Introduction
Head and neck cancer includes squamous cell
carcinoma that originates in the oral cavity, sinonasal cavity,
pharynx or larynx (1,2). These types of cancer are frequently
diagnosed at advanced stages with distant metastasis, leading to a
poor prognosis (3). In 2022, there
were 188,960 new cases of laryngeal squamous cell carcinoma (LSCC)
worldwide, and it was the second most prevalent type of head and
neck cancer, with 891,453 new cases globally (4).
Traditionally, research on LSCC has primarily
focused on genetic factors (5,6);
however, the role of epigenetic modifications in cancer development
is being increasingly recognized (7). Epigenetic modifications, including DNA
methylation, histone modification and regulation by non-coding
RNAs, frequently occur and significantly affect gene expression
(8,9). In mammals, DNA methylation,
particularly at CpG dinucleotides in gene promoters (10,11),
can lead to the silencing of genes, especially of tumor suppressor
genes (TSGs), thereby increasing the risk of carcinogenesis
(12,13). Given the variability of DNA
methylation patterns with age and cancer type, these epigenetic
markers offer promising avenues for diagnostic and therapeutic
interventions (14).
Junctional adhesion molecules (JAMs), located at the
intercellular junctions of endothelial and epithelial cells, serve
diverse roles in cancer (15).
JAM3, a member of this family, is implicated in several types of
cancer, including renal carcinoma, colorectal cancer and
cholangiocarcinoma (16–18); however, its roles in LSCC has not
been thoroughly investigated, necessitating further assessment. The
present study aimed to investigate the epigenetic regulation of
JAM3, and its impact on cell proliferation, migration and
invasion in LSCC, with the goal of exploring its potential as a
targeted therapeutic approach and diagnostic marker.
Materials and methods
Tissue samples
LSCC specimens were obtained from the Department of
Pathology, The First Hospital, Shanxi Medical University (Taiyuan,
China) and were pathologically diagnosed as squamous cell carcinoma
between May 2018 and December 2020. A total of 38 archived
formalin-fixed paraffin-embedded LSCC tissues and paired adjacent
normal mucosa (ANM) tissues were used in the present study for
immunohistochemistry (IHC). The age of patients ranged between 43
and 86 years old (mean ± SD age, 63.39±9.51 years), and the sex
ratio of was 1:12 female/male. No patients had undergone
chemotherapy or radiotherapy prior to surgical resection. The
present study received ethical approval (approval no.
KYLL-2023-180) from the Ethics Committee of The First Hospital,
Shanxi Medical University and was conducted in accordance with the
committee's guidelines.
Cell lines and culture
The LSCC cell line FD-LSC-1 [provided by Professor
Liang Zhou, Department of Otolaryngology-Head and Neck Surgery,
Eye, Ear, Nose and Throat Hospital, Fudan University, Shanghai,
China (19)], the head and neck
squamous cell carcinoma (HNSCC) cell lines FaDu (cat. no. TCHu132;
The Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences) and HN30 (provided by Professor Qiancheng Shen, Medicinal
Chemistry and Bioinformatics Center, Shanghai Jiao Tong University,
School of Medicine, Shanghai, China) and the normal human epidermal
cell line HaCaT (cat. no. GDC0106; China Center for Type Culture
Collection) were cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.). The LSCC cell line AMC-HN-8 (cat. no. HZ-5240HC; Shanghai
Huzhen Industrial Co., Ltd.) was maintained in RPMI-1640
(Pricella). Notably, the HaCaT cell line underwent authentication
via short tandem repeat profiling to confirm its identity (Table SI). All media were supplemented
with 10% fetal bovine serum (FBS; Shanghai ExCell Biology, Inc.),
100 IU/ml penicillin and 100 µg/ml streptomycin. Cell cultures were
maintained at a temperature of 37°C in an atmosphere containing 5%
CO2.
Plasmids and small interfering
(si)RNAs
The JAM3 coding region sequence was inserted
into p3×Flag-CMV-10 (Promega Corporation) to construct an
overexpression vector (p3×Flag-CMV-10-JAM3), and its functionality
was confirmed by Sanger sequencing. The empty p3×Flag-CMV-10 vector
served as the negative control (NC). In addition, siRNA targeting
JAM3 (si-JAM3-1, sense: 5′-GAGAGACUCAGCCCUUUAUTT-3′,
antisense: 5′-AUAAAGGGCUGAGUCUCUCTT-3′; si-JAM3-2, sense:
5′-CUGUACCAGUAGGCAAGAUTT-3′, antisense:
5′-AUCUUGCCUACUGGUACAGTT-3′) and NC siRNA (si-NC, sense:
5′-UUCUCCGAACGUGUCACGUTT-3′, antisense:
5′-ACGUGACACGUUCGGAGAATT-3′) were designed and synthesized by
Shanghai GenePharma Co., Ltd. The transfection was conducted
according to the manufacturer's instructions of
Lipofectamine® 3000 Transfection Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). The AMC-HN-8 and FD-LSC-1 cell
lines were cultured to 70–80% confluence in 6-well plates. Plasmids
were transfected at a final concentration of 1.5 µg/ml, and siRNA
was transfected at a concentration of 37.5 nM. Transfection was
performed at room temperature for 30 min, followed by media
replacement 6 h post-transfection. Further analyses were carried
out 24 h post-transfection.
Total RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from HaCaT, AMC-HN-8,
FD-LSC-1, FaDu and HN30 cells using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.), and cDNA was generated using a kit
(cat. no. AU341; TransGen Biotech Co., Ltd.) according to the
manufacturer's protocol on a ProFlex™ 3×32-well PCR System (Thermo
Fisher Scientific, Inc.). qPCR was performed using
PerfectStart® Green qPCR SuperMix (TransGen Biotech Co.,
Ltd.) on a LightCycler® 96 instrument (Roche
Diagnostics). The relative mRNA expression levels were calculated
using the 2−ΔΔCq method (20). The 18S ribosomal RNA was used
as an internal control. All procedures (initial denaturation at
94°C for 30 sec, followed by 40 cycles of denaturation at 94°C for
5 sec, annealing at 60°C for 5 sec and extension at 72°C for 10
sec) were performed according to the manufacturer's instructions.
The primer sequences were as follows: JAM3 forward,
5′-TCCAGCAATCGAACCCCAG-3′ and reverse,
5′-CTTGTCTGCGAATCCGTAATGAT-3′; and 18S forward,
5′-CCTGGATACCGCAGCTAGGA-3′ and reverse,
5′-GCGGCGCAATACGAATGCC-3′.
5-Aza-2′-deoxycytidine (5-Aza)
treatment
The AMC-HN-8 and FD-LSC-1 cells were treated with 5
µM 5-Aza (MilliporeSigma) for 72 h at 37°C, and were then collected
for further analysis, including RT-qPCR and western blotting.
DNA isolation and bisulfite
conversion
Genomic DNA from AMC-HN-8 and FD-LSC-1 cell lines
was isolated using a TIANamp Genomic DNA Kit (cat. no. DP304;
Tiangen Biotech Co., Ltd.) and was then bisulfite-treated with an
EZ DNA Methylation-Gold kit (cat. no. D5006; Zymo Research Corp.)
according to the manufacturer's instructions.
Bisulfite sequencing PCR (BSP)
Bisulfite-treated DNA was amplified using primers
specific for bisulfite sequencing PCR. The following primer
sequences targeting a CpG island within the JAM3 promoter
were used: Forward primer, 5′-GTTTATTGAAAGAGAATTTATGTGT-3′; reverse
primer, 5′-AAACAACCCCTAAAAAACAACAAC-3′. CpG island region
prediction and BSP primer design were performed using MethPrimer
1.0 software (21), with the
JAM3 sequence ranging from −2,000 to +500 from the
transcription start site input into the software. PCR was performed
using TransTaq HiFi PCR SuperMix I (TransGen Biotech, Co., Ltd.)
with an initial denaturation at 94°C for 5 min; followed by 40
cycles at 94°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec; and
a final extension step at 72°C for 1 min. The products were
analyzed using 1.5% agarose gel electrophoresis, and the gel was
visualized with DuRed staining (cat. no. R21868; 1:10,000; Shanghai
Saint-bio Biotechnology Co., Ltd.). Images were captured using a
gel imaging system (Azure Biosystems, Inc.). With the aid of a blue
LED transilluminator (Sangon Biotech Co., Ltd), the target DNA
bands were excised from the gel and were subsequently recovered
using SanPrep Column DNA Gel Extraction Kit (cat. no. B518131;
Sangon Biotech Co., Ltd). The recovered PCR products were cloned
into the T1 cloning vector (TransGen Biotech Co., Ltd.). The cloned
products were transformed and plated onto Luria-Bertani culture
plates containing 100 µg/ml penicillin; the plate surface had
already been coated with a mixture of 8 µl 500 mM isopropyl
β-D-1-thiogalactopyranoside and 40 µl 20 mg/ml X-gal. After
overnight incubation, 10 white colonies were selected and sent to
Sangon Biotech Co., Ltd for Sanger sequencing. Sequencing outcomes
were analyzed using BiQ Analyzer v2.02 software (22).
Cell Counting Kit 8 (CCK8) assay
After a 24-h transfection, 4,000 transfected
AMC-HN-8 or FD-LSC-1 cells were seeded per well in a 96-well plate.
Subsequently, the cells were incubated with medium containing 10%
CCK8 assay reagent (Shanghai Yeasen Biotechnology Co., Ltd.) for 1
h at 37°C and detected at 450 nm using a multi-mode microplate
reader (SpectraMax i3×; Molecular Devices, LLC) at 0, 24, 48, 72
and 96 h.
Colony formation assay
Transfected AMC-HN-8 or FD-LSC-1 cells were seeded
at 1,000 cells/well in a 6-well plate and incubated for 10–14 days.
The cells were then fixed with 4% paraformaldehyde (Invitrogen;
Thermo Fisher Scientific, Inc.) for 20 min and stained with 0.1%
crystal violet (Amresco, LLC) for 10 min at room temperature, and
the number of colonies (defined as groups containing >50 cells)
was counted using ImageJ 1.53k analysis software (National
Institutes of Health).
