Introduction
Pancreatic cancer is a highly malignant type of
cancer with a high metastatic rate. With the abundance of
pancreatic blood and lymphatic vessels, pancreatic cancer rapidly
grows; this also results in early invasion and metastasis, which
are associated with a poor prognosis, and eventually mortality
(1). Notably, pancreatic cancer is
difficult to diagnose in the early stages; therefore, it often
metastasizes and is resistant to drug therapy, thus making it
difficult to reduce the high mortality rate (2). Therefore, it is important to identify
biomarkers capable of early diagnosis and an appropriate
therapeutic target for pancreatic cancer.
The prognosis of patients with pancreatic cancer
largely depends on the cancer stage at the time of diagnosis.
CA19-9 is a widely used biomarker for pancreatic cancer prognosis;
however, its sensitivity and specificity are insufficient (3). The main challenge in improving the
recovery rate of patients with pancreatic cancer is the lack of
biomarkers for early detection.
KIN17 is a highly conserved gene in mammalian cells
containing an N-terminal zinc finger structure (27–50 nucleotides)
and a C-terminal KOW membrane (335–373 nucleotides), which is
located on human chromosome 10p15-p14 (4). KIN17 has been reported to participate
in cellular activities and physiological processes, including DNA
replication, RNA transcription and cell cycle regulation (5). Recently, KIN17 has been established as
having an essential role in various types of cancer. Notably, Dai
et al (6) and Huang et
al (7) demonstrated that KIN17
may serve an important role in promoting epithelial-mesenchymal
transition (EMT) in hepatocellular carcinoma and luminal-A breast
cancer.
Cancer metastasis, which is a main cause of
cancer-related death, can exacerbate the progression of tumor
development (8). EMT refers to the
phenotypic changes produced by epithelial cells during a specific
process of transformation into stromal cells, thereby endowing them
with increased invasiveness, which is a major factor in the
metastatic ability and drug resistance of cancer (9,10).
Autophagy is a mechanism of membrane-mediated degradation and
recovery that is crucial for cellular homeostasis, which can be
significantly upregulated by various physiological stimuli, such as
malnutrition, hypoxia, endoplasmic reticulum stress, and immune and
hormonal stimulation (11).
Autophagy serves a dual role in cancer progression because it can
hinder or promote cancer occurrence and development (12). Although the mechanism underling the
progression of autophagy is complex, the PI3K/AKT/mTOR pathway has
a role in suppressing autophagy progression (13). Wei et al (14) demonstrated that arenobufagin may
enhance autophagy and cell apoptosis through PI3K/AKT/mTOR pathway
inhibition, effectively suppressing the growth of pancreatic cancer
cells both in vitro and in vivo. Moreover, Qian et
al (15) reported that the
Qingyihuaji Formula could promote apoptosis and pancreatic cancer
autophagy by suppressing two signaling pathways: MAPK/ERK and
PI3K/AKT/mTOR. However, the role of KIN17 in these pathways has not
been studied in depth. Based on the aforementioned findings, it was
hypothesized that KIN17 may act as a regulator of the PI3K/AKT/mTOR
signaling pathway.
The present study systematically assessed KIN17
expression in clinical pancreatic cancer samples, and revealed how
KIN17 expression may affect the clinicopathological characteristics
and survival rates of patients. The study explored the functions of
KIN17 in terms of cell migration, invasion and autophagy in
pancreatic cancer. In summary, the present study highlights novel
findings regarding the effects of KIN17 on autophagy through the
PI3K/AKT/mTOR pathway, thus affecting the migration and invasion of
pancreatic cancer cells.
Materials and methods
Cell lines and cell culture
The human pancreatic cancer cell lines ASPC-1,
PANC-1, PACA-2 and BxPC-3 were purchased from Shanghai Gaining
Biological Technology Co., Ltd. PANC-1 and PACA-2 cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.) containing
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin (PS; Beyotime Institute of
Biotechnology), while ASPC-1 and BxPC-3 cells were cultured in 1640
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% FBS and 1% PS. The normal human pancreatic cell line, HPNE, was
obtained from iCell Bioscience, Inc. HPNE cells were cultured in
cell culture medium [DMEM: 70%; M3:BaseF (cat. no. M300F-500;
Incell Corporation LLC)]: 20%; FBS: 10%) with 1%
penicillin-streptomycin (cat. no. 15140-122; Gibco; Thermo Fisher
Scientific, Inc.). All cells were incubated at 37°C in an
atmosphere containing 5% CO2. For drug intervention,
PANC-1 and PACA-2 cells were treated with autophagy inhibitors or
an autophagy activator purchased from MedChemExpress, including 2
mM 3-methyladenine (3-MA; cat. no. (HY-19312), 10 µM chloroquine
(chloroquine; cat. no. HY-17589A) and 100 nM Rapamycin (Rapa; cat.
no. HY-10219). The PANC-1 and PACA-2 cells were incubated with the
inhibitors/activator for 24 h at 37°C.
Bioinformatics analysis
The Gene Expression Profiling Interactive Analysis
(GEPIA) server (http://gepia.cancer-pku.cn/) was used to obtain and
analyze KIN17 expression data from pancreatic cancer and normal
tissues based on The Cancer Genome Atlas (TCGA) and Gene Type
Tissue Expression projects, including 179 cancer and 171 normal
samples. In addition, expression data were downloaded and extracted
from three Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) datasets [GSE15471
(16,17), GSE71989 (18) and GSE62165 (19)], which compared the mRNA expression
between normal and pancreatic cancer tissues. Raw data were
preprocessed in R language (version 4.3.1; http://www.r-project.org). The expression matrix data
of GSE15471, GSE71989 and GSE62165 were obtained by probe
transformation according to the annotation information of the chip
platform, from which the expression value of KIN17 was extracted.
Correlation analysis of KIN17 expression and the expression of
genes in the Akt/mTOR pathway was performed using TCGA-PAAD
(pancreatic adenocarcinoma) data on the GEPIA server. The
Kaplan-Meier method was used to draw survival curves using the
‘survival’ (https://cran.r-project.org/package=survival) and
‘survminer’ (https://cran.r-project.org/package=survminer) packages
in R software version 4.3.1 (https://www.r-project.org).
Patient samples and
immunohistochemistry (IHC)
At total of 72 pairs of pancreatic cancer and normal
pancreatic tissue samples (mean patient age: 58 years; age range:
33–77 years) were provided by Shanghai Outdo Biotech Co., Ltd. The
clinical and pathological characteristics of the patients are shown
in Table I. Firstly, the tissue
array was placed in an oven, and baked at 63°C for 1 h, after which
dewaxing was completed in an automatic staining machine
(LEICAST5020; Leica Microsystems GmbH). The slide was placed in an
antigen repair instrument (PT Link; Dako; Agilent Technologies,
Inc.), and antigen repair was initiated by selecting the program.
After repair, the slides were placed in distilled water at room
temperature and allowed to cool naturally for >10 min.
Subsequently, the slides were rinsed in PBS, a diluted primary
antibody (KIN17; 1:200; cat. no. sc-32769; Santa Cruz
Biotechnology, Inc.) working solution was added, and the slides
were incubated overnight at 4°C. The next day, the slides were
warmed at room temperature for 45 min, washed with PBS, and put
into an automatic immunohistochemical staining system instrument
(Autostainer Link 48; Dako; Agilent Technologies, Inc.); the
corresponding programs were selected for blocking (with 3% hydrogen
peroxide, 10 min), secondary antibody incubation (10 min)
[EnVision™ FLEX+, Mouse, High pH, (Link); cat. no. K8002; Dako;
Agilent Technologies, Inc.) and DAB color development according to
the manufacturer's protocol at room temperature. Subsequently,
hematoxylin staining was performed for 1 min at room temperature.
The slides were immersed in 0.25% hydrochloric acid and alcohol
(400 ml 70% alcohol + 1 ml concentrated hydrochloric acid) for ~10
sec and were then rinsed with tap water for 5 min. Finally the
slides were dried at room temperature and sealed with neutral
resin. The tissue array was examined using an Aperio scanner
(Aperio ScanScope XT; Leica Microsystems GmbH). The tissues
underwent IHC, and KIN17 staining intensity [classified into four
levels, from 0 (negative) to 4 (strong)] and percentage of positive
cells (0–100%) was determined. Finally, the staining results were
scored by multiplying the intensity level and percentage, and
labelled as the rapid (Q) score. The median Q-score (Q=100) served
as the cut-off value to classify the patients into low (Q≤100) or
high (Q>100) KIN17 expression groups.
 | Table I.Association of KIN17 expression with
clinicopathological features in pancreatic cancer. |
Table I.
