Introduction
Colorectal cancer (CRC) ranks as the third most
frequently diagnosed cancer in both women and men and is a major
cause of cancer-related mortality globally (1). The survival outcomes of CRC remain
poor, partially owing to the presence of distant metastases
(2). Specifically, the 5-year
survival rate decreases to 8% for patients with CRC and advanced
metastasis (3). Despite
improvements in the diagnosis and treatment strategies for CRC at
an unparalleled pace in recent years, the prognosis of advanced CRC
remains poor, which is largely due to the mechanisms that underlie
the induction of CRC progression remaining unexplained (4). Due to the scarcity of powerful
therapeutic strategies in CRC, new pathways and targets against
this carcinoma need to be unearthed. A growing body of evidence has
shown that genetic and epigenetic alterations contribute to the
pathogenesis of CRC (5–7). Therefore, interrogating genetic and
epigenetic factors that drive tumor progression and constructing
more accurate models for prognostic prediction in CRC are urgently
needed.
N6-methyladenosine (m6A) modification is
garnering increasing interest as a prevalent epigenetic
modification of eukaryotic mRNA, affecting multiple steps of RNA
metabolism including RNA splicing, maturation, nuclear export and
translation (8–10). The m6A RNA modification
is a dynamic and reversible process coordinated by a
methyltransferase complex (m6A ‘writer’), demethylases
(m6A ‘erasers’) and m6A-binding proteins
(m6A ‘readers’) (11).
The m6A methylase complex is comprised of at least five
writer proteins [methyltransferase like-3 (METTL3), METTL14, WT1
associated protein (WTAP), vir-like m6A methyltransferase
associated and RNA binding motif protein 15/15B), among which
METTL3 protein serves a central role (12–15).
The erasers, human Alk B homolog-5 (ALKBH5) and fat mass and
obesity-associated gene (FTO), have m6A demethylation
activity to specifically remove the m6A modification
(16,17). METTL3, acting as an oncogene,
maintains SRY-box transcription factor 2 (SOX2) expression by
preventing SOX2 mRNA degradation via m6A/insulin like
growth factor 2 mRNA binding protein 2 (IGF2BP2) axis regulation,
thus contributing to the progression of CRC (18). Additionally, METTL3 facilitates the
progression of CRC by upregulating Janus kinase 1 and STAT3
expression through both m6A-dependent and -independent
mechanisms, consequently activating the phosphorylated-STAT3
signaling pathway (19). Moreover,
METTL3 is upregulated in human CRC and promotes CRC progression by
elevating MYC expression and epigenetically attenuating the
expression of the yippee-like 5 tumor suppressor, as well as
stabilizing cyclin-E1 mRNA via m6A mRNA modification of
the effectors (20–22). These studies unrevealed that
m6A modification and METTL3 regulation play pivotal
roles in tumorigenesis, tumor development and metastasis, and the
dysregulation of m6A is closely associated with the
development and pathogenesis of CRC. Nevertheless, m6A
modification play a central role in various cancer types and the
definite role of m6A in CRC remains obscure, and the
dysregulation of METTL3-mediated m6A modification in the
progression of CRC requires further investigation.
Matrix metallopeptidase (MMP)-9 and MMP2 participate
in angiogenesis by remodeling the extracellular matrix (ECM),
activating and deactivating ECM components by proteolysis cleavage,
and are thus involved in cancer cell proliferation and
differentiation (23). MMP9
inhibition significantly decreased primary tumor growth and the
incidence of metastasis in a surgical CRC orthotopic xenograft
model (24). A number of findings
have revealed that MMP9 plays a crucial role in tumor progression,
with the MMP9 expression level correlating with the grade and stage
of carcinoma (25–27). However, the relationship between
m6A-related factors and MMP9 in CRC progression outcomes
are not well defined.
The present study aimed to explore the role of m6A
modification and its key regulator, METTL3, in the progression of
CRC. The primary objectives were to assess the expression levels of
m6A and m6A-related regulators in CRC tissues and to investigate
the functional impact of METTL3 on CRC cell behavior. To achieve
these aims, the study employed a multi-faceted approach that
included both in vitro and in vivo experiments. The
in vitro component involved the use of cell cultures to
manipulate the expression levels of METTL3 and to observe the
subsequent effects on CRC cell proliferation, migration and
apoptosis. By combining these methods, the study aimed to provide a
comprehensive understanding of the METTL3/MMP9 axis in CRC and to
establish a foundation for the development of novel therapeutic
strategies targeting this axis. The results of these experiments
are expected to contribute significantly to the field by offering
insights into the molecular mechanisms underlying CRC and
potentially leading to more effective treatments.
Materials and methods
Cell culture
In total, three CRC cell lines (LS174T, HCT116 and
SW480) and two normal human colon mucosal epithelial cell lines
(NCM-460 and HIEC-6) were utilized in the present study, all
acquired from The Cell Bank of Type Culture Collection of The
Chinese Scientific Academy. The HCT116, SW480, NCM-460 and HIEC-6
cell lines were regularly maintained in RPMI-1640 medium with 10%
fetal bovine serum (FBS) and 1% penicillin-streptomycin (all from
Gibco; Thermo Fisher Scientific, Inc.) in a humidified incubator
set at 37°C and 5% CO2. Conversely, the LS174T cells
were cultured using Dulbecco's Modified Eagle's Medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS and 1%
penicillin-streptomycin.
Establishment of stable knockdown and
overexpression cell lines
To perform RNA interference, HCT116 and SW480 cells
were seeded in 60 mm tissue culture plates for 24 h prior to
transfection. METTL3 short hairpin (sh)RNA cloned into the pLKO.1
lentiviral vector (Addgene, Inc.) was obtained. Subsequently, the
vector (pLKO.1-METTL3) was co-transfected into CRC cells with the
psPAX2 and pmD2G (Addgene, Inc.) packing plasmids at a 4:3:1 ratio,
using the PolyJet reagent (SignaGen Laboratories) in serum-free
media. After 48 h transfection at 37°C, the cell culture
supernatant was harvested and filtered through a 0.22 µm membrane.
Following concentration using a 100 kD Millipore filter, the viral
solution was added to the cancer cell culture medium at a
multiplicity of infection of 3, allowing for a 24-h infection
period. Successfully transfected cells expressing shRNAs were
selected using 5 µg/ml puromycin (Sigma-Aldrich; Merck KGaA) and
maintained in RPMI-1640/10% FBS supplemented with 1 µg/ml puromycin
at 37°C and 5% CO2. The sequences of the shRNAs were as
follows: sh-METTL3, 5′-GCACTTGGATCTACGGAATCC-3′; and sh negative
control (NC), 5′-GTCCATCGAACTCAGTAGCT-3′.
To achieve METTL3 overexpression, HCT116 cells were
stably transfected with the pcDNA3/Flag-METTL3 plasmid (cat. no.
53739; Addgene, Inc.). HCT116 and SW480 cells were stably
transfected with the pcDNA3-MMP9 plasmid (Suzhou Hongxun
Biotechnologies Co., Ltd.). The transfection procedure was
conducted utilizing Lipofectamine 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer's instructions.
Briefly, the cells were grown in 6-well plates until they reached
90% confluency. Then, the cells were transfected with plasmid DNA
(5.0 mg per well), utilizing Lipofectamine 3000 (10 µl) to
facilitate transfection at 37°C. After 48 h, the cells were
subjected to trypsinization and subsequently transferred to 10-cm
culture dishes in medium supplemented with 300 mg/ml G418 (Gibco;
Thermo Fisher Scientific, Inc.) and maintained with 50 mg/ml G418.
