Worldwide, pancreatic cancer is the 7th most common
cause of cancer-related mortality, resulting in approximately
432,000 deaths per year, according to the 2018 GLOBOCAN study
(1). In Western countries alone,
the mortality rate associated with pancreatic cancer is ranked 4th
and is projected to be ranked 2nd by 2030 (1,2).
In the United States, 82% of pancreatic cancer cases lead to death
(3). Among the different types of
pancreatic cancers, pancreatic ductal adenocarcinoma (PDAC)
encompasses 90-95% of all cases (4,5).
According to the American Cancer Society, a patient with stage IIA
pancreatic cancer has a 5-year survival rate of approximately 5%
(6). These statistics indicate an
alarming increase in the incidence of and mortality associated with
pancreatic cancer. The poor prognosis of patients with pancreatic
cancer is due to a number of reasons, including late-stage
detection, a lack of sensitive and specific markers, as well as
ineffective imaging in the early stages (4,7).
p53 family isoform proteins include p53, p63 and
p73, all of which are evolutionarily conserved in humans and other
animals. In fact, the origins of p63 seems to go further than the
other two proteins (8-10).
In cancer research, p53 is known to be the ‘guardian of the genome’
with anti-proliferative properties that prevent the growth of
cancer. In 1997, the discovery of p63 and p73 genes, both of which
encode for p53-like sequence specific transcription factors with
similar functions, attracted scientists to investigate their role
in various types of cancer (11-15).
p63 and p73 have the potential to transactivate
target genes of p53, including BAX, NOXA and PUMA, which are
responsible for cell death, p21WAF1, responsible for
cell cycle arrest and cellular senescence, and 14-3-3σ, which is
pivotal in cell cycle arrest (8,12).
The transactivating (TA) isoforms of p63 and p73 transactivate
p53-target genes in response to anti-cancer drugs with
pro-apoptotic functions, while the
NH2-terminally-truncated deltaN (DN) isoforms exert a
dominant-negative behaviour against TA, and hence, are known to be
pro-oncogenic (16). It should be
noted that p53-dependent cell death requires the assistance of
TAp73 and/or TAp63, whereas TAp73/TAp63-dependent cell death
following DNA damage occurs without the need for p53(17).
With their controversial roles in cancer, interest
in the p53 family isoforms has intensified over the past decade.
Notably, research into p53 mutations in PDAC progression has
established a platform for exploration of their various
interactions with p63 and p73 (16,17). It is becoming increasingly clear
that the aggressive and chemoresistant traits of PDAC may not be
entirely elucidated by p53-driven mechanisms alone, but may
implicate specific isoforms of the p53 family (18-20).
Thus, this review discusses the role of the p53 family isoforms in
pancreatic cancer, with a perspective on factors conferring cancer