Transwell migration and invasion
assays
AMC-HN-8 or FD-LSC-1 cells transfected with
p3×Flag-CMV-10 or p3×Flag-CMV-10-JAM3, and si-NC or si-JAM3 were
suspended in serum-free medium and adjusted to 8×105
cells/ml, after which a 200-µl suspension was added to the upper
chamber (PET membrane; pore size, 8 µm; Corning, Inc.) in a 24-well
plate. DMEM or RPMI-1640 supplemented with 20% FBS was added to the
lower chamber. For the invasion assay, each membrane was coated
with Matrigel (Corning, Inc.) for 6 h at 37°C, and the cell density
was adjusted to 10×105 cells/ml. After incubation at
37°C for 48 h, the chambers were fixed with 4% paraformaldehyde for
20 min and stained with 0.1% crystal violet for 10 min at room
temperature. The number of cells that migrated to or invaded the
lower chamber was counted in eight light microscopic fields
(magnification, ×100).
Western blot analysis
Proteins were extracted from the AMC-HN-8 and
FD-LSC-1 cells using RIPA lysis buffer (Thermo Fisher Scientific,
Inc.) containing protease and phosphatase inhibitors (Thermo Fisher
Scientific, Inc.). The protein concentration was determined using
the BCA protocol (Shanghai Yeasen Biotechnology Co., Ltd.).
SDS-PAGE loading buffer (Shanghai Yeasen Biotechnology Co., Ltd.)
was added to the protein solution and heated at 100°C for 5 min.
Subsequently, 30–60 µg proteins were separated by SDS-PAGE on 10%
gels, and transferred onto PVDF membranes. The membranes were then
blocked in 10% non-fat milk (BD Biosciences) for 1.5 h at room
temperature to prevent non-specific binding, and were incubated
with primary antibodies targeting Flag (cat. no. F1804; 1:1,000;
mouse; MilliporeSigma), JAM3 (cat. no. bs-11086R; 1:1,000; rabbit;
BIOSS), large tumor suppressor kinase 1 (LATS1; cat. no.
17049-1-AP; 1:1,000; rabbit; Proteintech Group, Inc.),
phosphorylated (p)-LATS1 (Thr1079) (cat. no. 28998-1-AP; 1:5,000;
rabbit; Proteintech Group, Inc.), yes-associated protein 1 (YAP1;
cat. no. 13584-1-AP; 1:5,000; rabbit; Proteintech Group, Inc.),
p-YAP1 (Ser127) (cat. no. 13008S; 1:1,000; rabbit; Cell Signaling
Technology, Inc.) and β-actin (cat. no. HC201-02; 1:2,000; mouse;
TransGen Biotech Co., Ltd.) overnight at 4°C. After primary
antibody incubation, the membranes were washed and subsequently
incubated with appropriate HRP-conjugated secondary antibodies
[anti-rabbit (cat. no. HS101-01; 1:5,000) and anti-mouse (cat. no.
HS201-01; 1:5,000); both from TransGen Biotech Co., Ltd.)] for 2 h
at room temperature, based on the primary antibodies used.
Following secondary antibody incubation, protein bands were
visualized using enhanced chemiluminescence detection reagents
(cat. no. K-12045-D50; Advansta Inc.) and images were captured
using a MiniChemi 610 instrument (SinSage Technology, Co., Ltd.).
Band intensity was semi-quantified by densitometry using ImageJ
1.53k software to assess relative protein levels.
In vivo assay
Animal experiments were conducted in accordance with
the guidelines approved (approval no. DWLL-2024-027) by the
Research Ethics Committee for Animal Experimentation at The First
Hospital, Shanxi Medical University. Six specific pathogen-free
female BALB/c nude mice (age, 4–6 weeks; average weight, 13.15±0.17
g) were obtained from Beijing Vital River Laboratory Animal
Technology Co., Ltd. and were housed under controlled conditions.
The housing environment was maintained at a temperature of 22–24°C
and a relative humidity of 50–60%. Mice had ad libitum
access to food and water and were maintained under a 12-h
light/dark cycle. Following a 7-day acclimation period,
5×106 AMC-HN-8 cells were subcutaneously injected into
both flanks of each mouse. Tumor dimensions were measured twice
daily. Once tumors reached 5×5 mm, si-NC was administered into the
left tumor, and si-JAM3 into the right tumor at the same interval;
both types of siRNA were prepared at a concentration of 0.1 µg/µl.
The siRNA solution was injected directly into the tumor mass at a
volume of 100 µl per injection. This treatment was repeated every
other day for a total of seven injections. After seven injections,
the nude mice were anesthetized with isoflurane at an induction
concentration of 4–5% in oxygen for 1–3 min. Following the
induction phase, the isoflurane concentration was reduced to 1–2%
for an additional 1–2 min to ensure deep anesthesia. Once the loss
of the righting reflex and a lack of response to a toe pinch
confirmed deep anesthesia, cervical dislocation was promptly and
competently performed by trained personnel. The tumors were then
removed and weighed. The excised tumors were processed for further
analysis, including RT-qPCR and IHC. Tumor volume was calculated
using the following formula: Tumor volume=(length ×
width2)/2 (23).
Hematoxylin and eosin (H&E)
staining
Formalin-fixed (10% neutral buffered formalin at
room temperature for 24 h), paraffin-embedded 4-µm tissue sections
were deparaffinized in xylene and rehydrated through a graded
series of alcohol. The sections were then stained with hematoxylin
for 1 min, washed in water, differentiated in 1% acid alcohol for
30 sec. After a rinse in water, the sections were stained with
eosin for 1 min, dehydrated in increasing concentrations of
alcohol, cleared in xylene and mounted with a resinous medium. The
stained slides were examined under a light microscope to evaluate
tissue morphology, ensuring clarity of nuclear and cytoplasmic
details.
IHC staining
Formalin-fixed (10% neutral buffered formalin at
room temperature for 24 h), paraffin-embedded 4-µm tissue sections
were mounted on charged slides. Sections were deparaffinized in
xylene, rehydrated through a graded series of alcohol, and
submerged in 10 mM citrate buffer (pH 6.0) for antigen retrieval
using a pressure cooker for 2 min. After cooling and rinsing in
phosphate-buffered saline (PBS), endogenous peroxidase was quenched
with 3% hydrogen peroxide for 10 min at room temperature. Sections
were then blocked with 5% bovine serum albumin (BSA; Beijing
Solarbio Science & Technology Co., Ltd.) for 20 min at room
temperature to reduce non-specific binding, then incubated
overnight at 4°C with primary antibodies against Ki-67 (cat. no.
RMA-0731; Fuzhou Maixin Biotechnology Development Co., Ltd.), JAM3
(cat. no. bs-11086R; 1:200; rabbit; BIOSS), YAP1 (cat. no.
13584-1-AP; 1:500; rabbit; Proteintech Group, Inc.), E-Cadherin
(cat. no. 20874-1-AP; 1:2,000; rabbit; Proteintech Group, Inc.),
N-Cadherin (cat. no. 22018-1-AP; 1:2,000, rabbit; Proteintech
Group, Inc.) and Vimentin (cat. no. 5741S; 1:600; rabbit; Cell
Signaling Technology, Inc.). Following primary antibody incubation,
slides were washed with PBS and incubated with a biotinylated
secondary antibody (cat. no. PV-6000; Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd.) for 20 min at 37°C. Detection used an
avidin-biotin complex method with diaminobenzidine (cat. no.
ZLI-9019; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.) as the
chromogen, and sections were then counterstained with hematoxylin
for 1 min, differentiated in 1% acid alcohol for 30 sec and rinsed
in 0.1% ammonia water for another 30 sec at room temperature.
Slides were then dehydrated, cleared and mounted. Staining
intensity and cell positivity were independently evaluated by two
pathologists under a light microscope, with results semi-quantified
to assess protein expression levels. Images were acquired using the
PANORAMIC SCAN II (3DHISTECH, Ltd.), the results were analyzed
using CaseViewer version 2.3 and the expression levels were
recorded as H-Score (24).
Immunofluorescence staining
Immunofluorescence staining was performed on LSCC
cells cultured on glass coverslips. Cells were fixed and
permeabilized with cool methanol at −20°C for 10 min and blocked
with 5% BSA for 1 h at room temperature to prevent non-specific
binding. The cells were then incubated overnight at 4°C with
primary antibodies targeting YAP1 (cat. no. 13584-1-AP; 1:100;
rabbit; Proteintech Group, Inc.) diluted in the blocking solution.
After washing with PBS, cells were incubated with a CY3-conjugated
secondary antibody (cat. no. BA1032; 1:250; goat; Wuhan Boster
Biological Technology, Ltd.) for 1 h at room temperature in the
dark. Nuclei were stained with DAPI (Wuhan Boster Biological
Technology, Ltd.) for 2 min at room temperature, and coverslips
were mounted with an anti-fade mounting medium to preserve
fluorescence. Fluorescence microscopy (Leica Microsystems GmbH) was
used to examine and capture images of the cells, focusing on
protein expression and localization. Analysis of fluorescence
intensity and subcellular distribution was performed using ImageJ
1.53k to semi-quantify expression levels and patterns.
Bioinformatics analysis
The present study employed comprehensive
bioinformatics analyses to investigate gene expression and
methylation patterns associated with LSCC and HNSCC. For gene
expression, sequencing data from GSE216664, and microarray data
from GSE59102 (25) and GSE51985
(26) specific to laryngeal tissues
were analyzed using GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/). To
investigate the association between JAM3 expression and its
methylation status, microarray and corresponding methylation data
were examined from GSE33205 (27)
and GSE33202 (27) for HNSCC
through GEO2R. This relationship was further validated by analyzing
data from The Cancer Genome Atlas (TCGA; http://www.cancer.gov/tcga) (28) using the DNA Methylation Interactive
Visualization Database (DNMIVD; http://119.3.41.228/dnmivd/) (29). The methylation status of each CpG
site was quantified by calculating the β value, defined as the
ratio of the fluorescent signal from the methylated allele to the
sum of the signals from both the methylated and unmethylated
alleles. The β value ranges from 0 (completely unmethylated) to 1
(completely methylated), providing a continuous measure of DNA
methylation levels (30). Protein
expression data were analyzed using The University of Alabama at
Birmingham Cancer data analysis Portal (UALCAN; http://ualcan.path.uab.edu/index.html),
with data from the Clinical Proteomic Tumor Analysis Consortium
(CPTAC) and the International Cancer Proteogenome Consortium (ICPC)
(31). The protein expression
values were normalized to Z-values, which represent standard
deviations from the median across samples for the given cancer type
(31). Gene expression associations
were assessed via The cBio Cancer Genomics Portal (https://www.cbioportal.org/) (32) and survival analysis focusing on CpG
sites of the JAM3 promoter was conducted through MethSurv
(https://biit.cs.ut.ee/methsurv/)
(33). These tools facilitated a
detailed examination of the genetic and epigenetic factors
contributing to tumor biology in LSCC and HNSCC.