Clonal expansion was achieved through the isolation of single-cell
clones.
mRNA m6A
quantification
Total RNA was isolated using the TRIzol reagent
(Ambion; Thermo Fisher Scientific, Inc.). Subsequently, the
polyadenylated RNA was extracted from the total RNA using the
Dynabeads™ mRNA Purification Kit (Invitrogen; Thermo Fisher
Scientific, Inc.), with any contaminating rRNA removed by the
RiboMinus transcriptome isolation kit (Invitrogen; Thermo Fisher
Scientific, Inc.). The relative levels of m6A in the
mRNA were assessed utilizing an m6A RNA Methylation
Quantification Kit (Colorimetric; cat. no. P-9005; EpigenTek Group,
Inc.), according to the manufacturer's instructions. Briefly,
capture and detection antibodies were employed to measure the
m6A levels, which were determined at measuring the
absorbance at 450 nm. Each reaction was replicated three times to
quantify the relative absorbance.
RNA extraction, reverse transcription
(RT) and quantitative PCR (qPCR)
Total cellular RNA was isolated using the RNA
isolation kit (Shanghai Yeasen Biotechnology Co., Ltd.) and
subsequently reverse transcribed into cDNA with the PrimeScript™ RT
Reagent Kit, which included a gDNA Eraser (Takara Bio, Inc.). qPCR
was conducted using the TB Green® Premix Ex Taq™ (Takara
Bio, Inc.). The qPCR thermocycling conditions were as follows: 40
cycles at 37°C for 15 min, 60°C for 5 sec and 72°C for 30 sec. The
relative mRNA expression levels were calculated using the
2−∆∆Cq method (28). All
procedures followed the manufacturer's instructions and were
repeated three times. The primer (Sangon Biotech Co., Ltd.)
sequences were as follows: METTL3 forward,
5′-CTATCTGGCACTCGCAAGA-3′ and reverse,
5′-GCTTGAACCGTGCAACCACATC-3′; MMP9 forward,
5′-CCAATCACCACCATCCGTTG-3′ and reverse, 5′-CCTCGGGCAAATGTCTTACC-3′;
GAPDH forward, 5′-GTCTCCTCTGACTTCAACAGCG-3′ and reverse,
5′-ACCACCCTGTTGCTGTAGCCAA-3′; and 18S forward,
5′-GGAGTATGGTTGCAAAGCTGA-3′ and reverse,
5′-ATCTGTCAATCCTGTCCGTGT-3′.
m6A-modified RNA
immunoprecipitation
Stable knockdown METTL3 cell RNA was extracted, and
polyadenylated RNA was enriched using an mRNA purification kit
(Thermo Fisher Scientific, Inc.). This enriched RNA was then
treated with DNase I (Thermo Fisher Scientific, Inc.). Following
this, 100 µg global RNA was incubated with m6A or IgG
antibodies for immunoprecipitation using the Magna methylated RNA
immunoprecipitation (MeRIP) m6A kit (cat. no. 17-10499;
MilliporeSigma) according to the manufacturer's instructions. For
m6A RIP qPCR, the total RNAs were fragmented into 300-nucleotide
fragments after incubation in fragmentation buffer at 94°C for 30
sec and immunoprecipitated using anti-m6A antibody according to the
manufacturer's instructions. In total, one-tenth of the fragmented
RNAs were saved as input control, and the enrichment of m6A was
quantified using RT-qPCR.
RNA half-life assay and qPCR
To assess the degradation rate of MMP9 mRNA, cells
were treated with Actinomycin-D (Act-D; Sigma-Aldrich; Merck KGaA)
at a final concentration of 3 µg/ml at 37°C to inhibit new RNA
synthesis. At specified intervals, cell samples were collected and
analyzed using qPCR (as aforementioned). The MMP9 expression levels
were standardized against 18S RNA.
Protein stability assay
To measure protein stability, shNC and shMETTL3
transfected cells were seeded into 6-well plates and treated with
cycloheximide (Cayman Chemical Company; to inhibit RNA translation)
at 37°C at final concentration of 20 µg/ml for the indicated times.
Cells were collected and lysed in RIPA lysis buffer (Cell Signaling
Technology, Inc.). The expression of proteins was measured through
western blot analysis.
Cell viability assay
In total, 100 µl of culture medium containing
3×104 cells were seeded into 96-well plates and cultured
for 24 h. Subsequently, 10 µl Cell Counting Kit-8 (CCK-8) reagent
(Dojindo Laboratories, Inc.) was added to each well. Following an
additional 2-h incubation in a humidified environment at 37°C with
5% CO2, the absorbance of the cells at 450 nm was
measured using a spectrophotometer. Each assay was performed in
triplicate.
Cell migration and invasion
assays
To assess the migration capability, 1×106
cells per well were cultured in 6-well plates until a 80–90%
confluency was reached. A sterile 200 µl pipette tip was employed
to create straight uniform scratches. Cells were serum-starved
prior to and during the assay. Images were obtained by light
microscopy (Nikon C1 Eclipse; Nikon Corporation) and captured at
both 0 and 48 h, with each experimental condition tested in
triplicate. Image analysis was performed using ImageJ
(version1.42q; National Institutes of Health). The relative scratch
width was expressed as: (original scratch width-new scratch
width)/original scratch width ×100%.
For evaluating the invasion capability, Transwell
chambers pre-coated with Matrigel (BD Biosciences) at 37°C for 4 h
were utilized. The upper chamber contained the specific cells
(1×104 cells) in serum-free medium, while the lower
chamber contained complete medium supplemented with 10% FBS. Cells
that invaded the surface of the lower chamber after 24 h were then
fixed with 4% paraformaldehyde for 15 min and stained with 0.1%
crystal violet (Beijing Solarbio Science & Technology Co.,
Ltd.) for 3 min at room temperature. After air drying, the invaded
cells were imaged using a light microscope (Nikon C1 Eclipse; Nikon
Corporation) and quantified by counting cells in five randomly
selected fields. Image analysis was performed using ImageJ
(version1.42q; National Institutes of Health).
Apoptosis assays
To evaluate apoptosis, flow cytometry was employed
utilizing a FITC-Annexin V/PI detection kit (Wanleibio, Co., Ltd.).
Each well was seeded with 1×106 cells in 6-well plates,
followed by harvesting after 48 h of incubation at 37°C. The cells
were then stained with FITC-Annexin V and PI for 15 min at room
temperature, in the dark. Apoptotic cell percentages were
subsequently determined through FACS flow cytometry (FACS Canton
II; BD Biosciences) and the data were analyzed using FlowJo 8.6.3
(Tree Star, Inc.).
Protein isolation and western
blotting
Cellular proteins were extracted utilizing RIPA
buffer (cat. No. 9806; Cell Signaling Technology, Inc.)
supplemented with a protease and phosphatase inhibitor cocktail
(Sigma-Aldrich; Merck KGaA). Protein concentrations were quantified
using a BCA Protein Assay kit (Beyotime Institute of
Biotechnology). The proteins (20 µg per lane) were resolved on 10%
SDS-PAGE gels and subsequently transferred PVDF membranes. To block
non-specific binding, the membranes were incubated with 5% non-fat
milk in TBST (0.1% Tween-20) for 2 h at room temperature. Following
this, the membranes were incubated overnight at 4°C with primary
antibodies against METTL3 (1:2,000; cat. no. 86132S; Cell Signaling
Technology, Inc.), MMP9 (1:1,000; cat. no. ab76003; Abcam) and
GAPDH (1:5,000; cat. no. 5174S; Cell Signaling Technology, Inc.).