Association of KIN17 expression with
clinicopathological features in pancreatic cancer.
|
|
| Expression of
KIN17 |
|
|
|---|
|
|
|
|
|
|
|---|
| Characteristic | Total (n=72) | Low | High | χ2 | P-value |
|---|
| Age |
|
|
| 2.217 | 0.136 |
| <60
years | 37 | 18 | 19 |
|
|
| ≥60
years | 35 | 11 | 24 |
|
|
| Sex |
|
|
| 0.002 | 0.968 |
|
Male | 42 | 17 | 25 |
|
|
|
Female | 30 | 12 | 18 |
|
|
| Tumor size |
|
|
| 0.15 | 0.698 |
| ≤2
cm | 14 | 5 | 9 |
|
|
| >2
cm | 58 | 24 | 34 |
|
|
|
Differentiation |
|
|
| 0.062 | 0.803 |
|
Poor | 31 | 13 | 18 |
|
|
|
Well/Moderate | 41 | 16 | 25 |
|
|
| TNM stage |
|
|
| 0.02 | 0.888 |
| I and
II | 39 | 16 | 23 |
|
|
| III and
IV | 33 | 13 | 20 |
|
|
| T stage |
|
|
| 0.062 | 0.803 |
| T1,
2 | 31 | 13 | 18 |
|
|
| T3,
4 | 41 | 16 | 25 |
|
|
| Lymph node
metastasis |
|
|
| 5.957 | 0.015a |
| N0 | 23 | 14 | 9 |
|
|
| N1 +
pN2 | 49 | 15 | 34 |
|
|
| Distant
metastasis |
|
|
| 0.018 | 0.892 |
| M0 | 49 | 20 | 29 |
|
|
| M1 | 23 | 9 | 14 |
|
|
| Vascular
invasion |
|
|
| 1.952 | 0.162 |
| No | 40 | 19 | 21 |
|
|
|
Yes | 32 | 10 | 22 |
|
|
| Perineural
invasion |
|
|
| 1.245 | 0.265 |
| No | 31 | 13 | 18 |
|
|
|
Yes | 41 | 16 | 25 |
|
|
Small interfering RNA (siRNA)
transfection
The siRNA oligonucleotides specifically designed for
KIN17 (siKIN17) and the negative control siRNA (siNC) were
purchased from Suzhou GenePharma Co., Ltd. with the following
sequences: siNC, sense 5′-UUCUCCGAACGUGUCACGUTT-3′, antisense
5′-ACGUGACACGUUCGGAGAATT-3′; siKIN17#1, sense
5′-GCAGAAGCUACGCUGGUAUTT-3′, antisense 5′-AUACCAGCGUAGCUUCUGCTT-3′;
siKIN17#2, sense 5′-GGAAUUCCGAAAUGACUUUTT-3′, antisense
5′-AAAGUCAUUUCGGAAUUCCTT-3′; siKIN17#3, sense
5′-GCAACAUCUUCCAAGUCAATT-3′, antisense 5′-UUGACUUGGAAGAUGUUGCTT-3′.
PANC-1 and PACA-2 cells at 70% confluence were transfected with
siRNAs (150 pmol) using siRNA-mate (Suzhou GenePharma Co., Ltd.)
according to the manufacturer's protocol at room temperature. After
48 and 72 h, the cells were collected for reverse
transcription-quantitative PCR (RT-qPCR) and western blotting (WB)
to evaluate the corresponding mRNA and protein expression levels,
respectively.
RNA extraction and RT-qPCR
Total RNA was extracted from the cultured cells
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. RNA
quality, including concentration and purity, was evaluated using a
NanoDrop Spectrophotometer (NanoDrop; Thermo Fisher Scientific,
Inc.). Subsequently, cDNA was obtained from 1 µg RNA through RT
using PrimeScript™ RT Reagent Kit (cat. no. RR047Q; Takara
Biotechnology Co., Ltd.). Finally, mRNA expression was examined
using qPCR with an Applied Biosystems 7500 Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and using TB
Green® Premix Ex Taq™ (Tli RNaseH Plus) (cat. no.
RR420A; Takara Biotechnology Co., Ltd.) according to the
manufacturer's protocol. The qPCR thermal cycling conditions were
as follows: Initial denaturation at 95°C for 30 sec; followed by 60
cycles of denaturation at 95°C for 5 sec, annealing at 55°C for 30
sec and extension at 72°C for 30 sec. KIN17 expression was
normalized to GAPDH, using the 2−ΔΔCq method (20). The primer sequences used were as
follows: human KIN17, forward AGACGCTTTGGCACTAAAAGG and reverse
AGTGGCATTCATGTGGATGTG; human GAPDH, forward GCACCGTCAAGGCTGAGAAC
and reverse TGGTGAAGACGCCAGTGGA.
WB
Protein was extracted from pancreatic cancer cells
using RIPA buffer (Beyotime Institute of Biotechnology) and 20 µg
proteins were separated by SDS-PAGE on 8–15% gels, before being
transferred to PVDF membranes (MilliporeSigma). After blocking the
membranes in 5% milk for 60 min at room temperature, they were
incubated with the following primary antibodies: GAPDH (cat. no.
60004-1-Ig; 1:20,000), Vimentin (cat. no. 60330-1-Ig; 1:50,000),
Beclin1 (cat. no. 66665-1-Ig; 1:2,000), mTOR (cat. no. 28273-1-AP;
1:50,000), phosphorylated (P)-mTOR (cat. no. 67778-1-Ig; 1:5,000)
(all from Proteintech Group, Inc.), KIN17 (cat. no. sc-32769;
1:1,000; Santa Cruz Biotechnology, Inc.), E-cadherin (cat. no.
ab40772; 1:10,000), N-cadherin (cat. no. ab76011; 1:10,000), P62
(cat. no. ab109012; 1:10,000), ULK1 (cat. no. ab177472; 1:10,000),
PI3K (cat. no. ab40776; 1:5,000) (all from Abcam), LC3B (cat. no.
2775; 1:1,000), AKT (cat. no. 4691; 1:1,000), P-AKT (cat. no. 4060;
1:1,000) and P-ULK1 (cat. no. 6888; 1:1,000) (all from Cell
Signaling Technology, Inc.), and P-PI3K (cat. no. AF5905 1:500;
Beyotime Institute of Biotechnology) overnight at 4°C. The
membranes were then washed with 1X TBS-0.1% Tween-20 and incubated
with secondary antibodies for 1 h at room temperature. The
following secondary antibodies were used: Anti-rabbit IgG,
HRP-linked antibody (1:5,000; cat. no. SA-00001-2; Proteintech
Group, Inc.) and anti-mouse IgG, HRP-linked Antibody (1:5,000; cat.
no. SA-00001-1; Proteintech Group, Inc.). Finally, bands were
visualized using WesternLumaxLight Superior HRP substrate (cat. no.
310209; Zeta-Life) and visualized using a Tanon 5200 imaging system
(Tanon Science and Technology Co., Ltd.). The intensity of the
protein bands was semi-quantified using Image Lab software (5.2.1)
and the protein expression levels were normalized to the respective
GAPDH bands. All WB experiments were conducted in triplicate.
Cell migration and invasion
assays
During the logarithmic growth phase,
5×104 cells/well in serum-free medium were plated in the
upper chamber of a 8-µm Transwell system at room temperature, while
0.6 ml medium supplemented with 10% FBS was added to the lower
chamber in a 24-well plate. After incubation at 37°C for 24 h, the
translocated cells were fixed with 4% paraformaldehyde for 20 min,
followed by staining with 0.1% crystal violet solution at room
temperature for 30 min at room temperature. For quantification,
images from five random fields were captured and the cells were
counted under optical microscope (magnification, ×200). In the
invasion assay, the upper Transwell chamber was precoated with 1:8
diluted Matrigel (BD Biosciences) at 37°C for 1 h, and the cells
were cultured at 37°C incubator for 72 h, whereas the remaining
steps were the same as those performed in the migration assay. Each
experiment was conducted in triplicate.
Wound-healing assay
A total of 5×105 cells/well in a 6-well
plate were cultured without 10% FBS. When the cell fusion rate
reached 90%, a wound was generated using a 200-µl pipette tip to
draw a straight line at the bottom of the plate. Images of the
cells were captured using an optical microscope at 0 h, when the
wound was created, and at 24 h. Relative migration was calculated
as follows: Relative migration rate=area (0–24 h)/area at 0 h. Each
experiment was repeated three times.
Autophagy assay
Autophagy was detected using a CYTO-ID®
Autophagy Detection Kit (cat. no. ENZ-KIT175-0050; Enzo Life
Sciences, Inc.). After cultivating the cells on a 14×14 mm confocal
dish, the culture medium was carefully removed when the cells
reached a fusion level of 50–70%. Subsequently, the cells were
washed with 100 µl 1X Assay Buffer, and were incubated with CYTO-ID
Green Detection Reagent 2 for 30 min at room temperature. Finally,
the stained cells were analyzed using a confocal microscope.
GFP-mRFP-LC3 staining
The mRFP-GFP-LC3 lentiviral vector was purchased
from Suzhou GenePharma Co., Ltd., and PANC-1 and PACA-2 cells were
infected according to the manufacturer's instructions. The cells
stably expressing mRFP-GFP-LC3 were selected by puromycin (1 µg/ml)
and 1×104 stably expressing mGFP-RFP-LC3 cells/dish were
seeded into a confocal dish. After incubation for 20 h at room
temperature, the cells were transfected with siNC or siKIN17 as
aforementioned. The autophagosomes were labeled yellow (mRFP and
GFP) whereas autolysosomes were labeled red (mRFP only, and the
results in five independent fields were observed under a confocal
laser-scanning microscope (Olympus Corporation).
Statistical analysis
To ensure accuracy, all experiments were
independently repeated three times. Statistical analysis was
performed using either SPSS 27.0 statistical software (IBM
Corporation) or GraphPad Prism 8 software (Dotmatics). To analyze
the significant differences between two groups, a paired Student's
t-test was conducted for paired data, while an unpaired Student's
t-test was used for unpaired data. For comparisons among three or
more groups, one-way ANOVA with Tukey's multiple comparisons test
was applied. The χ2 test was used to determine the
association between KIN17 expression and the clinicopathological
variables of the samples. The survival curve was plotted using the
Kaplan-Meier method and data were compared using the log-rank test.
The relationship between the expression of two genes was analyzed
using Pearson's correlation coefficient. P<0.05 was considered
to indicate a statistically significant difference.