Statistical analysis
All in vitro experiments were repeated three
times. Differences between two groups were determined using an
unpaired two-tailed Student's t-test or paired two-tailed Student's
t-test. For comparisons involving more than two groups, one-way
ANOVA followed by Tukey's Honest Significant Difference test were
utilized. Pearson and Spearman correlation analyses were employed
to evaluate gene co-expression and the relationship between gene
expression and DNA methylation across the samples. Survival
analyses were conducted using Cox proportional-hazards models, as
performed by the MethSurv web tool, with model fit assessed using
log likelihood-ratio and Wald tests (33). Differences in survival rates were
visualized using Kaplan-Meier plots, with hazard ratio and
log-likelihood ratio test P-value included in the plots (33). All data were analyzed using GraphPad
Prism 7.0 (Dotmatics). P<0.05 was considered to indicate a
statistically significant difference.
Results
Downregulation of JAM3 in LSCC
Analysis of Gene Expression Omnibus (GEO) datasets,
including GSE216664, GSE59102 and GSE51985, demonstrated a
significant reduction in JAM3 mRNA expression in LSCC tumor
tissues compared with that in non-tumor counterparts (Fig. 1A). For a broader understanding of
JAM3 expression, further investigation into its protein levels
using data from the CPTAC and the ICPC accessed through UALCAN
revealed JAM3 was expressed at lower levels in HNSCC tumor tissues
compared with those in normal tissues, although the difference was
not statistically significant (Fig.
S1). Notably, the difference in JAM3 protein expression was
more pronounced and statistically significant in high-grade tumors
when compared with both normal and lower-grade tumors (Fig. 1B).
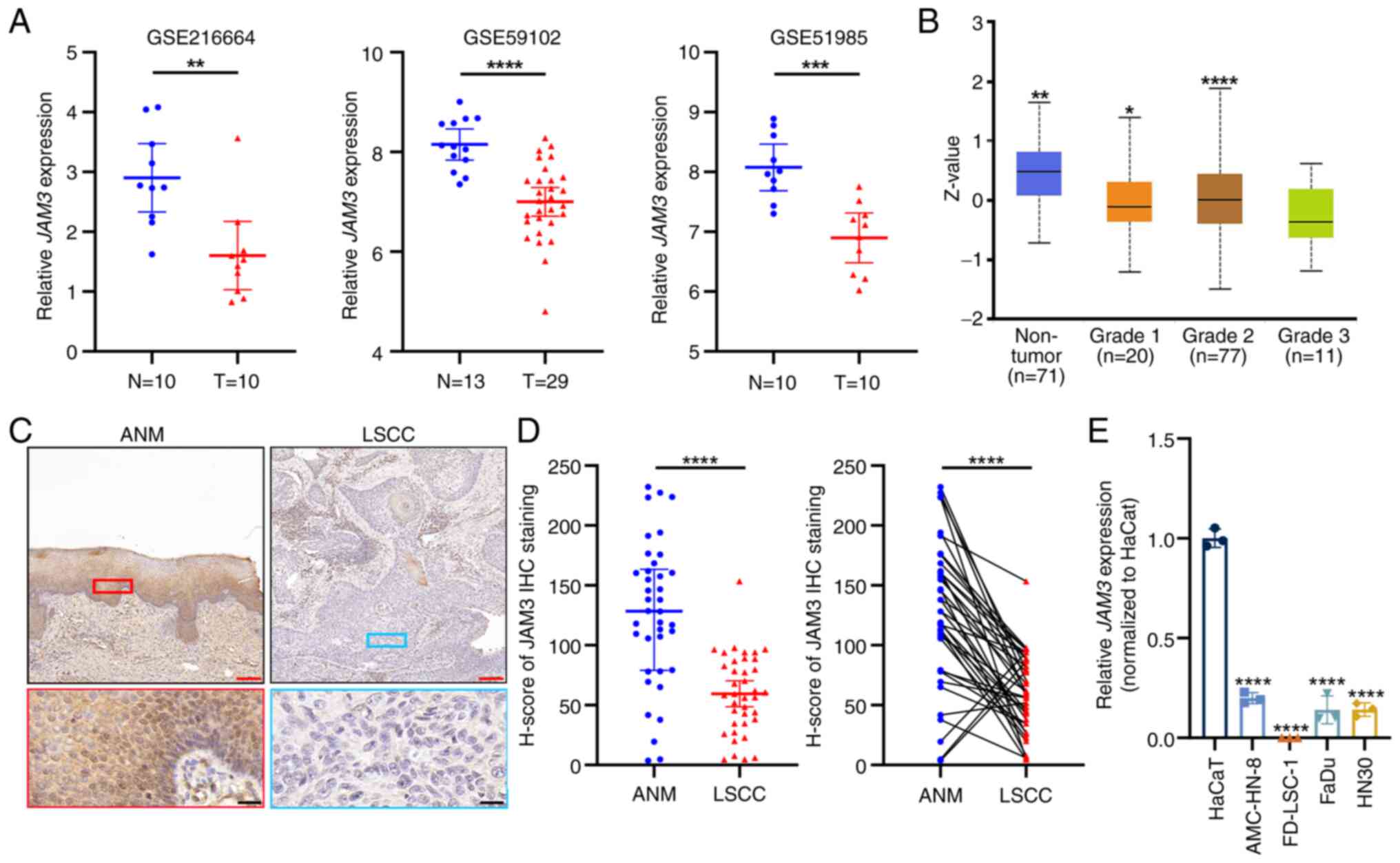 | Figure 1.JAM3 is downregulated in LSCC
tissues and samples at both mRNA and protein levels. (A) GSE216664,
GSE59102 and GSE51985 were used to analyze the different expression
levels of JAM3 between N and LSCC T tissues. For GSE216664
and GSE51985, N and T tissues were from the same LSCC patients; for
GSE59102, N tissues were ANM tissues from different patients with
LSCC. Data are presented as the mean with 95% CI indicated by error
bars. **P<0.01, ***P<0.001, ****P<0.0001. (B) Protein
levels of JAM3 in the Clinical Proteomic Tumor Analysis Consortium
and the International Cancer Proteogenome Consortium datasets among
different grades of HNSCC tissues. Data are presented as the mean
and range. *P<0.05, **P<0.01, ****P<0.0001 vs. Grade 3.
(C) Representative images of JAM3 IHC staining of LSCC tissues and
paired ANM tissues. Red scale bar, 150 µm; Black scale bar, 20 µm.
(D) IHC results of clinical tissues were analyzed by paired
two-tailed Student's t-test. Data are presented as the mean with
95% CI. ****P<0.0001. (E) mRNA expression levels of JAM3
in HaCaT, AMC-HN-8, FD-LSC-1, FaDu and HN30 cell lines. Data are
presented as the mean ± SD of three independent experiments.
****P<0.0001 vs. HaCaT. ANM, adjacent normal mucosa; IHC,
immunohistochemistry; JAM3, junctional adhesion molecule 3; LSCC,
laryngeal squamous cell carcinoma; N, non-tumor; T, tumor. |
To validate these findings at the protein level, IHC
was conducted on LSCC tissues and ANM tissues. The staining
highlighted the diminished expression of JAM3 in the tumor tissues
(Fig. 1C), with semi-quantification
supported by statistical analysis using paired two-tailed Student's
t-tests (Fig. 1D). Additionally,
further analysis of IHC H-scores did not reveal any significant
differences in JAM3 expression when comparing tumor samples based
on patient age (>60 years), presence of lymph node metastasis,
tumor grade or differentiation level (Fig. S2). These results suggested that
while JAM3 expression was generally lower in tumor tissues, its
levels were not significantly associated with these clinical
parameters in patients with LSCC; this may be due to the limited
number of clinical specimens. Another comparative mRNA expression
analysis in the HaCaT normal epidermal cell line, LSCC cell lines
(AMC-HN-8 and FD-LSC-1) and HNSCC cell lines (FaDu and HN30)
consistently showed a decrease in JAM3 expression in tumor
cells compared with that in HaCaT cells (Fig. 1E). In summary, these findings
indicated a consistent downregulation of JAM3 at both mRNA
and protein levels in LSCC, highlighting its potential role in the
pathology of this cancer type across diverse datasets and sample
types.
Aberrant hypermethylation of the JAM3
promoter is associated with reduced expression and a poor prognosis
in LSCC
To investigate the underlying causes of JAM3
downregulation in LSCC, the present study extended the analysis to
include broader TCGA and GEO datasets of HNSCC tissues. This
approach allowed for the use of larger datasets to strengthen the
understanding of epigenetic influences across related types of
cancer. Analysis of GEO datasets GSE33202 and GSE33205 revealed
that lower JAM3 expression in tumor tissues was
significantly negatively correlated with higher methylation levels
(Spearman's ρ=−0.31; Pearson's r=−0.34; Fig. 2A). This trend was corroborated by
data from TCGA, where similar negative correlations between
JAM3 expression and methylation levels were noted
(Spearman's ρ=−0.34; Pearson's r=−0.33), as analyzed using the
DNMIVD online tool (Fig. 2B).
Furthermore, prognosis analysis using MethSurv identified four
hyper-methylated CpG sites within the JAM3 gene promoter
(cg03640071, cg06250693, cg06726804 and cg27073337), each
significantly associated with poorer survival outcomes in HNSCC
(Fig. 2C). These findings suggested
that epigenetic mechanisms contributing to JAM3
downregulation in HNSCC could be relevant to LSCC, given the shared
pathophysiological characteristics of these types of cancer.