After three washes with PBST (0.1% Tween 20), the membranes were
incubated for 1 h at room temperature with a horseradish peroxidase
(HRP)-conjugated secondary antibody (1:1,000; cat. no. BA1055;
Boster Biological Technology). The signal was detected using ECL
western blotting detection reagents (Tanon Science and Technology
Co., Ltd.). Quantification of protein expression levels was
performed using ImageJ software (version 1.50; National Institutes
of Health).
Xenograft animal models
HCT116, METTL3 knockdown HCT116 and METTL3
overexpressing HCT116 cells (5×106 cells; n=5 mice per
group) were suspended in 100 µl PBS and Matrigel (Shanghai Yeasen
Biotechnology Co., Ltd.) at a 1:1 ratio. This cell mixture was then
subcutaneously injected into the flanks of 4-week-old nude mice
(weighing 14–16 g). All 15 mice were placed in SPF housing
conditions with a 12:12-h light/dark cycle and at a constant
temperature (22±2°C). Furthermore, the mice were given unrestricted
access to standard chow and water. Throughout the experimental
period, mice were monitored three times a week for tumor
development. The tumors were measured using calipers, with a humane
endpoint set at a tumor diameter of >2,000 mm. The tumor volume
was calculated using the following formula: Volume=W2 ×
L/2, where W represents the short diameter and L represents the
long diameter. The weights of the mice were rigorously monitored to
ensure that any decrease did not exceed 20%, thereby mitigating
potential suffering. No animals died before meeting the criteria
for humane endpoint euthanasia. At the end of the experiment (day
24), the mice were anesthetized through intraperitoneal
administration of sodium pentobarbital (50 mg/kg) and subsequently
sacrificed by cervical dislocation. Following euthanasia, the
xenograft tumors were carefully excised from the sacrificed mice
and weighed immediately. The experiments adhered to institutional
guidelines and ethical standards regarding euthanasia and death
verification. Some tumor specimens were used for RNA extraction
with TRIzol, followed by qPCR to assess the METTL3 and MMP9 mRNA
expression levels (as aforementioned). The remaining samples were
fixed with 10% (v/v) neutral-buffered formalin for 24 h at room
temperature, then transferred to 70% ethanol until they were
embedded in paraffin and sectioned at a 3 µm thickness. For
hematoxylin-eosin staining, the sections were treated with
hematoxylin for 2 min and eosin for 1 min.
Immunohistochemistry (IHC)
Antigen retrieval was performed using boiling sodium
citrate buffer (0.1 M, pH 4). After deparaffinization and hydration
of the paraffin-embedded tissue sections, endogenous peroxidase
activity was blocked with Peroxidase 1 blocking reagent (Biocare
Medical, LLC) for 10 min followed by blocking with Background
Sniper serum-free blocking reagent (Biocare Medical, LCC) for 15
min at room temperature. Tissue sections were incubated overnight
at 4°C with primary antibodies targeting METTL3 (1:50; cat. no.
86132S; Cell Signaling Technology, Inc.), MMP9 (1:1,000; cat. no.
ab76003; Abcam) and Ki67 (1:100; cat. no. ab16667; Abcam). Post
incubation, sections were washed with PBST (0.05% Tween-20) and
then treated with a HRP-conjugated rabbit secondary antibody
(1:5,000; cat. no. BM3894; Wuhan Boster Biological Technology,
Ltd.) at room temperature for 1 h. The sections were subsequently
developed using 0.05% 3-diaminobidine tetrahydrochloride for 10 sec
at room temperature, followed counterstaining with 10% Mayer's
hemoxylin for 4 min at room temperature. The IHC results were
analyzed by two experienced pathologists. For imaging, five random
fields at ×200 magnification under a light microscope (Leica
DMI4000B; Leica Microbiosystems GmbH) were selected.
Bioinformatics analysis
The Gene Expression Profiling Interactive Analysis
database (gepia.cancer-pku.cn/) served as the tool to assess the
mRNA expression levels of METTL3, METTL14, WTAP, ALKBH5 and FTO in
both CRC and normal tissues. For Kaplan-Meier analysis, the
log-rank test was employed. To determine the overall survival of
patients with CRC and varying levels of METTL3 expression,
Kaplan-Meier survival analyses were performed using the Kaplan
Meier plotter online tool (http://kmplot.com/analysis/) with default parameters.
The median value was set as the cut-off.
Statistical analysis
Each experiment was conducted a minimum of three
times, with representative outcomes illustrated. Data are presented
as the mean ± SD. One-way analysis of variance with Tukey's
multiple comparisons was used to identify significant differences
among three or more groups, while two groups were compared using
the unpaired Student's t-test. Data analysis was performed using
GraphPad Prism 8 software (Dotmatics). P<0.05 was considered to
indicate a statistically significant difference.
Results
Upregulation of METTL3 and
m6A is associated with clinicopathological features in
CRC
To investigate the role of m6A
modification in CRC in vitro, the m6A levels in
three CRC cell lines (LS174T, HCT116 and SW480) and two normal
human colon mucosal epithelial cell lines (HIEC-6 and NCM-460) were
quantified using the colorimetric m6A quantification
assay. The results demonstrated that the global mRNA m6A
levels were elevated in the CRC cell lines compared with the normal
cell lines (Fig. 1A). To assess the
expression profiles of m6A writers and erasers in CRC,
The Cancer Genome Atlas database was analyzed, and it was found
that METTL3 was upregulated in CRC clinical tissues (Fig. 1B). The RT-qPCR results further
indicated a significant increase in METTL3 expression in CRC cell
lines, particularly in HCT116 and SW480 cells, compared with the
normal epithelial cell lines (Fig.
1C). These findings suggest that METTL3, an RNA
methyltransferase, may play a crucial role in CRC progression.
Moreover, METTL3 expression was higher in advanced clinical stages
than in the early stages, showing a significant positive
association (Fig. 1D). Kaplan-Meier
survival curves indicated that higher METTL3 expression was
significantly associated with a lower survival rate among patients
with CRC (Fig. 1E). Thus, these
results indicate that METTL3 is highly expressed in CRC and is
associated with poor prognosis.