Results
Expression of KIN17 in pancreatic
cancer and its effects on overall survival rate
Bioinformatics analysis using TCGA database revealed
that the expression levels of KIN17 were upregulated in pancreatic
cancer tissues compared with those in the adjacent nontumor tissues
(Fig. 1A). In addition, the mRNA
expression levels of KIN17 were compared between normal and
pancreatic cancer tissues in the GSE15471 [expression analysis of
36 pancreatic ductal adenocarcinoma (PDAC) tumor tissues and
matching normal pancreatic tissue samples from patients with
pancreatic cancer], GSE71989 [8 normal pancreatic from healthy
controls and 14 PDAC tissues] and GSE62165 (118 PDAC samples and 13
normal samples from healthy controls) datasets from the GEO
database (Fig 1B-D). The expression
of KIN17 was significantly upregulated in pancreatic cancer tissues
compared with that in normal pancreatic tissues. Additionally, the
analysis of microarray slides containing 72 pairs of pancreatic
cancer tissues with adjacent tissues revealed that the KIN17
staining intensity was significantly higher in pancreatic cancer
tissues than in adjacent tissues (Fig.
1E). Notably, patients with pancreatic cancer and higher KIN17
expression were revealed to have poorer overall survival than those
with lower KIN17 expression, as determined by the Kaplan-Meier
survival analysis performed on data from 72 patients with
pancreatic cancer (Fig. 1F). In
addition, a GEO dataset (GSE62452; 69 pancreatic tumors and 61
adjacent non-tumor tissue from patients with PDAC) was used to
assess the association between pancreatic cancer survival and KIN17
expression levels; the Kaplan-Meier curves were plotted using the R
package survminer. The results showed that patients with pancreatic
cancer with high KIN17 expression had significantly lower overall
survival than those with low KIN17 expression (Fig. 1G). Clinical data from 72 patients
with pancreatic cancer were analyzed. As summarized in Table I, the high expression of KIN17 in
pancreatic cancer was positively associated with lymph node
metastasis (P=0.015). Tumor-Node-Metastasis stage has been reported
in the relevant literature as a risk factor for poor prognosis in
patients with pancreatic cancer (21). Subsequently, the mRNA and protein
expression levels of KIN17 were detected in normal pancreatic
epithelial HPNE cells, and four pancreatic cancer cell lines:
ASPC-1, PANC-1, PACA-2 and BxPC-3. The results of RT-qPCR and WB
showed that the mRNA and protein expression levels of KIN17
expression were both increased in pancreatic cancer cells compared
with those in HPNE cells, but there was no significant difference
in KIN17 expression between BxPC-3 and HPNE cells (Fig. 1H and I). These results indicated
that KIN17 may be upregulated in pancreatic cancer tissues and cell
lines, and that this increase in KIN17 expression is associated
with a shorter survival period.
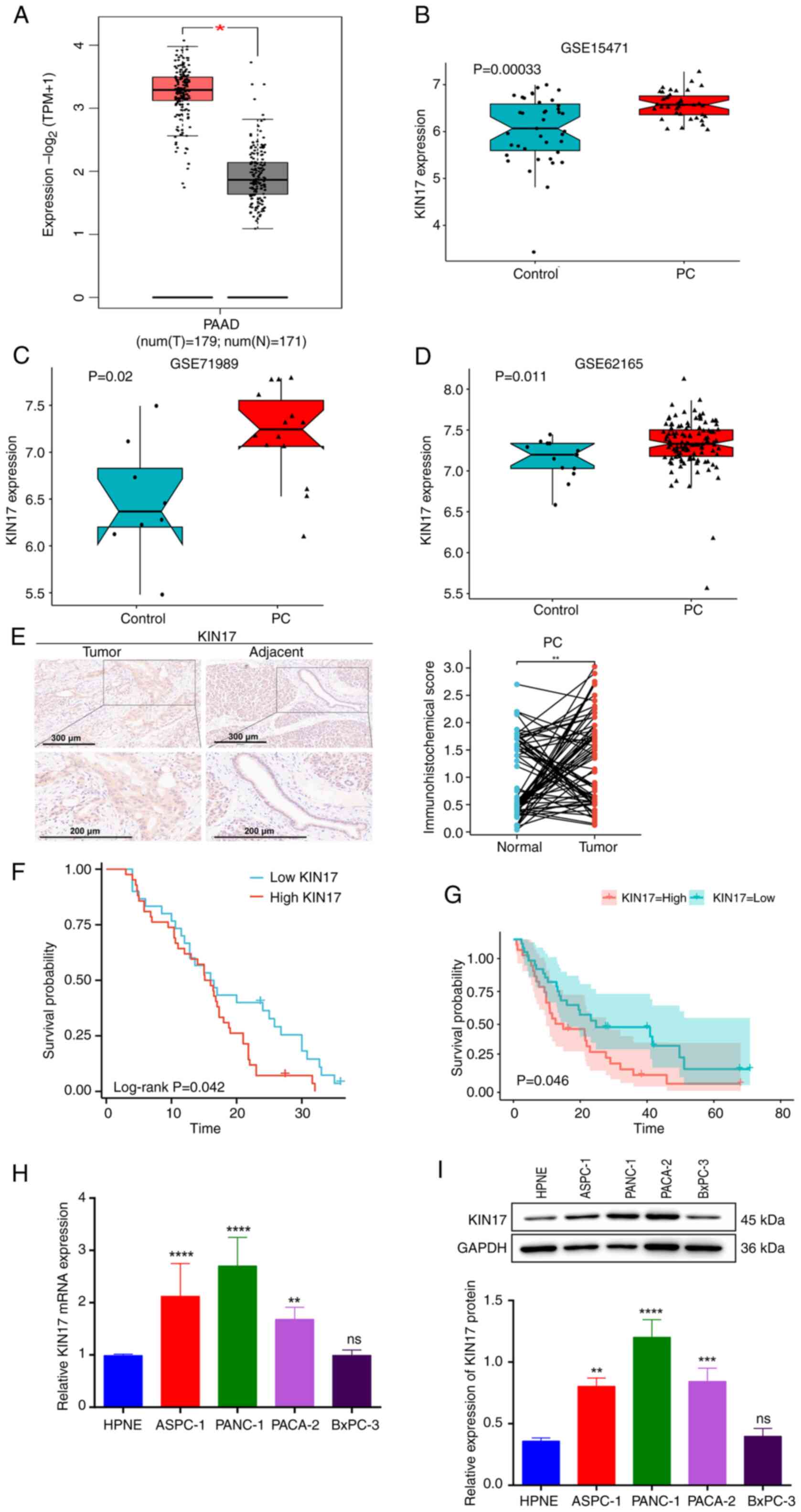 | Figure 1.KIN17 is highly expressed in
pancreatic cancer and is associated with overall survival. (A)
Expression of KIN17 in pancreatic cancer from The Cancer Genome
Atlas database. *P<0.05. (B) Differential expression of KIN17 in
pancreatic ductal adenocarcinoma tumors and matching normal
pancreatic tissue samples from patients with pancreatic cancer in
the GSE15471 dataset. (C) Differential expression of KIN17 in
normal pancreatic and PDAC tissues in the GSE71989 dataset. (D)
Differential expression of KIN17 in PDAC samples and control
samples in the GSE62165 dataset. (E) Representative KIN17
immunohistochemistry staining images, with semi-quantification in
72 pairs of human pancreatic cancer tissues and adjacent tissues.
Scale bars, 300 µm (upper image) and 200 µm (lower images).