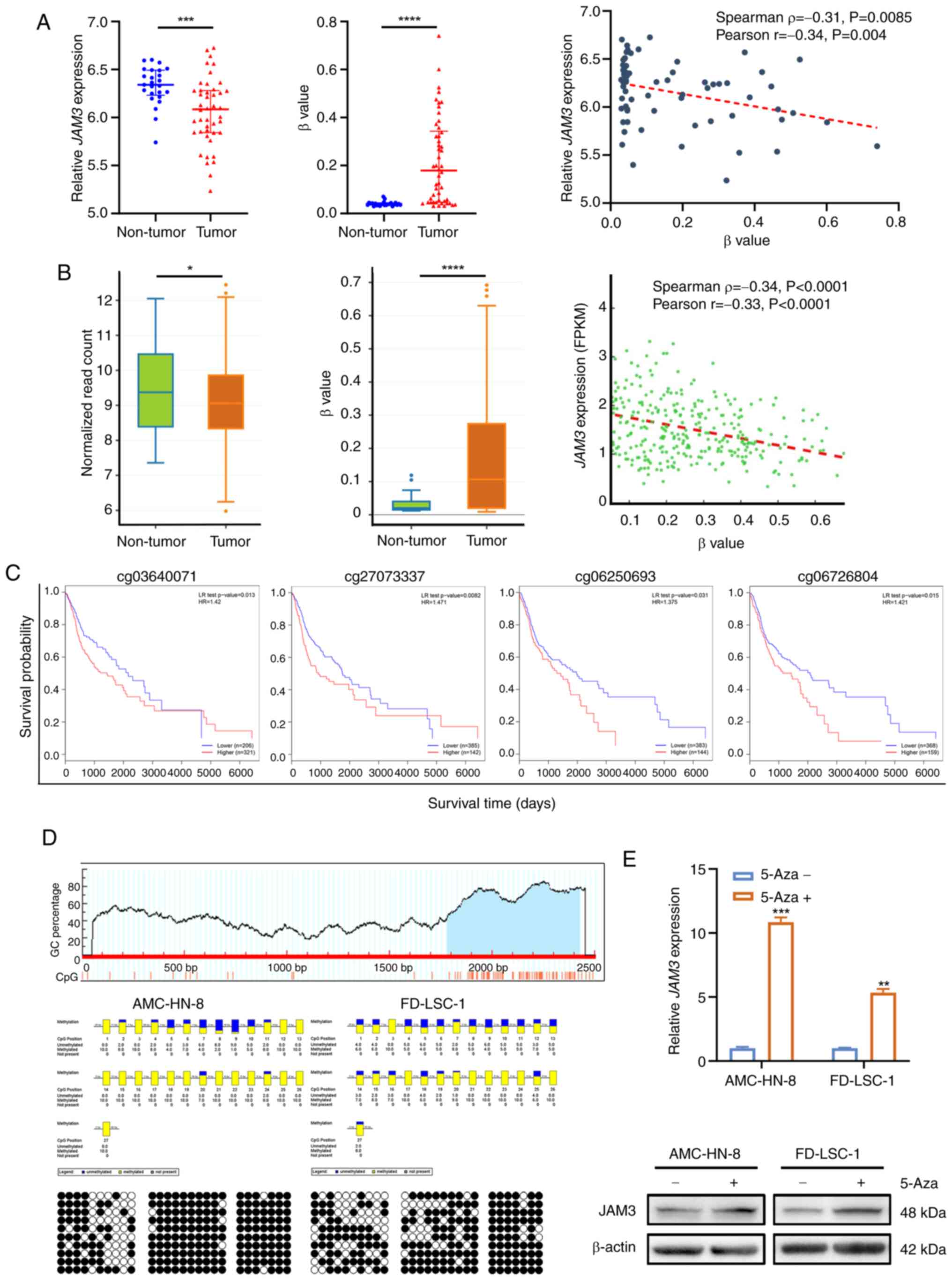 | Figure 2.Aberrant hypermethylation in the
JAM3 promoter is related to low expression of JAM3.
(A) Analysis of JAM3 expression and methylation levels, as
well as their correlation using the Gene Expression Omnibus
datasets GSE33202 and GSE33205. β value indicates the methylation
level of the CpG site. Data are presented as the mean with 95% CI.
***P<0.001, ****P<0.0001. (B) Analysis of JAM3
expression and methylation levels, as well as their correlation in
The Cancer Genome Atlas HNSCC samples. β value indicates the
methylation level of the CpG site. Data are presented as the mean
and range. *P<0.05, ****P<0.0001. (C) Kaplan-Meier plots,
generated using the MethSurv webtool highlighted the relationship
between methylation levels at certain JAM3 sites and overall
survival rates in patients with HNSCC. (D) CpG island (blue area)
predicted by MethPrimer tool were detected by bisulfite sequencing
PCR in AMC-HN-8 and FD-LSC-1 cell lines. Each row represents an
individual cloned allele. Black circles show methylated CpG sites
and white circles show unmethylated CpG sites. The percentage
methylation rate of each CpG site is shown by the blue-yellow
columns; blue indicates unmethylated and yellow indicates
methylated sites. (E) Reverse transcription-quantitative PCR and
western blotting showing restored expression of JAM3 in both
AMC-HN-8 and FD-LSC-1 cells treated with 5-Aza (5 µM) for 72 h.
Data are presented as the mean ± SD of three independent
experiments. **P<0.01, ***P<0.001 vs. the 5-Aza-group. 5-Aza,
5-Aza-2′-deoxycytidine; HNSCC, head and neck squamous cell
carcinoma; JAM3, junctional adhesion molecule 3; FPKM, fragments
per kilobase million; LR test, log-likelihood ratio test; HR,
hazard ratio. |
Further validation specific to LSCC involved
MethPrimer (21) analysis, which
identified a CpG-rich region (among −216 to +425 bp from
transcription start site) within the JAM3 promoter.
Sequencing of 10 clones from the T1 cloning vector containing the
CpG island region of AMC-HN-8 and FD-LSC-1 cell lines demonstrated
extensive methylation. In the AMC-HN-8 cell line, 16 of the 27
cytosine sites showed complete methylation, with others showing
partial methylation; the FD-LSC-1 cell line exhibited similar
patterns (Fig. 2D). Furthermore,
treatment with 5-Aza, a known DNA promoter methylation reverser,
restored JAM3 expression at both the mRNA and protein levels
in both LSCC cell lines (Fig.
2E).
Collectively, these results indicated that low
JAM3 expression may originate from aberrant hypermethylation
of the JAM3 promoter, impacting survival outcomes in HNSCC.
By integrating findings from broader HNSCC analyses and the similar
modulation of expression by methylation of JAM3 promoter in
LSCC cell lines, the potential of JAM3 methylation patterns
to inform on LSCC was identified, reinforcing its significance as a
prognostic biomarker and a target for therapeutic
interventions.
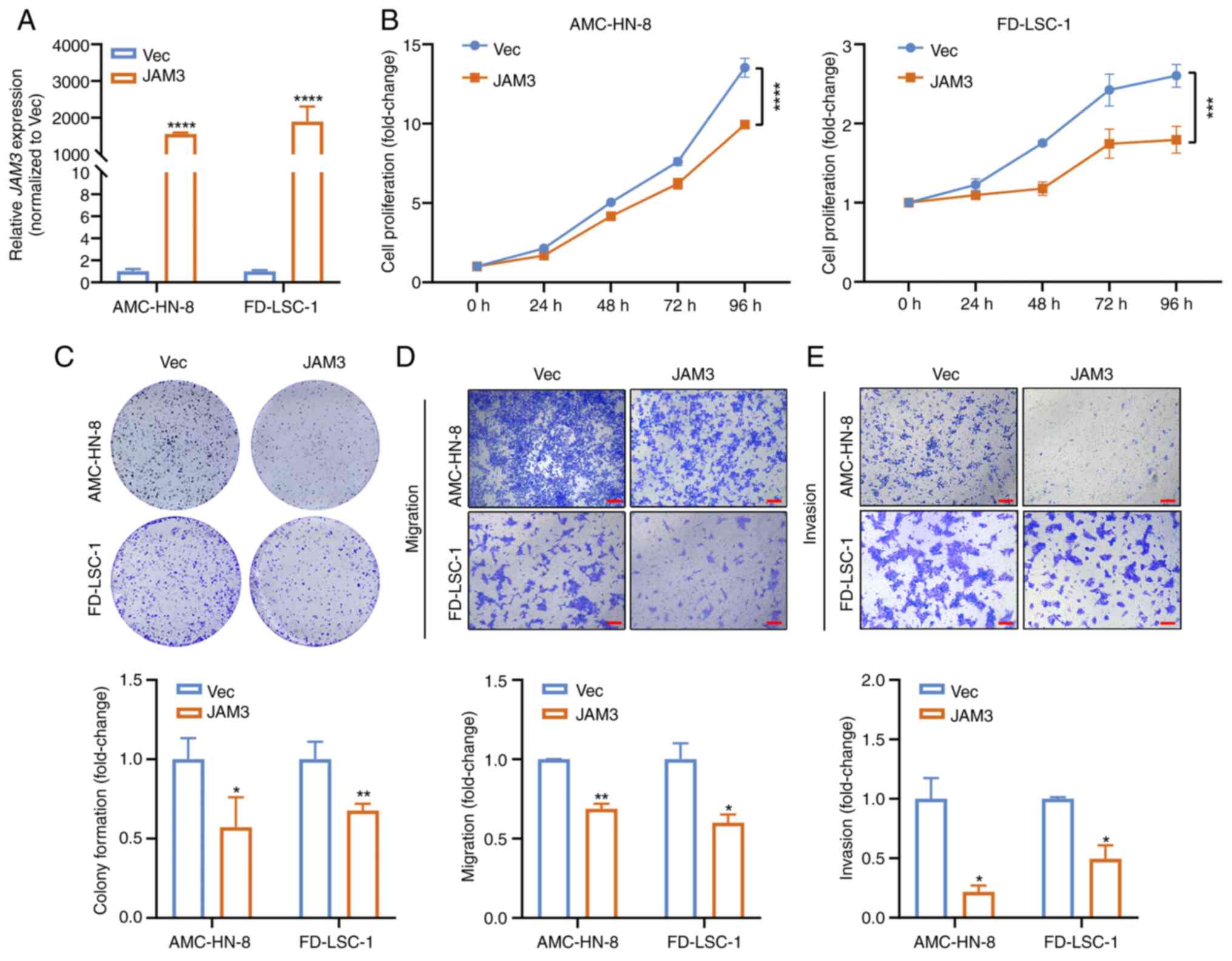
Impact of JAM3 overexpression and
knockdown on LSCC cell behavior
To elucidate the function of JAM3 in the
tumorigenesis of LSCC, experiments were conducted to assess its
impact on cancer cell behavior. AMC-HN-8 and FD-LSC-1 cells were
transfected with p3×Flag-CMV-10-JAM3 to induce overexpression of
the gene, or with p3×Flag-CMV-10 empty vector as a control.