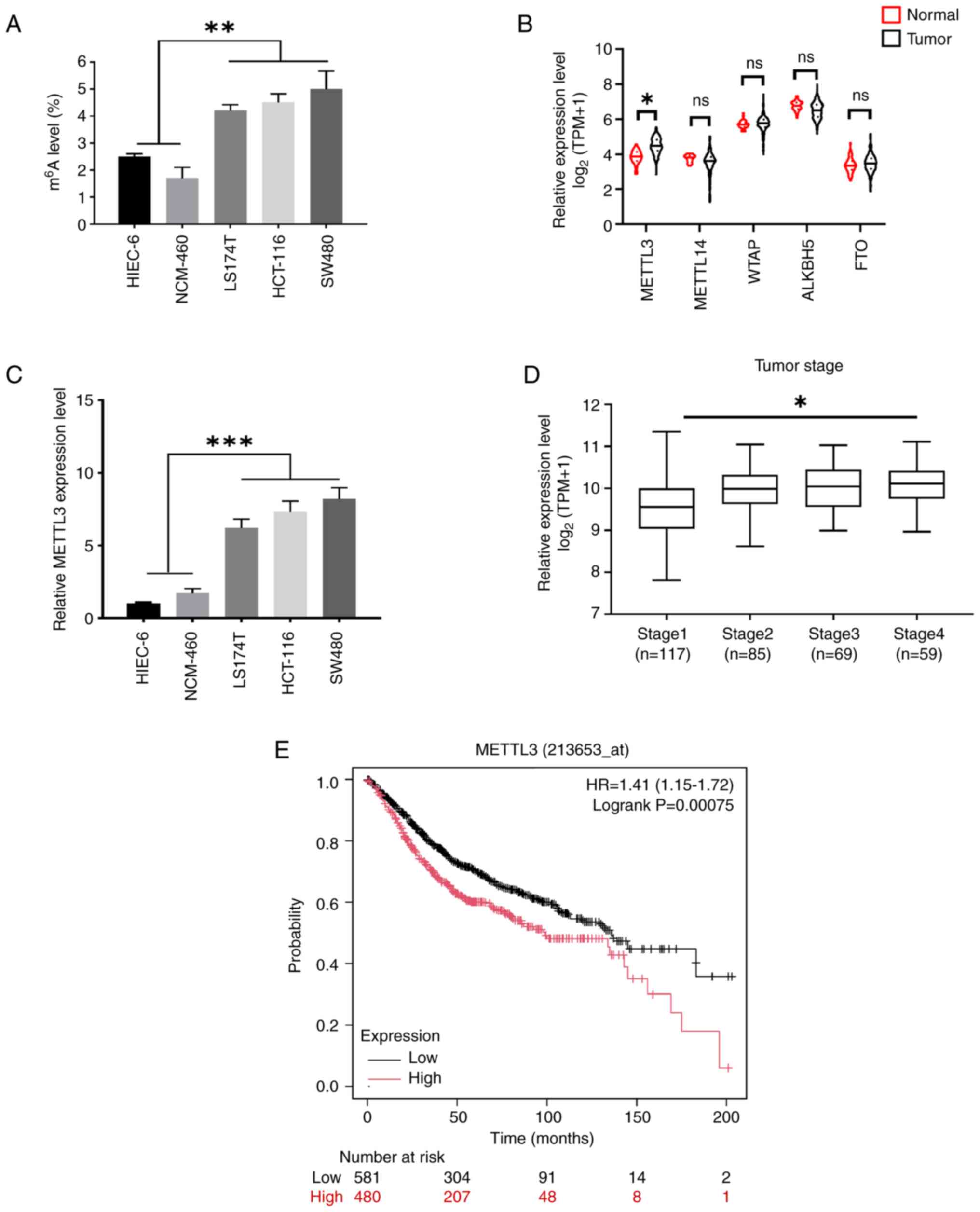 | Figure 1.Upregulation of m6A and
METTL3 in colorectal cancer. (A) Total mRNA m6A levels
in colorectal cancer cell lines compared with normal human colonic
epithelial cell lines was detected by
m6A-immunoprecipitated quantitative PCR. (B)
Bioinformatics analysis of METTL3 in tumor vs. normal colorectal
tissues in the Gene Expression Profiling Interactive Analysis
database. (C) The METTL3 expression levels in CRC cell lines
compared with normal human colonic epithelial cell lines. (D) The
METTL3 expression levels in tumors of different clinical stages of
CRC was depicted by box plot. (E) Kaplan-Meier overall survival
analysis of patients with CRC. Error bars, SD. *P<0.05,
**P<0.01, ***P<0.001. METTL3, methyltransferase-like 3; CRC,
colorectal cancer; m6A, N6-methyladenosine; TPM,
transcripts per million; ns, not significant; WTAP, WT1 associated
protein; ALKBH5, Alk B homolog-5; FTO, fat mass and
obesity-associated gene; HR, hazard ratio. |
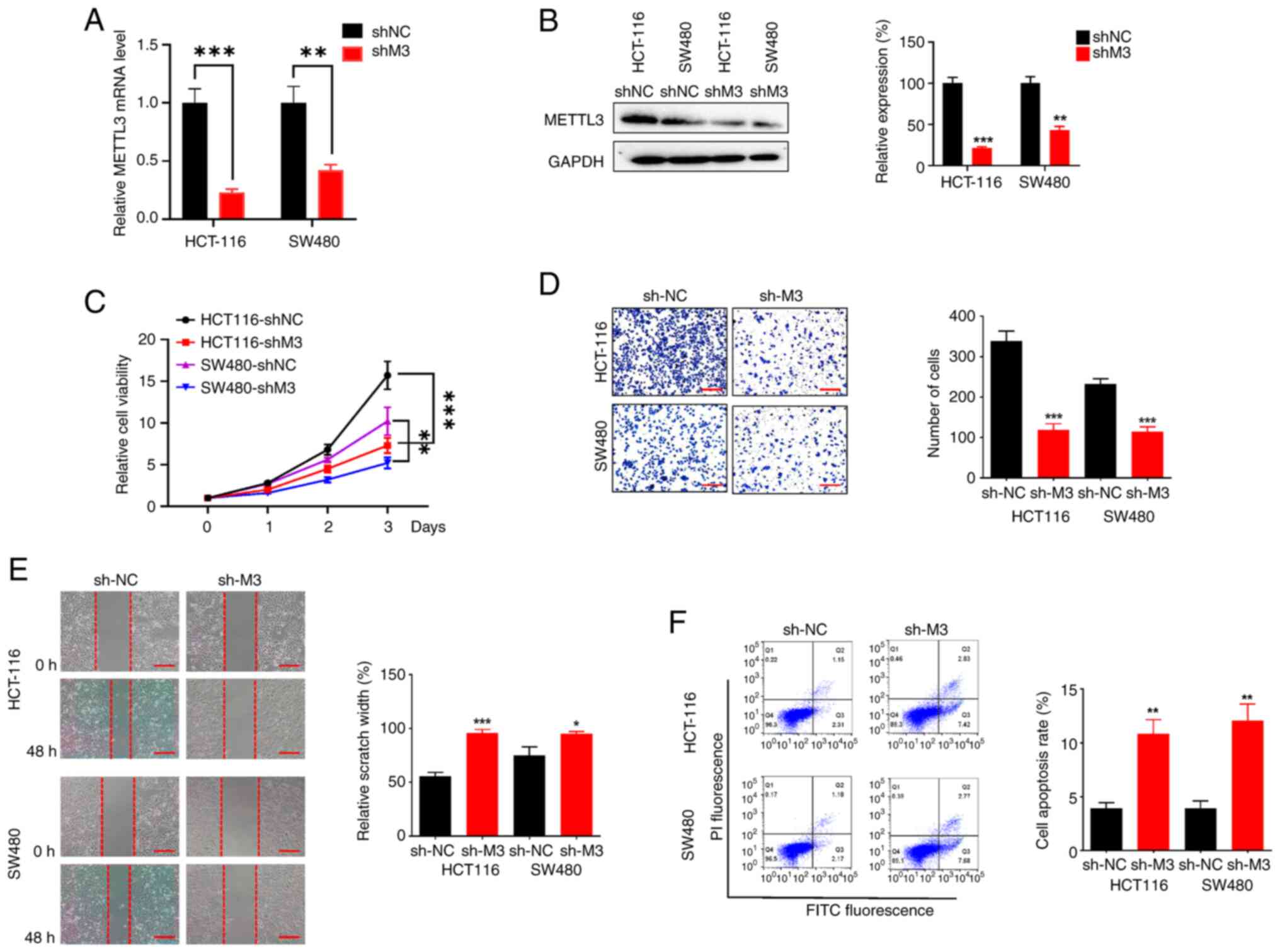
Knockdown of METTL3 blocks metastatic
characteristics in CRC
To explore the role of METTL3 in the progression of
CRC as suggested by the clinical data, the proliferation and
invasion capabilities of CRC cells were examined. For this purpose,
two stable CRC cell lines with METTL3 knockdown were established
using different shRNAs delivered via lentivirus. Validation of
METTL3 knockdown at the mRNA level showed a reduction of 25% in
HCT116 cells and 45% in SW480 cells (Fig. 2A). Correspondingly, the METTL3
protein levels were significantly lower in the knockdown cells
compared with the controls. Densitometric analysis confirmed a 20%
decrease in METTL3 protein in HCT116 cells and a 43% decrease in
SW480 cells (Fig. 2B). RT-qPCR and
western blotting therefore demonstrated the notable METTL3
knockdown efficiency of the lentivirus used. The stable
with/without METTL3 knockdown (shMETTL3 and shNC) HCT116 and SW480
cells were employed to investigate the influence of METTL3 on the
proliferation and migration capacity. The CCK-8 assay results
demonstrated a significant downregulation of the viability of the
shMETTL3 HCT116 and SW480 cells (Fig.