**P<0.01. (F) Analysis and comparison of overall survival rate
based on the KIN17 expression level in patients with pancreatic
cancer. (G) Survival curves for KIN17 levels in patients with
pancreatic cancer based on the GEO dataset (GSE62452). (H) Relative
mRNA expression levels of KIN17 in HPNE cells, and four different
pancreatic cancer cell lines (ASPC-1, PANC-1, PACA-2 and BxPC-3)
using reverse transcription-quantitative PCR. **P<0.01,
****P<0.0001 vs. HPNE. (I) KIN17 protein levels in HPNE cells,
and four different pancreatic cancer cell lines (ASPC-1, PANC-1,
PACA-2 and BxPC-3) detected using western blotting. **P<0.01,
***P<0.001, ****P<0.0001 vs. HPNE. GEO, Gene Expression
Omnibus; PAAD/PDAC, pancreatic ductal adenocarcinoma. |
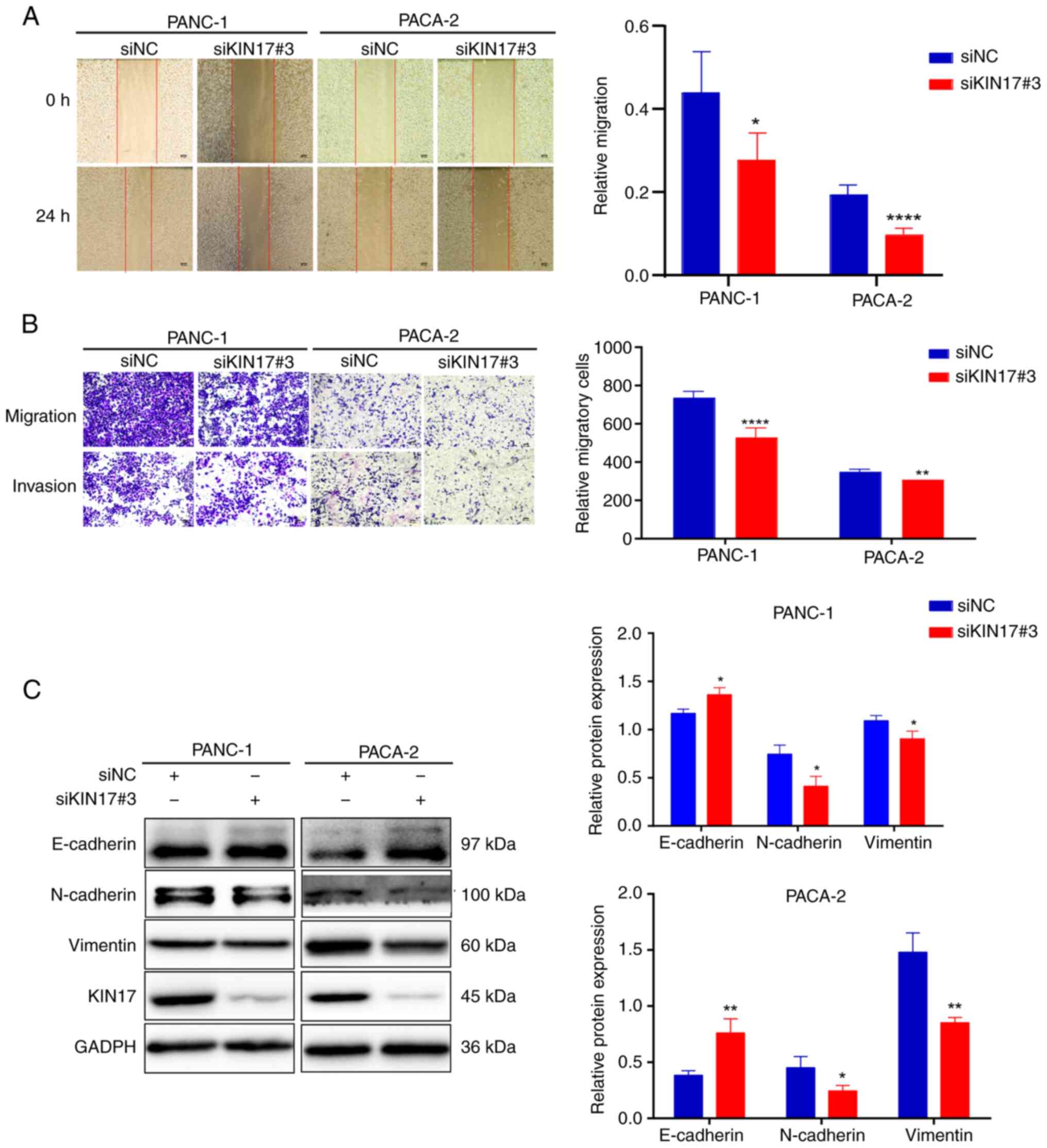
Effect of silencing KIN17 on cell
migration and invasion
Before exploring the role of KIN17, Cells were
transfected with three siRNAs specifically targeting KIN17 (siRNA
#1, #2 and #3) to knock down KIN17 expression, and the most
effective siRNA, siKIN17#3, was selected using RT-qPCR and WB
(Fig. S1), which was used in the
subsequent studies conducted in PANC-1 and PACA-2 cells. According
to our previous studies, using siRNAs to reduce KIN17 expression
has shown good knockdown efficiency in hepatocellular carcinoma
cells (6,22). In the present study, the siRNA
sequence siKIN17#3 had a relatively high knockdown efficiency for
KIN17 of ~80%. Wound-healing and Transwell assays were conducted to
evaluate the role of KIN17 in migration and invasion. The results
indicated that KIN17 knockdown reduced cell migration (Fig. 2A) and invasive ability (Fig. 2B) in PANC-1 and PACA-2 cells. It is
widely accepted that EMT makes tumor cells highly mobile and
invasive (23). In addition, it has
been shown that EMT is involved in pancreatic cancer metastasis
(24). The present study revealed
that knockdown of KIN17 upregulated the expression of epithelial
markers (E-cadherin), and downregulated the expression of
mesenchymal markers (N-cadherin and Vimentin) in PANC-1 and PACA-2
cells (Fig. 2C). These results
suggested that KIN17 suppression may lead to inhibition of the
migration and invasion of pancreatic cancer cells.
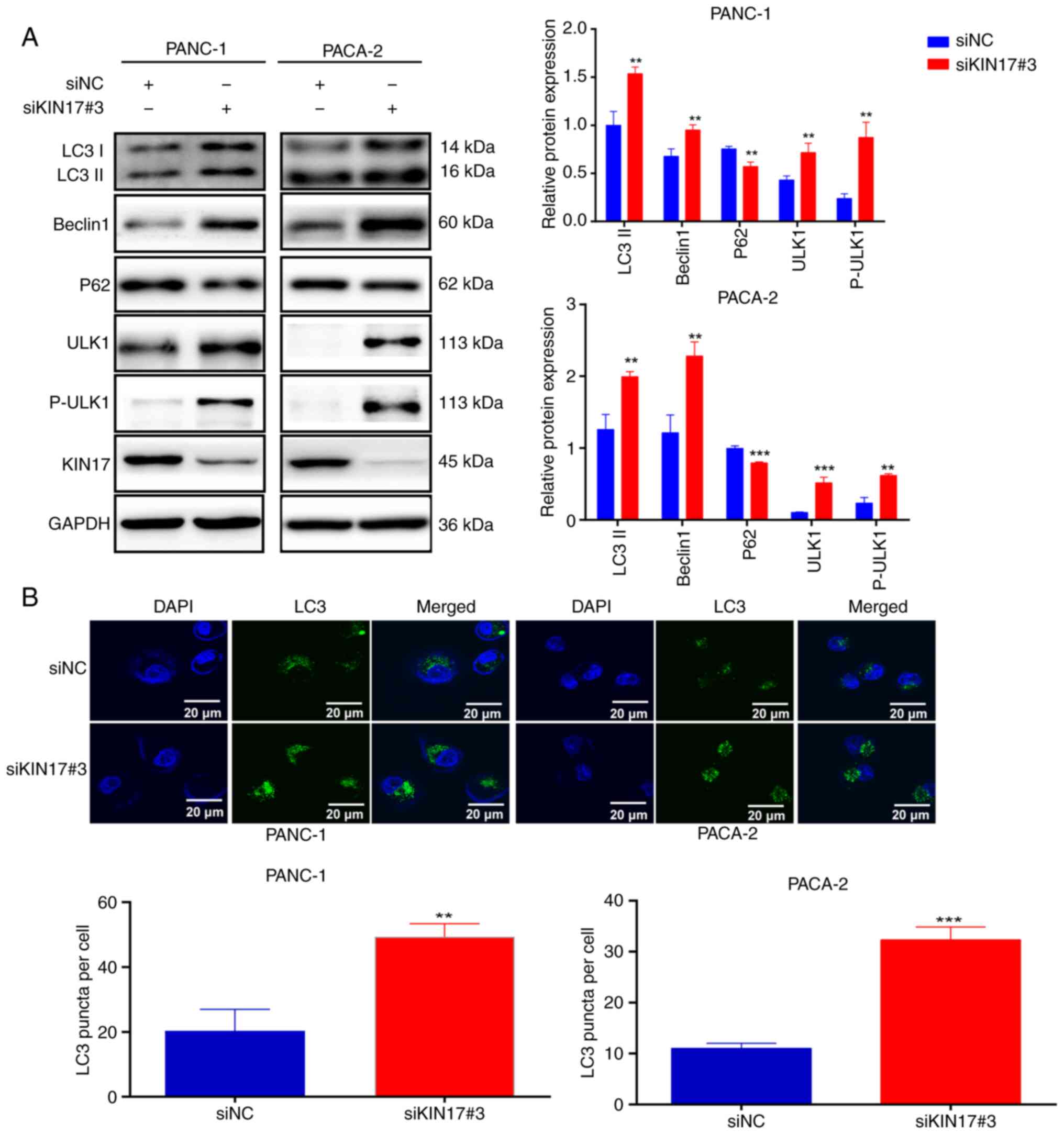
Promotion of autophagy by KIN17
knockdown
Previous studies have extensively explored the link
between autophagy and tumor metastasis (25,26).