JAM3 overexpression was confirmed by RT-qPCR (Fig. 3A). Cells overexpressing JAM3
exhibited a significant reduction in cell proliferation compared
with those transfected with the empty vector, as demonstrated using
a CCK8 assay (Fig. 3B). This
suppressive effect on cell proliferation was further supported by
results from the colony formation assay (Fig. 3C). Additionally,
JAM3-overexpressing cells displayed decreased migration and
invasion compared with that in the control group, as determined
using Transwell assays (Fig. 3D).
These findings indicated that JAM3 overexpression
significantly inhibited the proliferation, colony formation,
migration and invasion of LSCC cells, suggesting its potential role
as a TSG in LSCC.
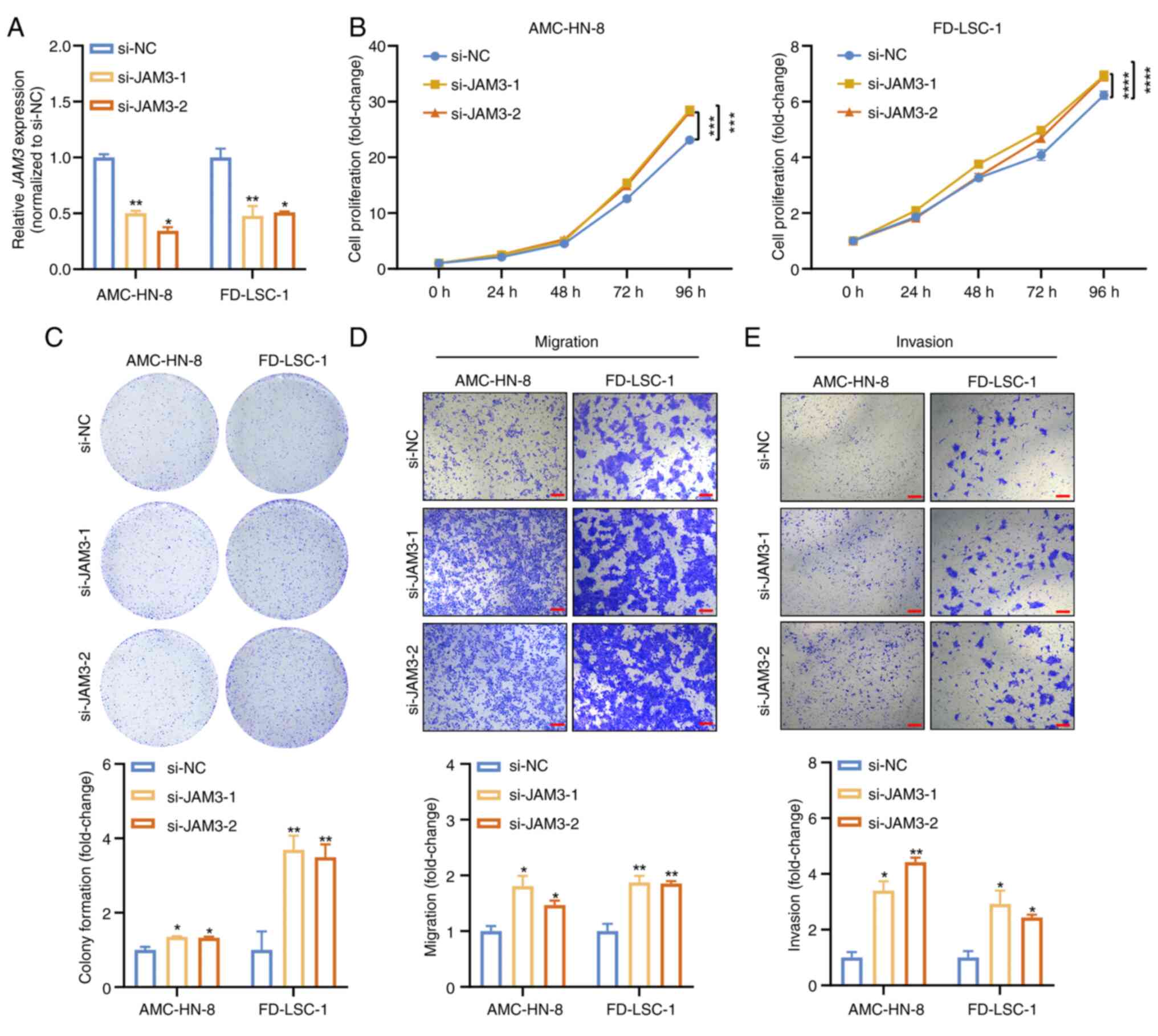
Conversely, knockdown experiments using siRNAs
targeting JAM3 (si-JAM3-1 and si-JAM3-2) highlighted its
critical regulatory role. Efficient knockdown was achieved, as
verified by RT-qPCR (Fig. 4A).
Cells with reduced JAM3 expression displayed enhanced
proliferative, migratory and invasive capabilities, as confirmed by
CCK8, colony formation and Transwell assays (Fig. 4B-E). These results suggested that
reduced JAM3 expression significantly augmented the
proliferation, migration and invasion of LSCC cells, further
establishing JAM3 as a pivotal tumor suppressor in LSCC.
Collectively, the present data indicated that
JAM3 served as a potent modulator of tumorigenic processes
in LSCC, where its expression levels directly influenced tumor cell
behavior. The dual experimental approach of overexpression and
knockdown elucidated the suppressive impact of JAM3 on LSCC
progression, offering valuable insights for potential therapeutic
strategies.
JAM3 modulates LSCC tumorigenesis via
the Hippo pathway
The investigation into the involvement of the Hippo
pathway in HNSCC was prompted by initial analyses conducted on the
cBio Cancer Genomics Portal platform, which revealed a weak
positive correlation between JAM3 and LATS1, a key
component of the Hippo pathway, in HNSCC (Fig. S3). These findings suggested that
JAM3 may interact with or influence the Hippo pathway, a
critical regulator of cell proliferation and apoptosis (34). To elucidate the molecular mechanisms
by which JAM3 influences LSCC tumorigenesis, the present
study further investigated its impact on key proteins within the
Hippo signaling pathway. This approach aimed to uncover how changes
in JAM3 expression affect pathway dynamics and,
consequently, tumor behavior.
Western blotting was performed on AMC-HN-8 and
FD-LSC-1 cells with either JAM3 overexpression or knockdown.
The protein levels of Flag, JAM3, p-LATS1 (Thr1079), total LATS1,
p-YAP1 (Ser127) and total YAP1 were examined, with β-actin serving
as a loading control. In cells with JAM3 overexpression,
there was a noticeable increase in total LATS1, p-LATS1 and p-YAP1,
indicating activation of the Hippo pathway (Fig. 5A). Conversely, JAM3 knockdown
resulted in reduced total LATS1, p-LATS1 and p-YAP1 expression,
suggesting that YAP1 activity and downstream signaling were
inhibited (Fig. 5A).