2C). Additionally, the Transwell and scratch assays
demonstrated that shMETTL3 cells exhibited reduced invasion
(Fig. 2D) and migration (Fig. 2E) capacities compared with shNC
cells. Flow cytometry further showed that METTL3 knockdown
increased apoptosis in CRC cell lines (Fig. 2F). Overall, these results suggest
that knocking down METTL3 decreases cell viability and migration
while promoting apoptosis in CRC cell lines.
MMP9 is regulated by METTL3-mediated
m6A modification
MMP9 plays a promotive role in various cancer types;
DNA methylation of the MMP9 gene has been shown to result in the
upregulation of MMP9 protein expression, thereby facilitating
cancer progression and metastasis (29). We hypothesized that METTL3 may
regulate the expression of MMP9 in an m6A-dependent
manner, further participating in CRC tumorigenesis via
post-transcriptional regulation. The level of MMP9 m6A
methylation in shMETTL3 and shNC cells were first examined by
employing the MeRIP assay. The results of the m6A
quantification assays implied that the MMP9 mRNA m6A
modification levels were higher in HCT116 shNC cells (Fig. 3A) and SW480 shNC cells (Fig. 3B) compared with shMETTL3 cells.
Moreover, western blotting showed that the expression of MMP9
protein was downregulated in shMETTL3 cells compared with shNC
cells in the two CRC cell lines (Fig.
3C). In METTL3-knockdown HCT116 and SW480 cells, the expression
level of MMP9 mRNA was also significantly reduced (Fig. 3D). Next, the mechanisms behind
METTL3-mediated regulation of MMP9 in CRC cells were explored by
determining the protein and mRNA stability. Results from the
western blotting analysis showed that the half-lives of MMP9
protein in shMETTL3 cells was comparable to that of shNC cells in
both the HCT116 (Fig. 3E) and SW480
(Fig. 3F) cell lines, suggesting
that decreased METTL3 expression was not related to protein
stability. Given the reduced mRNA levels of MMP9 in
METTL3-knockdown cells, we hypothesized that m6A
modification might influence MMP9 mRNA stability. After treating
cells with Act-D to assess mRNA abundance, the stability of mature
mRNA in shMETTL3 cells was significantly lower compared with their
shNC counterparts (Fig. 3G). This
suggested that the mRNA stability of MMP9 was decreased in
METTL3-knockdown CRC cells.
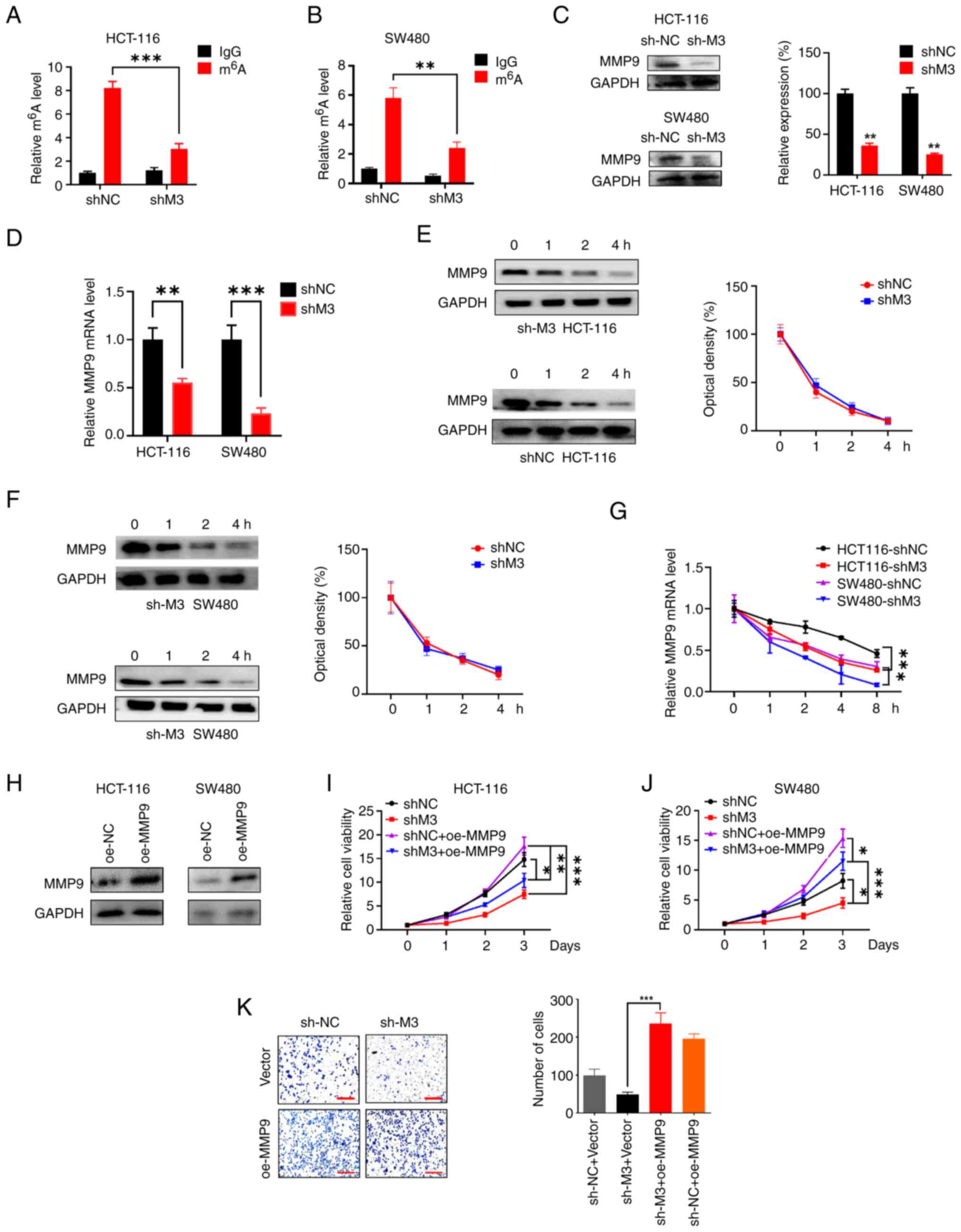 | Figure 3.METTL3 stabilizes MMP9 mRNA in a
m6A-dependent manner. MMP9 mRNA m6A levels of
stable METTL3 knockdown (A) HCT116 and (B) SW480 cell lines was
detected by m6A-immunoprecipitated qPCR. The (C) protein
and (D) mRNA levels of MMP9 in METTL3-knockdown HCT116 and SW480
cells. MMP9 protein expression levels in shNC and shMETTL3 (E)
HCT116 and (F) SW480 cell lines treated with cycloheximide (20
µg/ml) for the indicated time points were detected by western
blotting. (G) MMP9 mRNA expression levels in shNC and shMETTL3
cells treated with Actinomycin (5 µg/ml) for the indicated time
points were detected by qPCR. (H) Western blot analysis of MMP9
expression in oe-MMP9 cells was performed. The viability of (I)
HCT116 and (J) SW480 cells with or without METTL3 knockdown after
transfection with oe-MMP9 were determined by Cell Counting Kit-8
assay. (K) The invasion ability of HCT116 and SW480 cells with or
without METTL3 knockdown after transfection with oe-MMP9 was
determined by Transwell assay. Scale bars, 100 µm. Error bars, SD.