The present study assessed the levels of autophagy-related proteins
using WB following the knockdown of KIN17 in pancreatic cancer
cells. Notably, the depletion of KIN17 in PANC-1 and PACA-2 cells
resulted in a marked increase in the expression of
autophagy-related proteins, such as Beclin1, LC3II, ULK1 and
P-PULK1, and a significant decrease in P62 expression. The possible
reason for the simultaneous elevation of ULK1 and P-ULK1 when KIN17
was knocked down may be that when KIN17 is knocked down, pancreatic
cancer cells could initiate a compensatory mechanism to increase
the expression of downstream proteins to maintain normal
physiological functions. In this case, both ULK1 and P-ULK1 may be
elevated. Furthermore, phosphorylation is an important protein
modification that can change the activity, localization and
stability of proteins. When KIN17 is knocked down, the
phosphorylation level of the downstream protein ULK1 may change,
leading to the elevation of P-ULK1. At the same time, total protein
ULK1 may also be elevated for this reason (Fig. 3A). Furthermore, autophagosomes were
examined by observing the presence of GFP-LC3 points, revealing a
higher number of GFP-LC3 puncta in pancreatic cancer cells
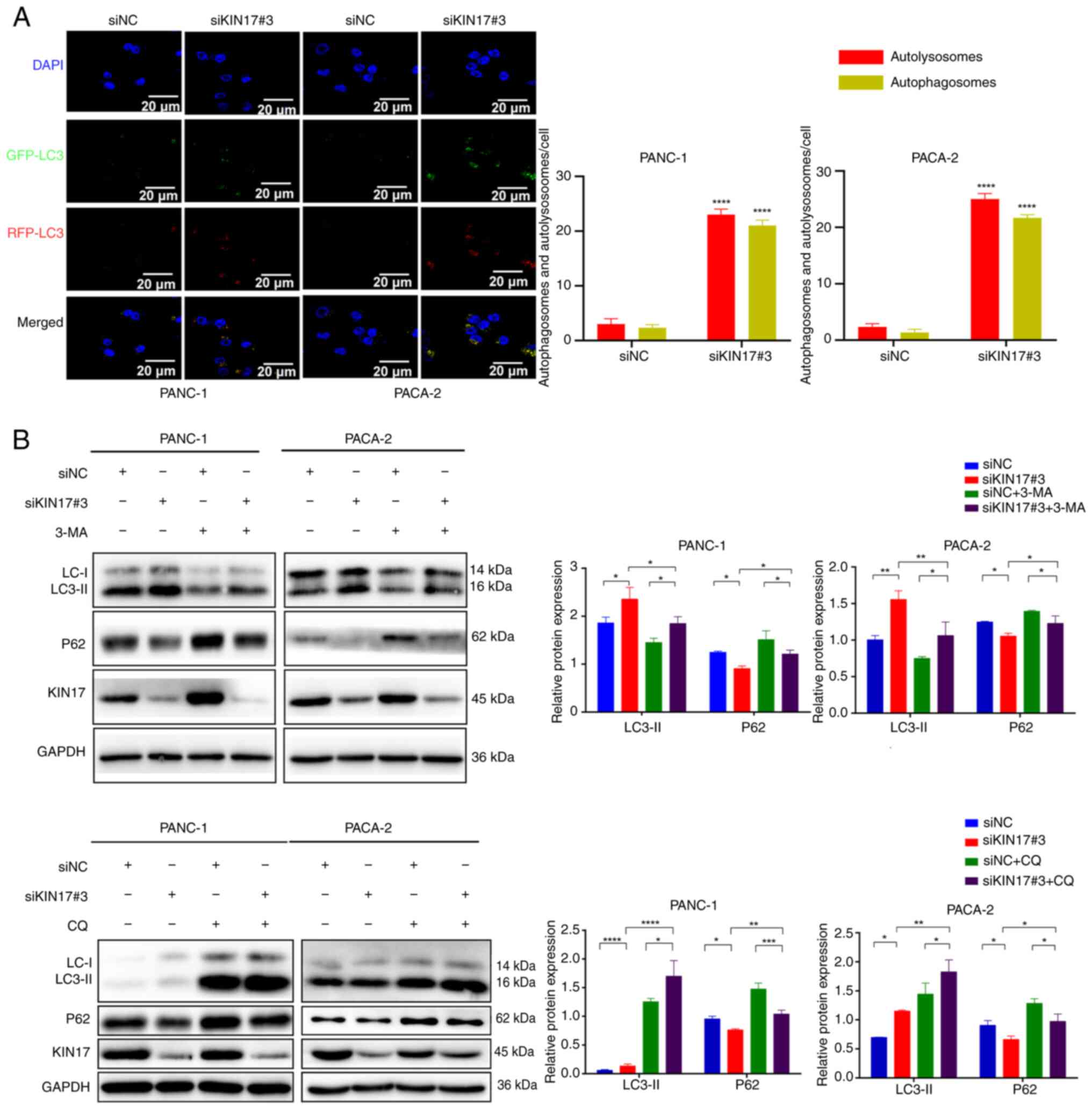
transfected with siKIN17 than in siNC cells (Fig. 3B). Next, the mRFP-GFP-LC3 dual
fluorescent lentivirus was used to monitor autophagosomes and
autolysosomes. After the formation of autolysosomes, their GFP
signals are susceptible to acidic conditions, whereas mRFP signals
are less affected. Therefore, in the merged figure, yellow dots
indicate autophagosomes and red dots indicate autolysosomes (fusion
of autophagosomes and lysosomes). The formation of both
autophagosomes and autolysosomes increased in PANC-1 and PACA-2
cells transfected with siKIN17, indicating increased autophagic
activity compared with in siNC cells (Fig. 4A). These findings suggested that
autophagosomes successfully fused with lysosomes instead of being
obstructed. In addition, autophagic flux was assessed by monitoring
the conversion of LC3I to LC3II. Two autophagy inhibitors, 3-MA and
CQ, were used in the present study. While 3-MA impedes the
formation of autophagosomes in the initial stages by deactivating
class III phosphatidylinositol 3-kinase (27), CQ increases lysosomal pH and hinders
the fusion of autophagosomes with lysosomes in later stages
(28). Comparative analysis
revealed that co-treatment with 3-MA significantly decreased KIN17
inhibition-dependent LC3-II protein expression, suggesting a
reversal of the autophagic process (Fig. 4B). Conversely, co-treatment with CQ
significantly increased KIN17 inhibition-dependent LC3-II protein
expression, indicating that 3-MA and CQ counteracted the
autophagy-promoting effects of KIN17 knockdown, this may be due to
3-MA inhibiting autophagosome formation and CQ inhibiting the
process of fusion of autophagosomes with lysosomes. Notably,
co-treatment with 3-MA and CQ reversed the expression pattern of
P62 compared with KIN17 knockdown alone, indicating their ability
to attenuate the effect of KIN17 knockdown on P62 degradation in
lysosomes. These results indicated that KIN17 knockdown may enhance
autophagy in pancreatic cancer cells.
Relationship between autophagy and
migration/invasion through KIN17 regulation
The induction of autophagy is considered to promote
the migratory and invasive capabilities of cancer cells (29). The present study examined the effect
of KIN17 inhibition on autophagy induction. Co-treatment of cells
with siKIN17#3 and the autophagy inhibitor 3-MA significantly
increased migration and invasion of PANC-1 and PACA-2 cells
compared with sole treatment with siKIN17#3, indicating reversal of
the anti-migratory and anti-invasive effects mediated by KIN17
knockdown (Fig. 5A and B). These
findings suggested that the inhibition of autophagy may counteract
the anti-migratory and anti-invasive effects of KIN17
knockdown.
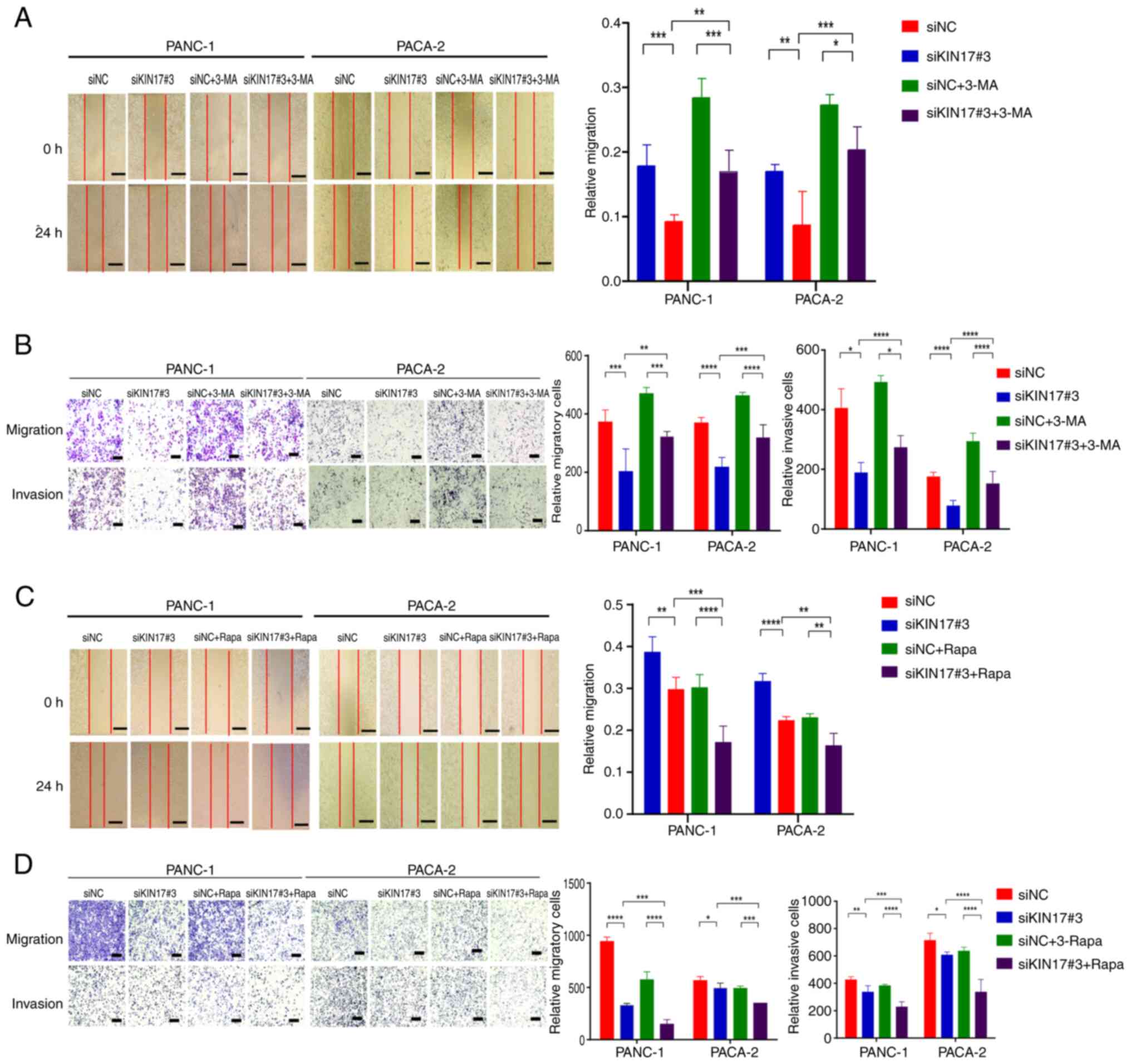 | Figure 5.Suppression of KIN17 hampers the
migration and invasion of pancreatic cancer cells mediated by
autophagy. (A) Evaluation of migration in PANC-1 and PACA-2 cells
through a wound-healing assay. Scale bar, 200 µm. (B) Assessment of
migration and invasion in PANC-1 and PACA-2 cells using Transwell
chambers. Scale bar, 200 µm. (C) Analysis of the migration of
PANC-1 and PACA-2 cells in a wound-healing assay. Scale bar, 200
µm. (D) Transwell assay examining the migration and invasion
potential of PANC-1 and PACA-2 cells treated with Rapa (100 nM).
Scale bar, 200 µm. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. 3-MA, 3-methyladenine; CQ, chloroquine; NC,
negative control; Rapa, rapamycin; si, small interfering. |
Subsequently, the present study investigated whether
simultaneous KIN17 knockdown and induction of autophagy could
further enhance the inhibition of migration and invasion in
pancreatic cancer cells. Rapa, a potent and specific mTOR inhibitor
with autophagy-inducing properties, was used in the present study.