aggressiveness and chemoresistance.
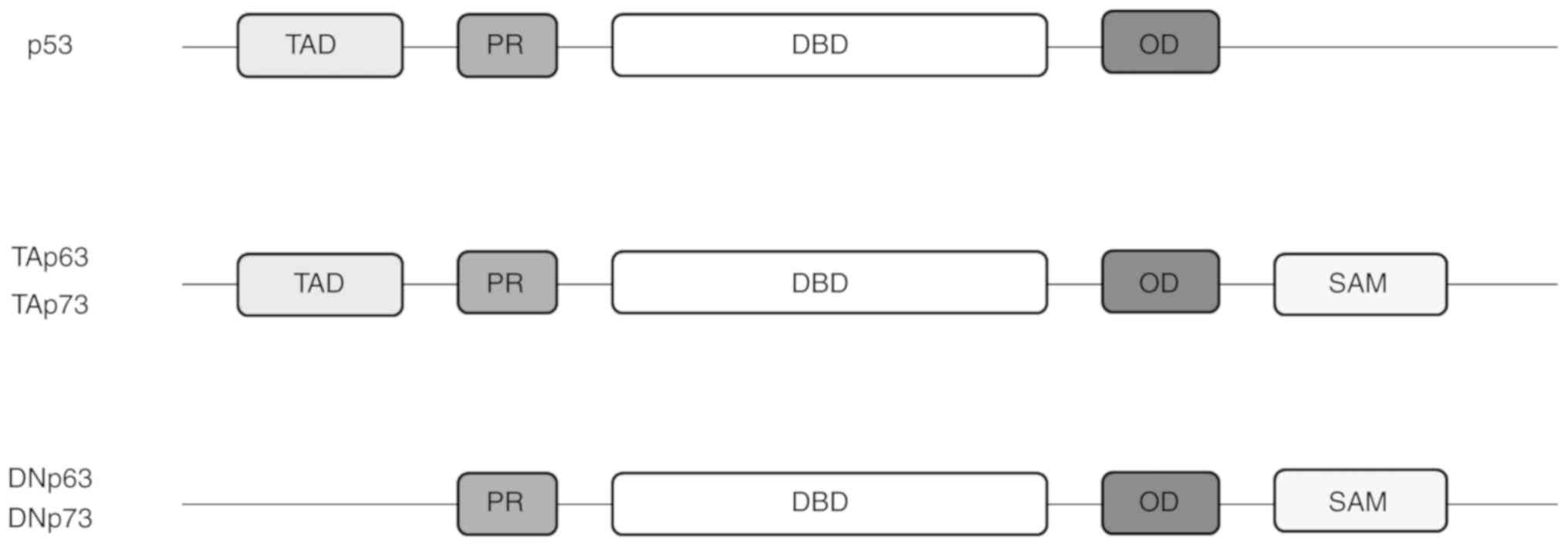
Structural similarity in proteins can be an
indication of the functional similarity of these proteins and p53
family isoforms are no exception (21,22) (Fig.
1). Isoforms of the p53 family have a similar build, with an
N-terminal transactivation domain (TAD) together with a
proline-rich domain (PR), a central, highly similar DNA-binding
domain (DBD), followed by a C-terminal
tetramerisation/oligomerisation domain (OD). p63 and p73 contain an
additional sterile alpha motif (SAM) domain and a
transcription-inhibition domain (TID), which are absent in p53
(23-28).
Unlike p53, p63 and p73 exist in distinct functional isoforms,
those containing a TAD and those without (13,28). Functionally, TAD seems to be
involved in DNA editing and repair pathways, as well as cellular
senescence (29); the SAM domain
is known to be involved in protein-protein interaction, as well as
in the stabilisation of p63 and p73 proteins (24,29,30); thus, it has been suggested that
p63 and p73 are more stable than p53 due to the presence of the SAM
domain (31).
The p53 gene codes for a nuclear transcription
factor that responds to genotoxic stress. Strong genotoxic stress
activates p53 and promotes cell cycle arrest, cellular senescence
and apoptosis, while mild genotoxic stress can activate pathways
responsible for repair mechanisms (17,20,32,33). On its own, p53 has 12 different
isoforms with similar or unique functions, as a result of alternate
splicing, the presence of diverse transcription promoters, as well
as multiple translation initiation sites (32,34,35).
In human cancer, p53 inactivation by mutation occurs
in >50% of cases, and therefore, it is known to be the most
common genetic alteration (36,37). These mutations commonly occur
within the DBD, resulting in the loss of p53 functions (38,39). In spite of this, the expression of
mutant p53 remains in cancerous cells, which is suggestive of
gain-of-function activities. In fact, cancers with the expression
of mutant p53 are known to develop more aggressive tumours with an
earlier onset, in comparison with cancers that are p53-null
(9,35,40,41). The missense mutations, R248H,
R273H and R175H, are p53 mutations with the highest frequencies in
human cancer (35,40,42,43). Certain mutations result in the
loss-of-function of remaining wild-type p53 (dominant-negative
effect), while others are known to exert a dominant-negative effect
on other tumour suppressors, such as TAp73 (35,44-52).
Gain-of-function activities of mutant p53 are
responsible for the enhanced tumourigenicity of pancreatic cancer.
This was first proven by Wolf et al (1984) through the
transfection of mutant p53 into p53-null tumour cells (58). A number of studies have since
demonstrated specific p53 gain-of-function activities, such as
metabolic changes, migration, promoting cell proliferation,
metastasis anti-apoptosis, invasion and angiogenesis (59-62).