*P<0.05, **P<0.01, ***P<0.001. m6A,
N6-methyladenosine; METTL3/M3, methyltransferase-like 3; MMP9,
matrix metallopeptidase 9; CRC, colorectal cancer; qPCR,
quantitative PCR; sh, short hairpin (RNA); NC, negative control;
oe, overexpression. |
To further characterize the role of MMP9, METTL3 and
MMP9 protein interactions were studied using a gain-and-loss
functional experiment system. For this purpose, stable
MMP9-overexpression HCT116 and SW480 cell lines were constructed
and western blotting was utilized to verify MMP9 expression
(Fig. 3H). The CCK-8 results showed
that downregulation of METTL3 attenuated the cell viability;
furthermore, overexpression of MMP9 fostered the sh-METTL3-induced
downregulation of cell viability in HCT116 cells (Fig. 3I). Similar results were observed in
SW480 cells, in which overexpression of MMP9 restored the
shMETTL3-induced downregulation of cell viability (Fig. 3J). Additionally, the Transwell assay
result showed that the MMP9 overexpression could prevent the
inhibition of cell invasion caused by METTL3-knockdown (Fig. 3K). Therefore, the results indicated
that METTL3 facilitates CRC tumorigenesis by enhancing the
expression of MMP9 through m6A modification and
downregulating the decay rate of MMP9 mRNA.
METTL3 facilitates CRC tumorigenesis
by enhancing the expression of MMP9 in vivo
Stable METTL3-overexpression HCT116 cells were
constructed and western blotting was utilized to verify METTL3
expression (Fig. 4A). To
investigate the role of METTL3 in vivo, a subcutaneous
xenotransplantation model was conducted to evaluate its
contribution to CRC progression. BALB/c nude mice were
subcutaneously injected with HCT116 cells with either upregulated
or downregulated METTL3 expression, thereby establishing CRC
xenograft models (Fig. 4B). After
24 days, the mice were sacrificed and the tumor tissues were
excised, weighed and imaged (Fig.
4C). The tumors from the METTL3 overexpression cells grew more
rapidly and the tumor weights were heavier compared with the
control cells, whereas knockdown of METTL3 significantly repressed
tumor growth compared with the control cells (Fig. 4D). The tumors derived from cells
with METTL3 overexpression exhibited significantly elevated METTL3
and MMP9 mRNA levels compared with the control group. Conversely,
tumors from the METTL3 knockdown cells showed a significant
reduction in METTL3 and MMP9 mRNA levels relative to the control
group (Fig. 4E). The IHC results
showed that the shMETTL3 HCT116 ×enograft exhibited lower
expression levels of METTL3 and MMP9 than the control xenografts,
while METTL3-overexpression HCT116 ×enograft exhibited higher
expression levels of METTL3 and MMP9 (Fig. 4F). In summary, the findings
indicated that the m6A modification facilitated by
METTL3 promoted CRC development by upregulating MMP9 expression
in vivo.
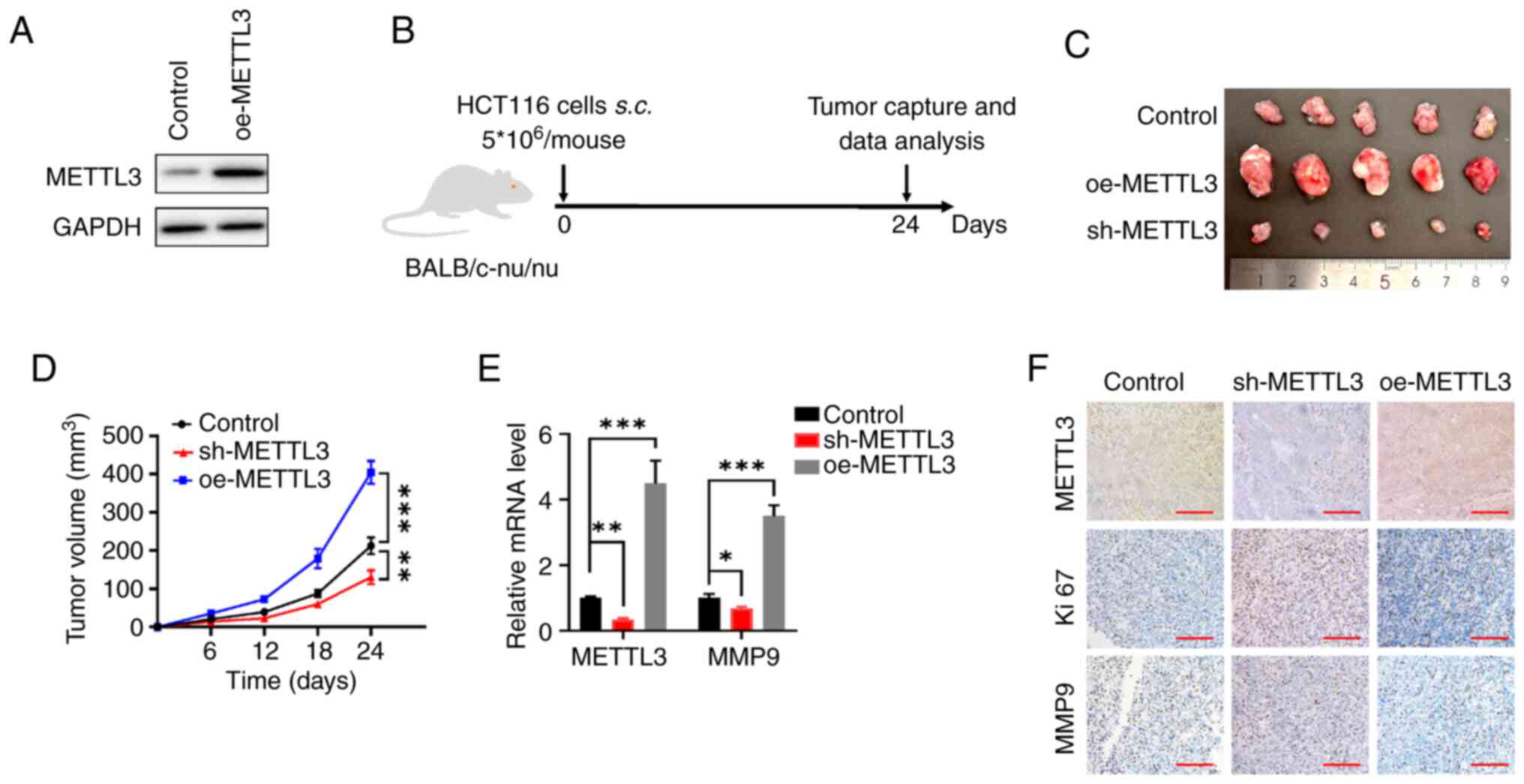 | Figure 4.METTL3 promotes colorectal cancer
cell progression in vivo. (A) Western blot analysis of
METTL3 was performed on HCT116 cells transfected with oe-METTL3.