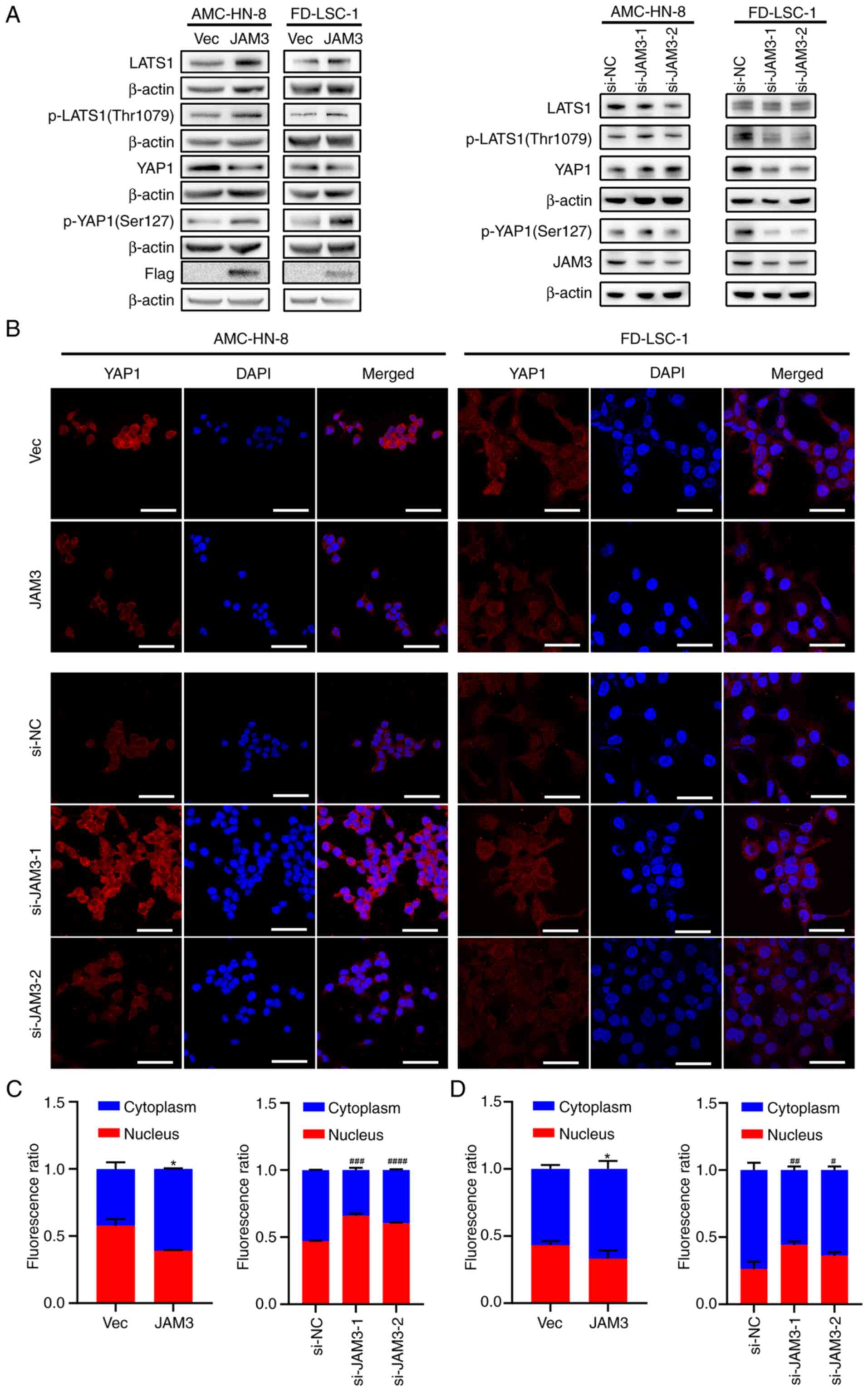 | Figure 5.JAM3 mediates laryngeal squamous cell
carcinoma tumorigenesis through the Hippo pathway. (A) Protein
levels of Flag, JAM3, p-LATS1 (Thr1079), LATS1, p-YAP1 (Ser127),
YAP1 and β-actin in AMC-HN-8 and FD-LSC-1 cells with overexpression
or knockdown of JAM3 were detected by western blotting. (B)
Expression of YAP1 in AMC-HN-8 and FD-LSC-1 cells after
transfection with the JAM3 overexpression plasmid or si-JAM3
were detected by confocal microscopy. Scale bar, 50 µm.
Fluorescence ratio of YAP1 in the cytoplasm and nucleus of (C)
AMC-HN-8 and (D) FD-LSC-1 cells was calculated by ImageJ and
analyzed by two-tailed Student's t-test or one-way ANOVA. Data are
presented as the mean ± SD of three independent experiments.
*P<0.05 vs. Vec; #P<0.05, ##P<0.01,
###P<0.001, ####P<0.0001 vs. si-NC
group. JAM3, junctional adhesion molecule 3; LATS1, large tumor
suppressor kinase 1; NC, negative control; p-, phosphorylated; si,
small interfering; Vec, p3×Flag-CMV-10 empty vector; YAP1,
yes-associated protein 1. |
To further elucidate the intracellular dynamics of
YAP1 following genetic manipulation of JAM3,
immunofluorescence assays were performed. Using confocal microscopy
(Fig. 5B), the subcellular
localization of YAP1 was observed in transfected cells.
Semi-quantitative analysis conducted with ImageJ 1.53k software
revealed a significant decrease in the nuclear accumulation of YAP1
in cells overexpressing JAM3, suggesting Hippo pathway
activation (Fig. 5C and D).
Conversely, cells with JAM3 knockdown displayed predominant
YAP1 localization in the nucleus, indicating suppression of the
pathway (Fig. 5C and D).
These findings underscored the role of JAM3
as a regulatory element in the Hippo pathway, influencing both the
phosphorylation status and the subcellular localization of YAP1 in
LSCC cells. These results suggested that JAM3 may serve as a
crucial modulator of cell proliferation and motility through its
effects on this signaling pathway.
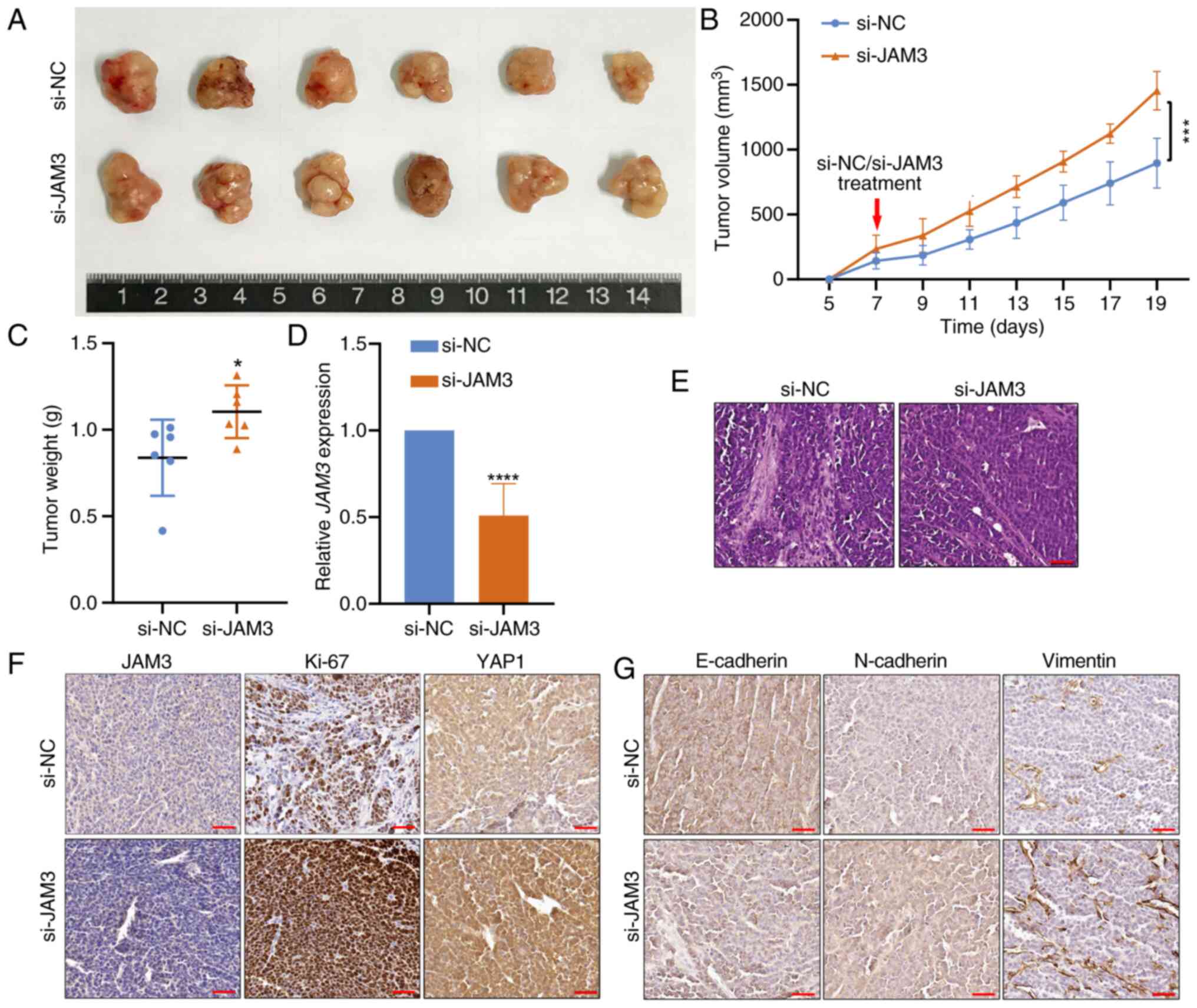
JAM3 knockdown enhances the
tumorigenicity of LSCC cells in vivo
To evaluate the tumor suppressor function of
JAM3 in an in vivo model, experiments were conducted
using nude mice. AMC-HN-8 cells were subcutaneously injected into
the mice, after which, si-JAM3 or si-NC was injected to observe the
effects on tumor growth and aggressiveness.
In these experiments, mice injected with si-JAM3
cells developed larger tumors than those in the si-NC group
(Fig. 6A). This finding was
confirmed by the significant increase in tumor volume and weight in
the si-JAM3 group, indicating a marked enhancement in tumorigenic
capacity associated with the suppression of JAM3 (Fig. 6B and C).
Further molecular analyses confirmed the knockdown
efficacy, with the RT-qPCR results showing significantly reduced
JAM3 expression in the tumors derived from the si-JAM3 group
(Fig. 6D); this result suggested
effective gene silencing was achieved. H&E staining
demonstrated structural changes in the xenograft tumors, including
an increase in LSCC tumor cell density after JAM3 knockdown
(Fig. 6E). Further analysis through
IHC clarified the cellular and molecular impacts of JAM3
knockdown. A reduction in JAM3 expression was detected, accompanied
by elevated levels of YAP1 and the proliferation marker Ki-67 in
the xenograft tumors (Fig. 6F).
Additionally, changes in epithelial-mesenchymal transition (EMT)
markers were evident; N-cadherin and Vimentin levels were higher,
whereas E-cadherin expression was reduced in xenograft tumors from
the si-JAM3 group (Fig. 6G). These
findings collectively supported the hypothesis that JAM3
silencing may promote the growth and aggressiveness of LSCC cells
in vivo by disrupting Hippo pathway signaling.
Discussion
The present findings demonstrated that JAM3
expression was suppressed through the hypermethylation of its
promoter, and this was revealed to be associated with poorer
patient outcomes. Moreover, the overexpression of JAM3
inhibited tumor-related behaviors by activating the Hippo pathway.
Conversely, JAM3 silencing promoted these oncogenic
behaviors in vitro and in vivo. The present study
provides deeper insights into the understanding of the role of
JAM3 as a TSG in the development and progression of LSCC,
and underscores its potential as a diagnostic and prognostic
biomarker. Overall, the current study aimed to elucidate the
mechanisms by which JAM3 modulates tumor dynamics, offering
promising directions for future therapeutic strategies.
Over the past 30 years, despite a decrease in
overall incidence, the survival rate of LSCC has decreased from 66
to 61% in the United States (35).