The results revealed that similar to KIN17 knockdown, siNC + Rapa
treatment led to decreased cell migration and invasion compared
with siNC alone (Fig. 5C and D).
Notably, the combined treatment of Rapa and siKIN17#3 significantly
amplified the suppression of cell migration and invasion induced by
siKIN17#3. In summary, these results indicated that the combined
inhibition of KIN17 and mTOR may effectively boost autophagic
activity, leading to synergistic anti-migratory and anti-invasive
effects in pancreatic cancer cells.
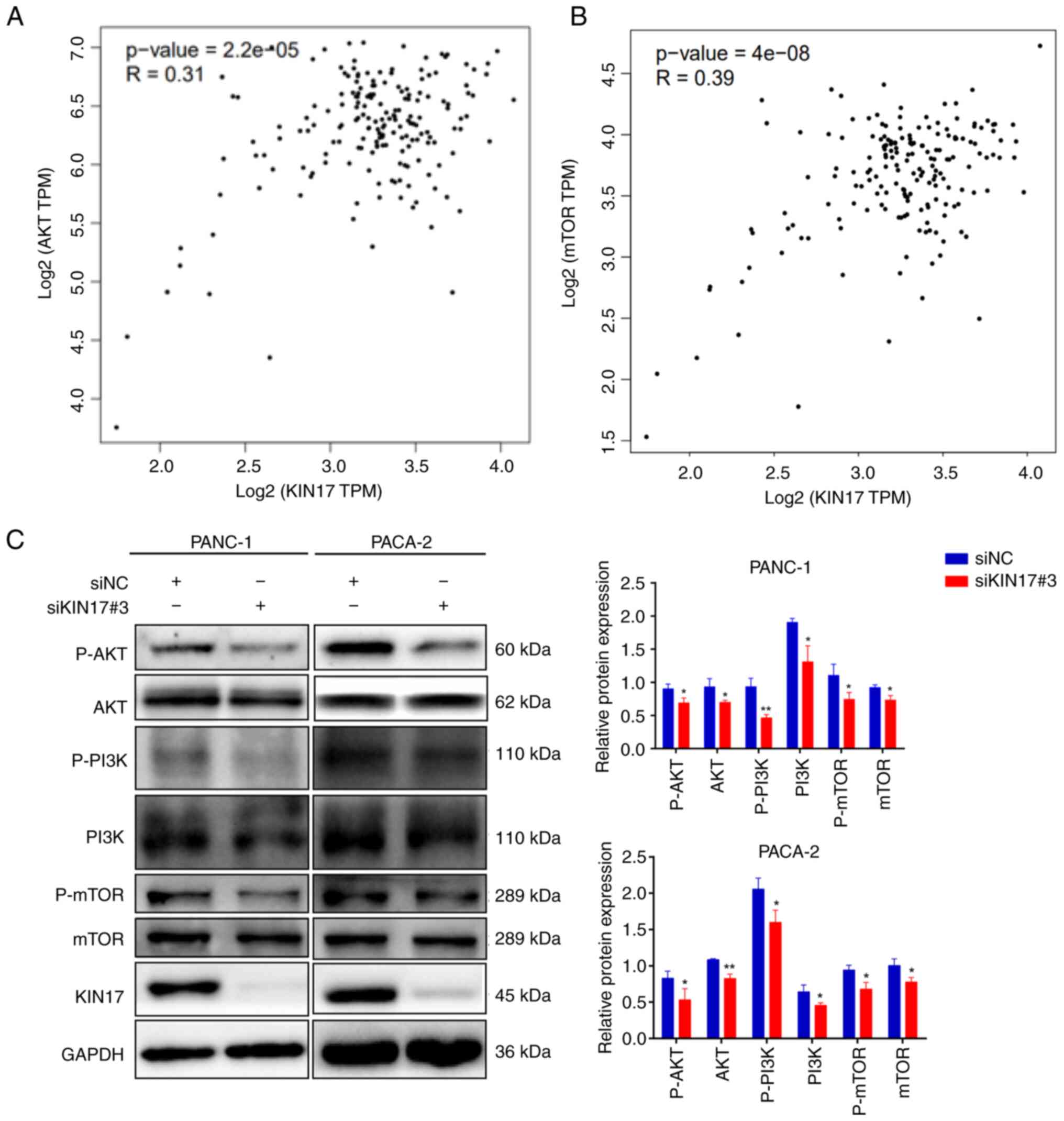
Role of KIN17 in the PI3K/AKT/mTOR
pathway
Increasing evidence has supported the involvement of
the PI3K/AKT/mTOR pathway in autophagy processes (30,31).
Through integrated analysis using the GEPIA database, the
correlation between KIN17 expression and the PI3K/AKT/mTOR
signaling pathway was investigated in pancreatic cancer. The
findings revealed a positive correlation between KIN17 expression
and AKT and mTOR expression (Fig.
6A-B). Results of WB confirmed that the mTOR, P-mTOR, PI3K,
P-PI3K, AKT and P-AKT expression levels were significantly
decreased in the PANC-1 and PACA-2 cells with KIN17 knockdown
(Fig. 6F). Knockdown of KIN17
decreased both total and phosphorylated proteins of the
PI3K/AKT/mTOR pathway, which may be due to a change in protein
stability; knockdown of KIN17 may affect the stability of proteins
in this pathway, leading to an increase in their degradation, which
results in a decrease in the total protein level. Meanwhile,
phosphorylation is a key factor in the stability of downstream
proteins, thus a decrease in total protein may be accompanied by a
decrease in phosphorylated proteins. Taken together, these results
suggested that KIN17 may act as a regulatory element within the
PI3K/AKT/mTOR pathway, validating the predictions from the GEPIA
data.
Discussion
Pancreatic cancer is one of the most aggressive
tumors worldwide, which is characterized by early metastasis and a
poor prognosis (32,33). Analyzing the mechanisms underlying
early metastasis of pancreatic cancer is of importance in managing
its advancement and enhancing patient outcomes. From a clinical
perspective, understanding the mechanisms of metastatic progression
in pancreatic cancer is crucial for refining current therapeutic
approaches. KIN17, a DNA- and RNA-binding protein, is upregulated
in various tumors, and its increased expression is associated with
unfavorable tumor prognoses, and the invasion and metastasis of
cancer cells (5,22). However, research on the role of
KIN17 in pancreatic cancer is limited, leaving its biological
function and the potential molecular underlying mechanisms unclear.
The present study performed a preliminary exploration of KIN17
involvement in pancreatic cancer metastasis, with the aim of
identifying a novel target for treating patients with pancreatic
cancer.
The present investigation detected increased KIN17
expression in pancreatic cancer tissue samples compared with that
in normal tissues. Using a prognostic model assay, it was
demonstrated that increased KIN17 expression was associated with
lymph node metastasis in pancreatic cancer. Furthermore, elevated
KIN17 levels were associated with a poor prognosis in patients with
pancreatic cancer. EMT serves a pivotal role in tumorigenesis and
metastasis, and is a physiological process marked by cytoskeletal
reorganization and extracellular matrix synthesis. EMT is
characterized by the transformation of epithelial cells into
stromal cells, facilitating their mobility within the cellular
matrix. This transformation involves reduced intercellular
adhesion, loss of polarity, and increased cell motility, invasion
and metastasis (34,35). Studies have indicated that EMT
induces malignant traits in cancer cells and enhances invasiveness,
cancer stem cell activity, and resistance to chemotherapy and
immunotherapy (36–40). Increasing evidence has underscored
the critical involvement of EMT in the initiation, progression,
invasion, migration, metastasis and drug resistance of pancreatic
cancer. Therefore, manipulating EMT regulation may emerge as a
novel therapeutic approach for the management of pancreatic cancer
(41–44). The present results demonstrated that
KIN17 inhibition notably upregulated E-cadherin expression, and
downregulated N-cadherin and Vimentin expression in pancreatic
cancer cells, thus implying that the inhibition of KIN17 may impede
EMT, thereby suppressing pancreatic cancer progression.
Autophagy is a dynamic safeguarding mechanism in
which large molecules and organelles are degraded within cells to
maintain body equilibrium. Most eukaryotic cells depend on
autophagy to regulate the stability of their internal environment.
Notably, autophagy serves a protective role in mitigating bodily
harm under specific stress conditions, such as ischemia and
hypoxia; however, excessive activation of autophagy can trigger
programmed cell death, a distinct form of cell death separate from
apoptosis and necrosis (45). The
role of autophagy in cancer cell death has sparked a debate. While
autophagy can sustain intracellular environmental stability by
breaking down harmful proteins or damaged organelles and fostering
normal cellular metabolism and regeneration, an excessive increase
in autophagy can induce autophagic cell death, impede cell
proliferation, degrade cancer proteins, thwart tumor initiation,
prevent metastasis and even increase chemotherapeutic sensitivity
(46). Autophagy and EMT are key
biological processes that influence cancer onset and progression,
with intricate interconnections between autophagy-related and
EMT-related signaling pathways. Previous research has revealed the
involvement of autophagy in both inducing and inhibiting EMT,
suggesting its potential to stimulate tumor metastasis through EMT
induction (47). Conversely, the
regulation of autophagy has been documented to prompt molecular
shifts from the stromal phenotype to the epithelial phenotype,
thereby impeding migration and invasion in various contexts
(48). The present study showed
that KIN17 knockdown led to an increased expression of classic
autophagy markers, such as LC3II, Beclin1 and ULK1, while
diminishing P62 levels. These results implied that KIN17 knockdown
may induce autophagy. To investigate the impact of autophagy on the
migration and invasion of pancreatic cancer cells, Rapa was
administered as an autophagy activator, and 3-MA as an autophagy
inhibitor to the cells, and the migration of both cell types was
subsequently assessed. The observations indicated that the
induction of autophagy may curb the migration and invasion of
pancreatic cancer cells, whereas autophagy inhibition yielded the
opposite outcomes. These results underscore the role of autophagy
in regulating EMT in pancreatic cancer cells.
