal Transduct Target Ther. 8:1982023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yang Y and Cao Y: The impact of VEGF on
cancer metastasis and systemic disease. Semin Cancer Biol. 86((Pt
3)): 251–261. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang L, Zhang M, Wang L, Li J, Yang T,
Shao Q, Liang X, Ma M, Zhang N, Jing M, et al: Identification of
CCL4 as an immune-related prognostic biomarker associated with
tumor proliferation and the tumor microenvironment in clear cell
renal cell carcinoma. Front Oncol. 11:6946642021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Rezayatmand H, Razmkhah M and
Razeghian-Jahromi I: Drug resistance in cancer therapy: The
Pandora's Box of cancer stem cells. Stem Cell Res Ther. 13:1812022.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zheng HC: The molecular mechanisms of
chemoresistance in cancers. Oncotarget. 8:59950–59964. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Gong L, Zhang Y, Liu C, Zhang M and Han S:
Application of radiosensitizers in cancer radiotherapy. Int J
Nanomedicine. 16:1083–1102. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ashraf-Uz-Zaman M, Shahi S, Akwii R, Sajib
MS, Farshbaf MJ, Kallem RR, Putnam W, Wang W, Zhang R, Alvina K, et
al: Design, synthesis and structure-activity relationship study of
novel urea compounds as FGFR1 inhibitors to treat metastatic
triple-negative breast cancer. Eur J Med Chem. 209:1128662021.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ashraf-Uz-Zaman M, Sajib MS, Cucullo L,
Mikelis CM and German NA: Analogs of penfluridol as
chemotherapeutic agents with reduced central nervous system
activity. Bioorg Med Chem Lett. 28:3652–3657. 2018. View Article : Google Scholar : PubMed/NCBI
|