This concerning trend is largely due to the fact that the majority
of patients are diagnosed at a late stage (3) and underscores the need to improve the
understanding of the molecular mechanisms underlying LSCC
tumorigenesis. This improved understanding may enable early
diagnosis and increase the accuracy of treatment, improving the
quality of life of patients.
JAM proteins, part of the immunoglobulin (Ig)
superfamily, contain two extracellular Ig-like domains and one
intracellular PDZ-binding motif (36), which are integral to cell-cell
contact and migration (37,38), processes crucial for early tumor
metastasis (39). Research has
identified JAM3 as a TSG, often silenced by methylation in
cancer, such as esophageal and colorectal cancer (17). JAM3 has also been identified
as a potential DNA methylation marker in cholangiocarcinoma and
cervical lesions (39,40). Although JAM3 has been
reported and characterized in several tumors, its role in LSCC
remains unclear. The present study observed a pronounced
downregulation of JAM3 in LSCC tumor tissues, both at the
mRNA and protein levels, which was corroborated by public dataset
analyses and IHC analysis of LSCC clinical specimens. Notably,
according to public dataset analyses, lower JAM3 protein levels
were observed in higher-grade HNSCC tumors compared with those in
normal and lower-grade tumors, suggesting a potential link between
JAM3 downregulation and poor tissue differentiation. However, in
the LSCC clinical specimens assessed, IHC confirmed a general
downregulation of JAM3 in tumor tissues compared with that in
normal tissues, but did not show a significant difference across
different tumor grades. This observation underscores the need for
further exploration of the role of JAM3 expression across various
stages of LSCC progression, especially because of the limited
sample size of the present study. Furthermore, a negative
correlation between JAM3 mRNA expression and promoter
methylation indicated that high methylation levels may contribute
to JAM3 depletion in LSCC. This was supported by extensive
methylation observed in BSP data and the successful restoration of
JAM3 expression following treatment with the demethylating
agent 5-Aza. Additionally, higher methylation at several CpG sites
within the JAM3 promoter was associated with poorer patient
outcomes, underscoring the importance of monitoring JAM3
methylation as a potential early diagnostic and prognostic
biomarker in HNSCC. These findings also suggested that JAM3
methylation may act as a biomarker in LSCC, which deserves further
exploration.
To deeply understand the function of JAM3 in
LSCC, the present study conducted comprehensive in vivo and
in vitro experiments. The findings indicated that
JAM3 overexpression inhibited LSCC cell proliferation,
migration and invasion, whereas its knockdown promoted these
oncogenic behaviors. Specifically, in vivo results
demonstrated enhanced proliferation, invasion and migration of
AMC-HN-8 cells with JAM3 knockdown, as evidenced by
increased Ki-67, N-cadherin and Vimentin staining, as well as
decreased E-cadherin staining in xenograft tumors. These results
collectively suggested that JAM3 may function as a TSG in
LSCC.
The Hippo pathway is critical in cancer development,
with previous studies highlighting the amplification of YAP1
and TAZ in 14% of HNSCC cases, and their association with
adverse clinical outcomes, including tumor recurrence and
resistance to therapy (41,42). In this context, the present study
revealed that JAM3 was positively correlated with
LATS1, a key regulator of YAP1/TAZ, which can promote the
phosphorylation of the downstream protein YAP1, and reduce the
activation of its target genes related to proliferation, EMT and
other hallmarks of cancer (43,44).
This association was important because it implies that JAM3
may exert tumor-suppressive effects through modulation of the Hippo
pathway. Further molecular investigations revealed that JAM3
overexpression in LSCC cells increased LATS1 levels and its
phosphorylation, and enhanced phosphorylation of YAP1, indicating
that the Hippo pathway was activated. JAM-A, a member of the JAM
family, has been shown to activate the Hippo pathway by sensing
cell-cell contact and promoting LATS1 activation (45). JAM3 likely enhances these processes
by improving cell-cell adhesion and supporting the role of JAM-A in
organizing Hippo pathway components. By facilitating the activity
of JAM-A, JAM3 overexpression could stabilize LATS1 expression and
enhance its phosphorylation, further promoting Hippo pathway
activation. Conversely, JAM3 knockdown led to reduced
phosphorylation of these proteins and increased nuclear
accumulation of YAP1, suggesting suppression of the Hippo pathway.
IHC analysis in mouse models reinforced these findings, showing
elevated YAP1 expression following JAM3 silencing, which
aligned with decreased pathway activity. This mechanism is
supported by similar observations in breast cancer research, where
JAM3 depletion has been shown to lead to increased YAP/TAZ
nuclear translocation and activation (46), highlighting a potential universal
role for JAM3 in regulating the Hippo pathway across various
types of cancer. Despite the significant findings, the present
study has some limitations, particularly the lack of direct
exploration into whether JAM3 expression influences tumor
differentiation and its potential as a methylation biomarker in
LSCC. Further research in this area is essential, not only to
improve understanding of the role of JAM3, but also to
establish its potential as a prognostic biomarker in LSCC. This
would require more detailed mechanistic studies to elucidate the
specific pathways through which JAM3 exerts its effects.
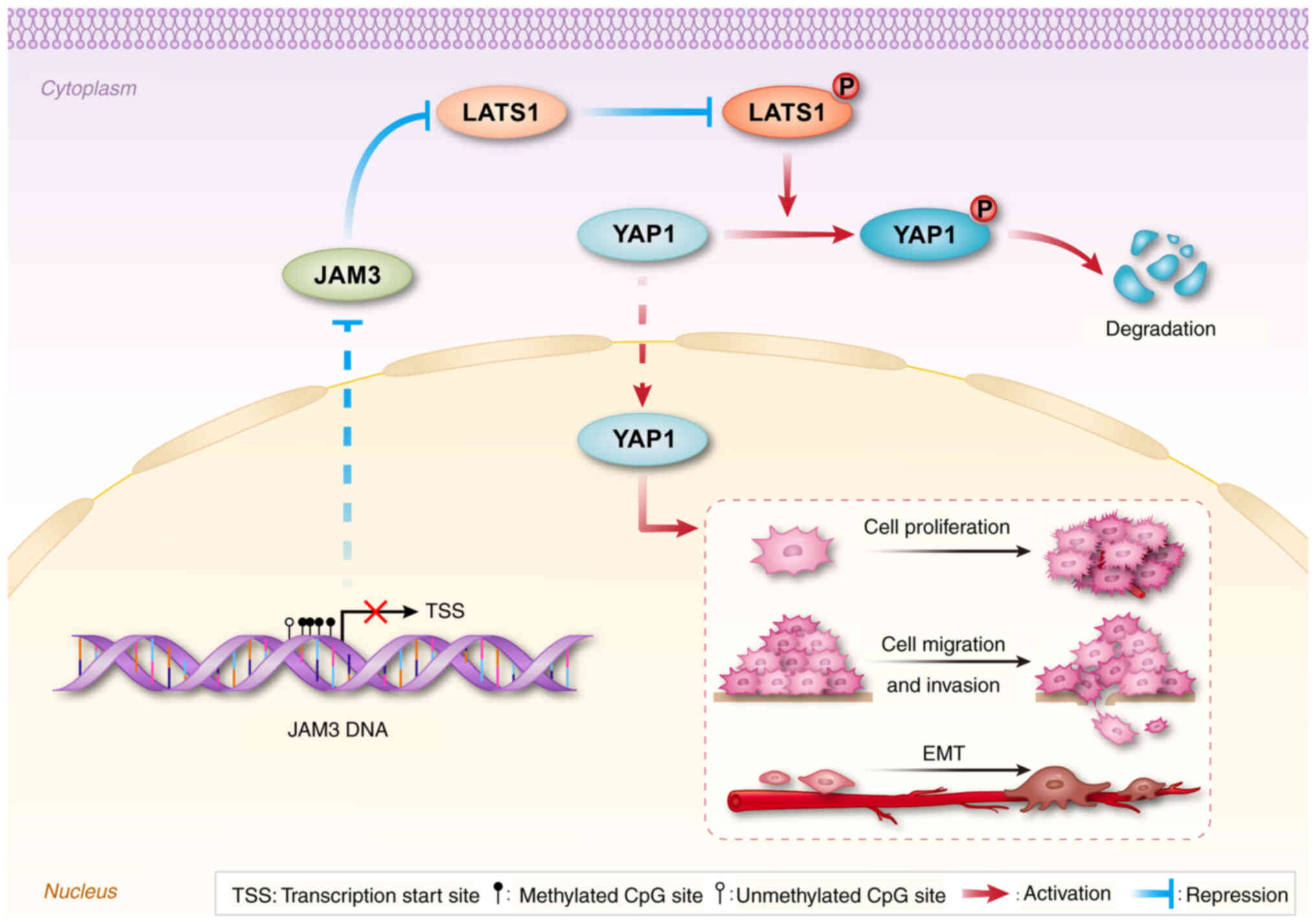
In conclusion, the present study substantiated the
role of JAM3 in the pathogenesis of LSCC. Aberrant
hypermethylation of the JAM3 promoter was demonstrated to be
associated with decreased JAM3 expression and poorer
clinical outcomes in patients with LSCC. The present findings
suggested that JAM3 may function as a TSG, inhibiting tumor
growth and progression, potentially through its regulatory effects
on the Hippo pathway in LSCC (Fig.
7). These interactions highlight JAM3 not only as a key player
in tumor dynamics but also as a promising prognostic biomarker for
LSCC. Further investigations into the mechanisms underlying the
effects of JAM3 could provide deeper insights into its
tumor-suppressive activities and pave the way for novel therapeutic
approaches aimed at enhancing JAM3 expression to mitigate
LSCC progression. This pioneering study on the role of JAM3
in LSCC expands possibilities for developing targeted treatments,
which could significantly improve prognosis and patient
outcomes.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors extend their gratitude to Miss Yujia
Guo (Shanxi Key Laboratory of Otorhinolaryngology Head and Neck
Cancer, The First Hospital, Shanxi Medical University) for her
contributions to the schematic diagram in Fig. 7. The authors would also like to
thank Professor Tao Bai (Department of Pathology, The First
Hospital, Shanxi Medical University) for providing the LSCC samples
essential for this study.