The correlation between KIN17 expression and the
mTOR pathway, derived from the GEPIA database, prompted an
exploration of the novel role of KIN17 in autophagy modulated by
the PI3K/AKT/mTOR pathway (49,50).
The PI3K/AKT/mTOR signaling pathway serves as a pivotal metabolic,
proliferative and survival regulatory pathway within cells, and is
crucial for maintaining cellular equilibrium. This pathway affects
tumor development and progression by influencing cellular autophagy
in various cancer types, such as laryngeal squamous cell carcinoma,
glioblastoma and cervical cancer (51–54).
Previous investigations have highlighted the significant role of
this pathway in autophagy regulation, from inhibiting autophagy
initiation to regulating autophagy processes and terminating
autophagy, making it a focal point in autophagy regulation
mechanisms (55–58). WB revealed that P-PI3K, P-AKT and
P-mTOR levels were decreased following KIN17 knockdown. Previous
findings have confirmed that PI3K/AKT/mTOR is one of the most
mutated signaling pathways in human malignancies, including
pancreatic cancer, which is activated by various factors such as
cytokines, hormones and growth factors (49,50).
Nie et al (59) reported
that ALDH1A3 can activate the PI3K/AKT/mTOR signaling pathway and
its downstream target, peroxisome proliferator-activated receptor
γ, thus promoting pancreatic cancer metastasis in vitro and
in vivo. Huang et al (60) revealed that MSI2 may regulate
NLK-mediated EMT and the PI3K/AKT/mTOR pathway to promote
pancreatic cancer progression. Likewise, Shao et al
(61) reported that CPA4
overexpression could contribute to the aggressive clinical stage of
patients with pancreatic cancer, and may promote EMT in
vitro by activating the PI3K/AKT/mTOR signaling pathway.
Consistent with the aforementioned findings, the present study
suggested that KIN17 knockdown may induce autophagy via the
PI3K/AKT/mTOR-mediated signaling pathway in pancreatic cancer
cells. However, a single gene can regulate multiple pathways. A
limitation of the present study is that other pathways were not
considered, which may affect the interpretation of the relationship
between KIN17, autophagy and the PI3K/AKT/mTOR pathway. Further
analysis is needed to understand this regulatory mechanism in more
detail.
In summary, the present study demonstrated that
KIN17 inhibition may significantly impede cell migration and
invasion by stimulating autophagy and suppressing the PI3K/AKT/mTOR
pathway. These results not only shed light on the molecular role of
KIN17 in autophagy but also underscore the potential of KIN17 as a
promising novel target for the prognosis and treatment of
pancreatic cancer. However, the present study has some limitations.
First, the mechanism of KIIN17 in regulating autophagy should be
further studied. Second, the study mainly concentrated on cellular
level research and lacks support from animal models or clinical
trials, making the research conclusions not comprehensive enough.
Further research is needed to remedy these limitations and fully
elucidate these issues.
In conclusion, the present study detected high KIN17
expression levels in pancreatic cancer, which were related to lymph
node metastasis and poor prognosis. In addition, downregulation of
KIN17 levels was shown to significantly suppress migration and
invasion via PI3K/AKT/mTOR signaling-mediated autophagy. These
results suggested that KIN17 may be a novel biomarker for
pancreatic cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was funded by the Guangdong Basic and Applied Basic
Research Foundation (grant no. 2023A1515010235), the Start-up Fund
for High-level Talents in the Affiliated Hospital of Guangdong
Medical University (grant no. 51301Z20200007), the Medical Science
and Technology Research Project of Guangdong Province (grant nos.
A2023168 and B2021180), the Discipline Construction Project of
Guangdong Medical University (grant nos. 4SG21266P and 4SG21276P)
and the Guangdong Medical University (grant no. 2XK22015). The
funders played no role in the study design, data collection and
analysis, manuscript preparation, or publication decisions.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
TZ and QL designed this study and conducted the data
analysis, while YY conducted the data search, and extracted and
analyzed the data. JT drafted the manuscript and performed data
analysis. QL and LTC provided key revisions to the manuscript for
important content and interpreted the data. XL provided the
technical support and designed study. HC and LC performed clinical
data analysis. QL and YY confirm the authenticity of all the raw
data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The requirement for ethics approval was waived by
the ethics committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
qPCR
|
quantitative PCR
|
|
IHC
|
immunohistochemistry
|
|
3-MA
|
3-methyladenine
|
|
CQ
|
chloroquine
|
|
GEO
|
Gene Expression Omnibus
|
|
WB
|
western blotting
|
References
|
1
|
Joshi VB, Gutierrez Ruiz OL and Razidlo
GL: The cell biology of metastatic invasion in pancreatic cancer:
Updates and mechanistic insights. Cancers (Basel). 15:21692023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Springfeld C, Jäger D, Büchler MW, Strobel
O, Hackert T, Palmer DH and Neoptolemos JP: Chemotherapy for
pancreatic cancer. Presse Med. 48:e159–e174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Irajizad E, Kenney A, Tang T, Vykoukal J,
Wu R, Murage E, Dennison JB, Sans M, Long JP, Loftus M, et al: A
blood-based metabolomic signature predictive of risk for pancreatic
cancer. Cell Rep Med. 4:1011942023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pinon-Lataillade G, Masson C,
Bernardino-Sgherri J, Henriot V, Mauffrey P, Frobert Y, Araneda S
and Angulo JF: KIN17 encodes an RNA-binding protein and is
expressed during mouse spermatogenesis. J Cell Sci. 117((Pt 16)):
3691–3702. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang X, Dai Z, Li Q, Lin X, Huang Q and
Zeng T: Roles and regulatory mechanisms of KIN17 in cancers
(Review). Oncol Lett. 25:1372023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai Z, Huang Q, Huang X, Zhu C, Zahid KR,
Liu T, Li Q, Wu C, Peng M, Xiao X, et al: KIN17 promotes cell
migration and invasion through stimulating the TGF-β/Smad2 pathway
in hepatocellular carcinoma. Mol Carcinog. 62:369–384. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang Q, Zahid KR, Chen J, Pang X, Zhong
M, Huang H, Pan W, Yin J, Raza U, Zeng J, et al: KIN17 promotes
tumor metastasis by activating EMT signaling in luminal-A breast
cancer. Thorac Cancer. 12:2013–2023. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heerboth S, Housman G, Leary M, Longacre
M, Byler S, Lapinska K, Willbanks A and Sarkar S: EMT and tumor
metastasis. Clin Transl Med. 4:62015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang Y, Hong W and Wei X: The molecular
mechanisms and therapeutic strategies of EMT in tumor progression
and metastasis. J Hematol Oncol. 15:1292022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goldsmith J, Levine B and Debnath J:
Autophagy and cancer metabolism. Methods Enzymol. 542:25–57. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou M, Xu W, Wang J, Yan J, Shi Y, Zhang
C, Ge W, Wu J, Du P and Chen Y: Boosting mTOR-dependent autophagy
via upstream TLR4-MyD88-MAPK signalling and downstream NF-κB
pathway quenches intestinal inflammation and oxidative stress
injury. EBioMedicine. 35:345–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei X, Yang J, Mao Y, Zhao H, Si N, Wang H
and Bian B: Arenobufagin inhibits the phosphatidylinositol
3-kinase/protein kinase B/mammalian target of rapamycin pathway and
induces apoptosis and autophagy in pancreatic cancer cells.
Pancreas. 49:261–272. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qian X, Bi QY, Wang ZN, Han F, Liu LM,
Song LB, Li CY, Zhang AQ and Ji XM: Qingyihuaji Formula promotes
apoptosis and autophagy through inhibition of MAPK/ERK and
PI3K/Akt/mTOR signaling pathway on pancreatic cancer in vivo and in
vitro. J Ethnopharmacol. 307:1161982023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Idichi T, Seki N, Kurahara H, Yonemori K,
Osako Y, Arai T, Okato A, Kita Y, Arigami T, Mataki Y, et al:
Regulation of actin-binding protein ANLN by antitumor miR-217
inhibits cancer cell aggressiveness in pancreatic ductal
adenocarcinoma. Oncotarget. 8:53180–53193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Badea L, Herlea V, Dima SO, Dumitrascu T
and Popescu I: Combined gene expression analysis of whole-tissue
and microdissected pancreatic ductal adenocarcinoma identifies
genes specifically overexpressed in tumor epithelia.
Hepatogastroenterology. 55:2016–2027. 2008.PubMed/NCBI
|
|
18
|
Jiang J, Azevedo-Pouly AC, Redis RS, Lee
EJ, Gusev Y, Allard D, Sutaria DS, Badawi M, Elgamal OA, Lerner MR,
et al: Globally increased ultraconserved noncoding RNA expression
in pancreatic adenocarcinoma. Oncotarget. 7:53165–53177. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Janky R, Binda MM, Allemeersch J, Van den
Broeck A, Govaere O, Swinnen JV, Roskams T, Aerts S and Topal B:
Prognostic relevance of molecular subtypes and master regulators in
pancreatic ductal adenocarcinoma. BMC Cancer. 16:6322016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Akerberg D, Ansari D and Andersson R:
Re-evaluation of classical prognostic factors in resectable ductal
adenocarcinoma of the pancreas. World J Gastroenterol.
22:6424–6433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang X, Dai Z, Zeng B, Xiao X, Zahid KR,
Lin X, Liu T and Zeng T: KIN17 functions in DNA damage repair and
chemosensitivity by modulating RAD51 in hepatocellular carcinoma.