(B) Flow chart of the in vivo experimental design. (C)
Xenograft assay was performed using HCT116 cells transfected with
sh-METTL3, oe-METTL or empty vector (pcDNA3; control). (D)
Quantitative analysis of the xenograft tumor volume. (E) The METTL3
and MMP9 mRNA levels in tumor tissues expressing sh-METTL3 or
oe-METTL3 or the control HCT116 cells. (F) Expression of METTL3,
Ki67 and MMP9 was detected by immunohistochemistry of
paraffin-embedded tissues. Scale bars, 100 µm. Error bars, SD.
*P<0.05, **P<0.01, ***P<0.001. METTL3,
methyltransferase-like 3; MMP9, matrix metallopeptidase 9; sh,
short hairpin (RNA); NC, negative control; oe, overexpression;
s.c., subcutaneous. |
Discussion
RNA modification has been steadily revealed for
decades and at least 170 different post-transcriptional RNA
modifications have been identified, among which the most common RNA
modifications are m6A, m1A, m5C,
hm5C, Ψ and 5-methoxyuracil (30,31).
In eukaryotic cells, m6A methylation is recognized as
the most prevalent reversible modification occurring
post-transcriptionally in RNA, constituting ~50% of total
methylated ribonucleotides and 0.1–0.5% of all adenosine residues
in the entire cellular RNA population (32). The results of the present study
demonstrated that the m6A levels of CRC cell lines (HIEC-6 and
NCM-460) account for >0.5%, while colorectal normal cell lines
(LS174T, HCT-116 and SW480) account for 0.2%, consistent with this
finding. What's more, the global mRNA m6A levels were
highly elevated in CRC cell lines compared with colorectal normal
cell lines, indicating a relationship between m6A
modification and CRC progression. Numerous studies have
demonstrated that METTL3 is aberrantly expressed in a number of
tumor types and is closely associated with the development of
tumors (33–35). In the present study, it was found
that METTL3 was significantly upregulated in CRC, in agreement with
previous studies. More notably, in the xenograft model analysis,
overexpression of METTL3 effectively promoted subcutaneous tumor
growth in nude mice, and vice versa. This corroborative evidence
further confirmed that dysregulation of METTL3 may be involved in
CRC progress. The findings also imply that METTL3 plays a pivotal
role in promoting CRC malignant progression. This suggests that
developing modulators targeting m6A and METTL3 could
offer promising new therapeutic strategies for combating CRC.
M6A modification is, in general,
functionally interpreted by m6A reader proteins
(36). The reader proteins can
recognize and bind m6A modifications, allowing them to
facilitate the gene regulatory functions of m6A
(33). Previous research has shown
that IGF2BP1-3 and YTH N6-methyladenosine RNA binding protein F1
function as m6A readers, recognizing m6A
modifications and thereby enhancing the stability of the
corresponding mRNA (37). The
results of the present study showed that METTL3 could stabilize
MMP9 mRNA in a m6A-dependent manner via Act-D analysis,
indicating the interplay between the m6A reader and the
m6A modification of MMP9 mRNA requires further
investigation. To comprehend the biological significance of RNA
m6A modifications, it is essential to identify
m6A modification sites across the entire transcriptome.
The mechanism by which the m6A site on MMP9 mRNA affects
the mRNA longevity via methylation by METTL3 needs to be further
explored.
MMP9, a zinc-dependent proteolytic enzyme, possesses
the capability to break down ECM components, playing a crucial role
in various pathophysiological processes (38). Formerly described transcriptional
control of MMP9 consists of histone modifications and microRNA
(39,40). In the present study, it was firstly
found that MMP9 mRNA could be methylated and that METTL3 could
catalyze MMP9 mRNA m6A methylation and further promote
its expression through enhancing the m6A-modified MMP9
mRNA stability, thus affecting the migration and proliferation of
CRC in vitro. Furthermore, the critical involvement of
METTL3 in CRC was verified experimentally using the subcutaneous
transplantation model, which suggested a possible therapeutic
intervention for cancer progression by targeting the novel MMP9
epigenetic modification.
However, the present study has certain limitations.
The number of animals used in the study was limited to 5 per group.
A small sample size can restrict the statistical power of the
study, making it difficult to detect significant effects or
differences. Furthermore, the present study did not incorporate
clinical specimens, which limits the ability to empirically
validate the findings. Without direct evidence from patient
samples, the applicability of the results to real-world clinical
scenarios remains uncertain. Additionally, only HCT-116 cell lines
were utilized in the animal experiments. The deficiency of
additional cell lines for validation restricts the ability to
confirm the mechanisms proposed in the present study. Without
corroborating evidence from other relevant models, the findings
regarding role of METTL3 in promoting tumorigenesis via MMP9
remains less robust. Future investigations should prioritize the
inclusion of a broader range of cell lines and models to enhance
the robustness and applicability of the results.
In summary, the results of the present study
indicated METTL3 was frequently upregulated in CRC and closely
related to the clinicopathological features. Moreover, upregulation
of METTL3 promoted CRC cell proliferation and tumorigenesis by
enhancing MMP9 expression and it was discovered that the
m6A modification of MMP9 may be involved in the
molecular mechanisms of these observed functional behaviors in CRC.
The results of the present study therefore indicated that
concurrently targeting METTL3 and MMP9 could offer novel
therapeutic avenues for the treatment of CRC.
Acknowledgements
Not applicable.
Funding
This study was financially supported by Jiaxing City and
Provinces to Build Medical Key Disciplines-Oncology (grant no.
2023-SSGJ-001) and National Oncology Clinical Key Specialty (grant
no. 2023-GJZK-001).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JC performed most of the experiments and data
analysis. JC, HW, TZ and JW assisted in the in vivo or in
vitro experiments. JC and ZC wrote the original manuscript
draft. JW contributed to experimental guidance. JC, JW and ZC
designed the project and revised and edited the manuscript. All
authors have read and approved the final version of the manuscript.
JC and ZC confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Animal experiments were approved by the Animal
Experimental Ethics Committee of the First Affiliated Hospital of
Jiaxing University (Jiaxing, China; approval no. JXYY2024-021).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Wagle NS, Cercek A, Smith RA
and Jemal A: Colorectal cancer statistics, 2023. CA Cancer J Clin.
73:233–254. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wolf D, Salcher S and Pircher A: The
multivisceral landscape of colorectal cancer metastasis:
Implications for targeted therapies. J Clin Invest.
134:e1783312024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Association NHCOTPROCSOOCM: National
Health Commission guidelines for diagnosis and treatment of
colorectal cancer 2023 in China (English version). Chin J Cancer
Res. 35:197–232. 2023.PubMed/NCBI
|
|
4
|
Bien J and Lin A: A review of the
diagnosis and treatment of metastatic colorectal cancer. Jama-J Am
Med Assoc. 325:2404–2405. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miranda E, Bianchi P, Destro A, Morenghi
E, Malesci A, Santoro A, Laghi L and Roncalli M: Genetic and
epigenetic alterations in primary colorectal cancers and related
lymph node and liver metastases. Cancer. 119:266–276. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clemens AW, Lin S, Jain S, Su YH and Song
W: Detection of colorectal cancer-associated genetic and epigenetic
alterations in urine of patients with CRC. Cancer Res. 75:abs.