Funding
This study was funded by the Research Project of The First
Hospital of Shanxi Medical University (136 Special Projects; grant
no. Y2022136029).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YJ, CZ and HH contributed to the conceptualization
of the study. YJ designed and performed most of the experiments,
with assistance from JL, JS, XW, LZ, YL and XG. YJ, JL and JS
sorted and analyzed the data. YJ, XW, LZ and YL contributed to the
animal experiments. CZ and HH designed the experiments and
supervised the study. YJ wrote the manuscript, and HH reviewed and
edited it. YJ and HH confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The present study involving clinical samples
received ethics approval (approval no. KYLL-2023-180) from the
Ethics Committee of The First Hospital, Shanxi Medical University
and was conducted in strict accordance with the committee's
guidelines. Due to the retrospective nature of the study and the
use of archived samples, the requirement for informed consent was
waived by the Ethics Committee, in line with The Declaration of
Helsinki. All patient data were thoroughly anonymized to ensure
confidentiality. Animal experiments (approval no. DWLL-2024-027)
were conducted according to the Health Guide for the Care and Use
of Laboratory Animals and were approved by the Research Ethics
Committee for Animal Experimentation at The First Hospital, Shanxi
Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chow LQM: Head and neck cancer. N Engl J
Med. 382:60–72. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnson DE, Burtness B, Leemans CR, Lui
VWY, Bauman JE and Grandis JR: Head and neck squamous cell
carcinoma. Nat Rev Dis Primer. 6:922020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhat AA, Yousuf P, Wani NA, Rizwan A,
Chauhan SS, Siddiqi MA, Bedognetti D, El-Rifai W, Frenneaux MP,
Batra SK, et al: Tumor microenvironment: An evil nexus promoting
aggressive head and neck squamous cell carcinoma and avenue for
targeted therapy. Signal Transduct Target Ther. 6:122021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cossu AM, Mosca L, Zappavigna S, Misso G,
Bocchetti M, De Micco F, Quagliuolo L, Porcelli M, Caraglia M and
Boccellino M: Long non-coding RNAs as important biomarkers in
laryngeal cancer and other head and neck tumours. Int J Mol Sci.
20:34442019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Verro B, Saraniti C, Carlisi D,
Chiesa-Estomba C, Maniaci A, Lechien JR, Mayo M, Fakhry N and
Lauricella M: Biomarkers in laryngeal squamous cell carcinoma: The
literature review. Cancers (Basel). 15:50962023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lyu H, Huang J, He Z and Liu B: Epigenetic
mechanism of Survivin dysregulation in human cancer. Sci China Life
Sci. 61:808–814. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inbar-Feigenberg M, Choufani S, Butcher
DT, Roifman M and Weksberg R: Basic concepts of epigenetics. Fertil
Steril. 99:607–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moore LD, Le T and Fan G: DNA methylation
and its basic function. Neuropsychopharmacology. 38:23–38. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mattei AL, Bailly N and Meissner A: DNA
methylation: A historical perspective. Trends Genet. 38:676–707.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li D, Da L, Tang H, Li T and Zhao M: CpG
methylation plays a vital role in determining tissue- and
cell-specific expression of the human cell-death-inducing
DFF45-like effector A gene through the regulation of Sp1/Sp3
binding. Nucleic Acids Res. 36:330–341. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smith J, Sen S, Weeks RJ, Eccles MR and
Chatterjee A: Promoter DNA hypermethylation and paradoxical gene
activation. Trends Cancer. 6:392–406. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schübeler D: Epigenomics: Methylation
matters. Nature. 462:296–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martìn-Padura I, Lostaglio S, Schneemann
M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A,
Ruco L, Villa A, et al: Junctional adhesion molecule, a novel
member of the immunoglobulin superfamily that distributes at
intercellular junctions and modulates monocyte transmigration. J
Cell Biol. 142:117–127. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, Yin A, Zhang W, Zhao F, Lv J, Lv J
and Sun J: Jam3 promotes migration and suppresses apoptosis of
renal carcinoma cell lines. Int J Mol Med. 42:2923–2929.
2018.PubMed/NCBI
|
|
17
|
Zhou D, Tang W, Zhang Y and An HX: JAM3
functions as a novel tumor suppressor and is inactivated by DNA
methylation in colorectal cancer. Cancer Manag Res. 11:2457–2470.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi Y, Feng X, Zhang Y, Gao J, Bao W, Wang
J and Bai JF: Downregulation of JAM3 occurs in cholangiocarcinoma
by hypermethylation: A potential molecular marker for diagnosis and
prognosis. J Cell Mol Med. 28:e180382024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu CP, Zhou L, Gong HL, Du HD, Tian J, Sun
S and Li JY: Establishment and characterization of a novel
HPV-negative laryngeal squamous cell carcinoma cell line, FD-LSC-1,
with missense and nonsense mutations of TP53 in the DNA-binding
domain. Cancer Lett. 342:92–103. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li LC and Dahiya R: MethPrimer: Designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bock C, Reither S, Mikeska T, Paulsen M,
Walter J and Lengauer T: BiQ analyzer: Visualization and quality
control for DNA methylation data from bisulfite sequencing.
Bioinformatics. 21:4067–4068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Vleeschauwer SI, van de Ven M, Oudin A,
Debusschere K, Connor K, Byrne AT, Ram D, Rhebergen AM, Raeves YD,
Dahlhoff M, et al: OBSERVE: Guidelines for the refinement of rodent
cancer models. Nat Protoc. 19:2571–2596. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Detre S, Jotti GS and Dowsett M: A
‘quickscore’ method for immunohistochemical semiquantitation:
Validation for oestrogen receptor in breast carcinomas. J Clin
Pathol. 48:876–878. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Figueiredo DLA, Chao BMP and Figueiredo
FNDS: Larynx cancer: Search for molecular markers. Arch Head Neck
Surg. 48:e004320192019. View Article : Google Scholar
|
|
26
|
Lian M, Fang J, Han D, Ma H, Feng L, Wang
R and Yang F: Microarray gene expression analysis of tumorigenesis
and regional lymph node metastasis in laryngeal squamous cell
carcinoma. PLoS One. 8:e848542013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stansfield JC, Rusay M, Shan R, Kelton C,
Gaykalova DA, Fertig EJ, Califano JA and Ochs MF: Toward
signaling-driven biomarkers immune to normal tissue contamination.
Cancer Inform. 15:15–21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng M, Brägelmann J, Schultze JL and
Perner S: Web-TCGA: An online platform for integrated analysis of
molecular cancer data sets. BMC Bioinformatics. 17:722016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding W, Chen J, Feng G, Chen G, Wu J, Guo
Y, Ni X and Shi T: DNMIVD: DNA methylation interactive
visualization database. Nucleic Acids Res. 48:D856–D862. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bibikova M, Lin Z, Zhou L, Chudin E,
Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, Vollmer E, et al:
High-throughput DNA methylation profiling using universal bead
arrays. Genome Res. 16:383–393. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cerami1 E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Modhukur V, Iljasenko T, Metsalu T, Lokk
K, Laisk-Podar T and Vilo J: MethSurv: A web tool to perform
multivariable survival analysis using DNA methylation data.
Epigenomics. 10:277–288. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fu M, Hu Y, Lan T, Guan KL, Luo T and Luo
M: The Hippo signalling pathway and its implications in human
health and diseases. Signal Transduct Target Ther. 7:3762022.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mandell K and Parkos C: The JAM family of
proteins. Adv Drug Deliv Rev. 57:857–867. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang TW, DeMarco RA, Mrsny RJ, Gurney A,
Gray A, Hooley J, Aaron HL, Huang A, Klassen T, Tumas DB and Fong
S: Characterization of huJAM: Evidence for involvement in cell-cell
contact and tight junction regulation. Am J Physiol Cell Physiol.
279:C1733–C1743. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mandicourt G, Iden S, Ebnet K,
Aurrand-Lions M and Imhof BA: JAM-C regulates tight junctions and
integrin-mediated cell adhesion and migration. J Biol Chem.
282:1830–1837. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lauko A, Mu Z, Gutmann DH, Naik UP and
Lathia JD: Junctional adhesion molecules in cancer: A paradigm for
the diverse functions of cell-cell interactions in tumor
progression. Cancer Res. 80:4878–4885. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jia X, Zhao C, Chen Q, Du Y, Huang L, Ye
Z, Ren X, Wang S, Lee C, Tang Z, et al: JAM-C maintains VEGR2
expression to promote retinal pigment epithelium cell survival
under oxidative stress. Thromb Haemost. 117:750–757. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sanchez-Vega F, Mina M, Armenia J, Chatila
WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia
S, et al: Oncogenic signaling pathways in the cancer genome atlas.
Cell. 173:321–337.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eun YG, Lee D, Lee YC, Sohn BH, Kim EH,
Yim SY, Kwon KH and Lee JS: Clinical significance of YAP1
activation in head and neck squamous cell carcinoma. Oncotarget.
8:111130–111143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Q, Wang M, Hu Y, Zhao E, Li J, Ren L,
Wang M, Xu Y, Liang Q, Zhang D, et al: MYBL2 disrupts the Hippo-YAP
pathway and confers castration resistance and metastatic potential
in prostate cancer. Theranostics. 11:5794–5812. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang Z, Zhang Z, Zhou C, Liu L and Huang
C: Epithelial-mesenchymal transition: The history, regulatory
mechanism, and cancer therapeutic opportunities. MedComm (2020).
3:e1442022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fan S, Smith MS, Keeney J, O'Leary MN,
Nusrat A and Parkos CA: JAM-A signals through the Hippo pathway to
regulate intestinal epithelial proliferation. iScience.
25:1043162022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Muñoz-Galván S, Felipe-Abrio B,
Verdugo-Sivianes EM, Perez M, Jiménez-García MP, Suarez-Martinez E,
Estevez-Garcia P and Carnero A: Downregulation of MYPT1 increases
tumor resistance in ovarian cancer by targeting the Hippo pathway
and increasing the stemness. Mol Cancer. 19:72020. View Article : Google Scholar : PubMed/NCBI
|