Hum Cell. 37:1489–1504. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Santamaria PG, Moreno-Bueno G, Portillo F
and Cano A: EMT: Present and future in clinical oncology. Mol
Oncol. 11:718–738. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luu T: Epithelial-mesenchymal transition
and its regulation mechanisms in pancreatic cancer. Front Oncol.
11:6463992021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang M, Liu S, Chua MS, Li H, Luo D, Wang
S, Zhang S, Han B and Sun C: SOCS5 inhibition induces autophagy to
impair metastasis in hepatocellular carcinoma cells via the
PI3K/Akt/mTOR pathway. Cell Death Dis. 10:6122019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao GS, Gao ZR, Zhang Q, Tang XF, Lv YF,
Zhang ZS, Zhang Y, Tan QL, Peng DB, Jiang DM and Guo QN: TSSC3
promotes autophagy via inactivating the Src-mediated PI3K/Akt/mTOR
pathway to suppress tumorigenesis and metastasis in osteosarcoma,
and predicts a favorable prognosis. J Exp Clin Cancer Res.
37:1882018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Mao W, Liu Y, Ding J, Wang J, Yu
Z, Huang R, Yang S, Sun Y and Dong P: 3-MA enhanced
chemosensitivity in cisplatin resistant hypopharyngeal squamous
carcinoma cells via inhibiting beclin −1 mediated autophagy. Curr
Pharm Des. 27:996–1005. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu J, Yang KC, Go NE, Colborne S, Ho CJ,
Hosseini-Beheshti E, Lystad AH, Simonsen A, Guns ET, Morin GB and
Gorski SM: Chloroquine treatment induces secretion of
autophagy-related proteins and inclusion of Atg8-family proteins in
distinct extracellular vesicle populations. Autophagy.
18:2547–2560. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Holm TM, Bian ZC, Manupati K and Guan JL:
Inhibition of autophagy mitigates cell migration and invasion in
thyroid cancer. Surgery. 171:235–244. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peng Y, Wang Y, Zhou C, Mei W and Zeng C:
PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we
making headway? Front Oncol. 12:8191282022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang L, Shi J, Liu S, Huang Y, Ding H,
Zhao B, Liu Y, Wang W, Yang J and Chen Z: RAC3 inhibition induces
autophagy to impair metastasis in bladder cancer cells via the
PI3K/AKT/mTOR pathway. Front Oncol. 12:9152402022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen X, Zeh HJ, Kang R, Kroemer G and Tang
D: Cell death in pancreatic cancer: From pathogenesis to therapy.
Nat Rev Gastroenterol Hepatol. 18:804–823. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ren B, Cui M, Yang G, Wang H, Feng M, You
L and Zhao Y: Tumor microenvironment participates in metastasis of
pancreatic cancer. Mol Cancer. 17:1082018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu W and Kang Y: Epithelial-mesenchymal
plasticity in cancer progression and metastasis. Dev Cell.
49:361–374. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baum B, Settleman J and Quinlan MP:
Transitions between epithelial and mesenchymal states in
development and disease. Semin Cell Dev Biol. 19:294–308. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dongre A and Weinberg RA: New insights
into the mechanisms of epithelial-mesenchymal transition and
implications for cancer. Nat Rev Mol Cell Biol. 20:69–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dardare J, Witz A, Merlin JL, Bochnakian
A, Toussaint P, Gilson P and Harle A: Epithelial to mesenchymal
transition in patients with pancreatic ductal adenocarcinoma:
State-of-the-art and therapeutic opportunities. Pharmaceuticals
(Basel). 14:7402021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Debaugnies M, Rodriguez-Acebes S, Blondeau
J, Parent MA, Zocco M, Song Y, de Maertelaer V, Moers V, Latil M,
Dubois C, et al: RHOJ controls EMT-associated resistance to
chemotherapy. Nature. 616:168–175. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lengrand J, Pastushenko I, Vanuytven S,
Song Y, Venet D, Sarate RM, Bellina M, Moers V, Boinet A, Sifrim A,
et al: Pharmacological targeting of netrin-1 inhibits EMT in
cancer. Nature. 620:402–408. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Joshi PJ, Chawla A, Memari P, Stansfi J,
Idowu M, Sima AP and Grossman SR: Role of C terminal binding
proteins (CtBP) in pancreatic adenocarcinoma (PDAC). J Clin Oncol.
36 (Suppl 4):S3202018. View Article : Google Scholar
|
|
41
|
Castellanos JA, Merchant NB and
Nagathihalli NS: Emerging targets in pancreatic cancer:
Epithelial-mesenchymal transition and cancer stem cells. Onco
Targets Ther. 6:1261–1267. 2013.PubMed/NCBI
|
|
42
|
Tao J, Yang G, Zhou W, Qiu J, Chen G, Luo
W, Zhao F, You L, Zheng L, Zhang T and Zhao Y: Targeting hypoxic
tumor microenvironment in pancreatic cancer. J Hematol Oncol.
14:142021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wong CH, Lou UK, Fung FK, Tong JHM, Zhang
CH, To KF, Chan SL and Chen Y: CircRTN4 promotes pancreatic cancer
progression through a novel CircRNA-miRNA-lncRNA pathway and
stabilizing epithelial-mesenchymal transition protein. Mol Cancer.
21:102022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Amaravadi R, Kimmelman AC and White E:
Recent insights into the function of autophagy in cancer. Genes
Dev. 30:1913–1930. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qiang L, Zhao B, Ming M, Wang N, He TC,
Hwang S, Thorburn A and He YY: Autophagy regulates tumor growth and
metastasis. bioRxiv. Nov 3–2023.(Epub ahead of print). doi:.
https://doi.org/10.1101/2023.10.31.564991
|
|
47
|
Wu J, Chen X, Liu X, Huang S, He C, Chen B
and Liu Y: Autophagy regulates TGF-β2-induced
epithelial-mesenchymal transition in human retinal pigment
epithelium cells. Mol Med Rep. 17:3607–3614. 2018.PubMed/NCBI
|
|
48
|
Zhu H, Gan X, Jiang X, Diao S, Wu H and Hu
J: ALKBH5 inhibited autophagy of epithelial ovarian cancer through
miR-7 and BCL-2. J Exp Clin Cancer Res. 38:1632019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mortazavi M, Moosavi F, Martini M,
Giovannetti E and Firuzi O: Prospects of targeting PI3K/AKT/mTOR
pathway in pancreatic cancer. Crit Rev Oncol Hematol.
176:1037492022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Stanciu S, Ionita-Radu F, Stefani C,
Miricescu D, Stanescu S II, Greabu M, Ripszky Totan A and Jinga M:
Targeting PI3K/AKT/mTOR signaling pathway in pancreatic cancer:
From molecular to clinical aspects. Int J Mol Sci. 23:101322022.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mohite R and Doshi G: Elucidation of the
role of the epigenetic regulatory Mechanisms of PI3K/AKT/mTOR
signaling pathway in human malignancies. Curr Cancer Drug Targets.
24:231–244. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gao W, Guo H, Niu M, Zheng X, Zhang Y, Xue
X, Bo Y, Guan X, Li Z, Guo Y, et al: circPARD3 drives malignant
progression and chemoresistance of laryngeal squamous cell
carcinoma by inhibiting autophagy through the PRKCI-Akt-mTOR
pathway. Mol Cancer. 19:1662020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sun X, Shu Y, Xu M, Jiang J, Wang L, Wang
J, Huang D and Zhang J: ANXA6 suppresses the tumorigenesis of
cervical cancer through autophagy induction. Clin Transl Med.
10:e2082020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zheng X, Li W, Xu H, Liu J, Ren L, Yang Y,
Li S, Wang J, Ji T and Du G: Sinomenine ester derivative inhibits
glioblastoma by inducing mitochondria-dependent apoptosis and
autophagy by PI3K/AKT/mTOR and AMPK/mTOR pathway. Acta Pharm Sin B.
11:3465–3480. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dai H, Hu W, Zhang L, Jiang F, Mao X, Yang
G and Li L: FGF21 facilitates autophagy in prostate cancer cells by
inhibiting the PI3K-Akt-mTOR signaling pathway. Cell Death Dis.
12:3032021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wei R, Xiao Y, Song Y, Yuan H, Luo J and
Xu W: FAT4 regulates the EMT and autophagy in colorectal cancer
cells in part via the PI3K-AKT signaling axis. J Exp Clin Cancer
Res. 38:1122019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Farhan M, Silva M, Xingan X, Zhou Z, Zheng
W and Ciriolo MR: Artemisinin inhibits the migration and invasion
in uveal melanoma via inhibition of the PI3K/AKT/mTOR signaling
pathway. Oxid Med Cell Longev. 2021:99115372021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang H, Li Z, Wang Z, Zhang X, Dai X, Zhou
G and Ding Q: Histocompatibility minor 13 (HM13), targeted by
miR-760, exerts oncogenic role in breast cancer by suppressing
autophagy and activating PI3K-AKT-mTOR pathway. Cell Death Dis.
13:7282022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nie S, Qian X, Shi M, Li H, Peng C, Ding
X, Zhang S, Zhang B, Xu G, Lv Y, et al: ALDH1A3 accelerates
pancreatic cancer metastasis by promoting glucose metabolism. Front
Oncol. 10:9152020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Huang L, Sun J, Ma Y, Chen H, Tian C and
Dong M: MSI2 regulates NLK-mediated EMT and PI3K/AKT/mTOR pathway
to promote pancreatic cancer progression. Cancer Cell Int.
24:2732024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shao Q, Zhang Z, Cao R, Zang H, Pei W and
Sun T: CPA4 promotes EMT in pancreatic cancer via stimulating
PI3K-AKT-mTOR signaling. Onco Targets Ther. 13:8567–8580. 2020.
View Article : Google Scholar : PubMed/NCBI
|