1561. 2015.https://doi.org/10.1158/1538-7445.AM2015-1561
View Article : Google Scholar
|
|
7
|
Nosho K, Kawasaki T, Ohnishi M, Suemoto Y,
Kirkner GJ, Zepf D, Yan L, Longtine JA, Fuchs CS and Ogino S:
mutation in colorectal cancer: Relationship with genetic and
epigenetic alterations. Neoplasia. 10:534–541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang XL, Liu BY, Nie Z, Duan L, Xiong Q,
Jin Z, Yang C and Chen Y: The role of m6A modification in the
biological functions and diseases. Signal Transduct Tar. 6:742021.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roundtree IA, Luo GZ, Zhang Z, Wang X,
Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, et al: YTHDC1
mediates nuclear export of N6 - methyladenosine
methylated mRNAs. Elife. 6:e313112017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu ZM, Huo FC, Zhang J, Shan HJ and Pei
DS: Crosstalk between m6A modification and alternative splicing
during cancer progression. Clin Transl Med. 13:e14602023.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fang Z, Mei WT, Qu C, Lu J, Shang L, Cao F
and Li F: Role of m6A writers, erasers and readers in cancer. Exp
Hematol Oncol. 11:452022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ping XL, Sun BF, Wang L, Xiao W, Yang X,
Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al: Mammalian WTAP is
a regulatory subunit of the RNA N6-methyladenosine
methyltransferase. Cell Res. 24:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang
Z, Cheng T, Gao M, Shu X, Ma H, et al: VIRMA mediates preferential
m6A mRNA methylation in 3′UTR and near stop codon and
associates with alternative polyadenylation. Cell Discov. 4:102018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patil DP, Chen CK, Pickering BF, Chow A,
Jackson C, Guttman M and Jaffrey SR: m(6)A RNA methylation promotes
XIST-mediated transcriptional repression. Nature. 537:369–373.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng G, Dahl JA, Niu Y, Fedorcsak P,
Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, et al: ALKBH5
Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and
Mouse Fertility. Mol Cell. 49:18–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG and He C: 6-Methyladenosine in
nuclear RNA is a major substrate of the obesity-associated FTO. Nat
Chem Biol. 7:885–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN,
Chen ZH, Zeng ZL, Wang F, Zheng J, et al: METTL3 facilitates tumor
progression via an mA-IGF2BP2-dependent mechanism in colorectal
carcinoma. Mol Cancer. 18:1122019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun Y, Gong W and Zhang S: METTL3 promotes
colorectal cancer progression through activating JAK1/STAT3
signaling pathway. Cell Death Dis. 14:7652023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiang S, Liang XL, Yin S, Liu J and Xiang
Z: N6-methyladenosine methyltransferase METTL3 promotes colorectal
cancer cell proliferation through enhancing MYC expression. Am J
Transl Res. 12:1789–1806. 2020.PubMed/NCBI
|
|
21
|
Zhou D, Tang W, Xu Y, Xu Y, Xu B, Fu S,
Wang Y, Chen F, Chen Y, Han Y and Wang G: METTL3/YTHDF2 m6A axis
accelerates colorectal carcinogenesis through epigenetically
suppressing YPEL5. Mol Oncol. 15:2172–2184. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu W, Si Y, Xu J, Lin Y, Wang JZ, Cao M,
Sun S, Ding Q, Zhu L and Wei JF: Methyltransferase like 3 promotes
colorectal cancer proliferation by stabilizing CCNE1 mRNA in an
m6A-dependent manner. J Cell Mol Med. 24:3521–3533. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mondal S, Adhikari N, Banerjee S, Amin SA
and Jha T: Matrix metalloproteinase-9 (MMP-9) and its inhibitors in
cancer: A minireview. Eur J Med Chem. 194:1122602020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marshall DC, Lyman SK, McCauley S,
Kovalenko M, Spangler R, Liu C, Lee M, O'Sullivan C, Barry-Hamilton
V, Ghermazien H, et al: Selective Allosteric Inhibition of MMP9 is
efficacious in preclinical models of ulcerative colitis and
colorectal cancer. PLoS One. 10:e01270632015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Choi SH, Lee HJ, Jin YB, Jang J, Kang GY,
Lee M, Kim CH, Kim J, Yoon SS, Lee YS and Lee YJ: MMP9 Processing
of HSPB1 Regulates Tumor Progression. PLoS One. 9:e855092014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee MA, Park JH, Rhyu SY, Oh ST, Kang WK
and Kim HN: Wnt3a expression is associated with MMP-9 expression in
primary tumor and metastatic site in recurrent or stage IV
colorectal cancer. BMC Cancer. 14:1252014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bauer L, Takacs A, Slotta-Huspenina J,
Langer R, Becker K, Novotny A, Ott K, Walch A, Hapfelmeier A and
Keller G: Clinical significance of NOTCH1 and NOTCH2 expression in
gastric carcinomas: An immunohistochemical study. Front Oncol.
5:942015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boonsongserm P, Angsuwatcharakon P,
Puttipanyalears C, Aporntewan C, Kongruttanachok N, Aksornkitti V,
Kitkumthorn N and Mutirangura A: Tumor-induced DNA methylation in
the white blood cells of patients with colorectal cancer. Oncol
Lett. 18:3039–3048. 2019.PubMed/NCBI
|
|
30
|
Barbieri I and Kouzarides T: Role of RNA
modifications in cancer. Nat Rev Cancer. 20:303–322. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen Z, Zhao P, Li FY, Wang Y, Smith AI,
Webb GI, Akutsu T, Baggag A, Bensmail H and Song J: Comprehensive
review and assessment of computational methods for predicting RNA
post-transcriptional modification sites from RNA sequences. Brief
Bioinform. 21:1676–1696. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roundtree IA, Evans ME, Pan T and He C:
Dynamic RNA Modifications in gene expression regulation. Cell.
169:1187–1200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeng C, Huang W, Li Y and Weng H: Roles of
METTL3 in cancer: Mechanisms and therapeutic targeting. J Hematol
Oncol. 13:1172020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen H, Pan Y, Zhou Q, Liang C, Wong CC,
Zhou Y, Huang D, Liu W, Zhai J, Gou H, et al: METTL3 Inhibits
Antitumor Immunity by Targeting m6A-BHLHE41-CXCL1/CXCR2
Axis to promote colorectal cancer. Gastroenterology. 163:891–907.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wei X, Huo Y, Pi J, Gao Y, Rao S, He M,
Wei Q, Song P, Chen Y, Lu D, et al: METTL3 preferentially enhances
non-mA translation of epigenetic factors and promotes
tumourigenesis. Nat Cell Biol. 24:1278–1290. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zaccara S, Ries RJ and Jaffrey SR:
Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell
Biol. 20:608–624. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang H, Weng H, Sun W, Qin X, Shi H, Wu
H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al: Recognition of RNA
N6-methyladenosine by IGF2BP proteins enhances mRNA
stability and translation. Nat Cell Biol. 20:285–295. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mizuno R, Kawada K, Itatani Y, Ogawa R,
Kiyasu Y and Sakai Y: The role of tumor-associated neutrophils in
colorectal cancer. Int J Mol Sci. 20:5292019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Song Z, Yang L, Hu W, Yi J, Feng F and Zhu
L: Effects of histone H4 hyperacetylation on inhibiting MMP2 and
MMP9 in human amniotic epithelial cells and in premature rupture of
fetal membranes. Exp Ther Med. 21:5152021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pirooz HJ, Jafari N, Rastegari M,
Fathi-Roudsari M, Tasharrofi N, Shokri G, Tamadon M, Sazegar H and
Kouhkan F: Functional SNP in microRNA-491-5p binding site of MMP9
3-UTR affects cancer susceptibility. J Cell Biochem. 119:5126–5134.
2018. View Article : Google Scholar : PubMed/NCBI
|