Studies on MiaPaca-2 pancreatic cancer cell lines, which contain
the R273H mutation of p53, have demonstrated that this mutation is
responsible for increased proliferation, increased colony formation
and drug resistance (36,57,63-65).
Platelet-derived growth factor receptor β (PDGFRβ) has been
identified as a downstream mediator of mutant p53 in MiaPaca-2 and
BxPC-3 pancreatic cancer cell lines, as well as in murine
pancreatic cancer models. This PDGFRβ-mutant p53 axis is believed
to increase pancreatic cancer cell growth (56). Mutant p53 has also been known to
manipulate the pancreatic cell autophagy mechanism, resulting in
increased nutrient uptake and a higher growth rate (66).
The mechanisms though which the different p53
hotspot mutations exhibit the gain-of-function properties are
diverse. Family isoforms of p53 are structurally similar to p53,
particularly in the DNA-binding domain, which allows a p53 target
genes to interact with p63 and p73 to mediate responses, such as
cell cycle arrest, cellular responses to stress and apoptosis. A
subset of p53 hotspot mutations are capable of inactivating p63 and
p73 by forming complexes with them. These interactions, which
become possible through the conformational changes in the
DNA-binding domain of mutant p53, result in gain-of-function
properties, such as metastasis, invasion, migration and
chemoresistance (21,48,67).
Another mechanism involves physical interaction
between mutant p53 and transcription factors, such as NF-Y to
mediate target gene expression by altering cell cycle regulation,
since the DNA binding sequence of NF-Y is present in the regulatory
region of genes involved in the cell cycle (68), as well as interactions with other
transcription factors, such as E2F transcription factor 1 (E2F1),
vitamin D receptor and nuclear factor (NF)-κB (69). Zhang et al (2013) (62), through their in vivo and
in vitro evaluation of mutant p53 knockin mice, discovered
that mutant p53 is capable of driving the Warburg effect. This
phenomenon is likely driven by the activation of ROCK signalling,
which promotes the translocation of GLUT1 to the plasma membrane
(62).
Mutant p53 is also known to enhance tumourigenicity
and genomic instability by forming complexes with proteins, such as
MRE11 (R175H), a DNA nuclease (70), and topoisomerase 1 (R273H), which
is responsible for maintaining DNA topology (71). The mutant p53-ATM complex is
responsible for inactivating DNA damage responses and leads to
chromosomal translocations and cell cycle arrest (in the case of
R273H mutation) (72). Brosh
and Rotter (2009) demonstrated that mutant p53 is capable of
up- or downregulating various genes involved in tumourigenesis,
such as NF-κB2, vascular endothelial growth factor receptor
(VEGFR), Myc, Fos, insulin-like growth factor 2 (IGF2), insulin
like growth factor 1 receptor (IGF1R) and early growth response
protein 1 (EGR1) (61). This is
possible due to the DNA-binding ability of mutant p53 in a DNA
structure-selective mode. It has a high affinity for the AT-rich
regions and is shown to bind selectively with high affinity B
conformation DNA (73,74).
Interaction with miRNA is another mechanism through
which mutant p53 exerts its effect, by either inducing or
repressing its functions. MicroRNA (miR)-155, which has been shown
to repress zinc finger protein 652 (ZNF652), and miR-27a, which has
shown to repress EGFR, are both suppressed by mutant p53. Through
the repression of EGFR, mutant p53 is capable of stimulating cell
proliferation and tumourigenesis by promoting sustained
EGFR-induced ERK1/2 activation (75,76). Table
I summarises the various mechanisms for mutant p53
gain-of-function.
The p63 protein consists of at least six variants,
three of which contain a TA domain, and the remaining without (DN
domain) (8). These isoforms
regulate a wide range of target genes with opposing regulatory
effects. However, their role in cancer remains ambiguous (77). The view that TAp63 is a tumour
suppressor, while DNp63 acts as an oncogene is not always
applicable (51,77). For instance, the study by Flores
et al (2005) concluded that p63 heterogeneity leads to the
development of spontaneous tumours (78), while Keyes et al (2006)
came to the opposite conclusion (79). The TA and DN isoforms of p63 play
various roles in normal cells, as well as in cancerous ones; TAp63
is responsible for glycolysis through liver kinase B1 (LKB1)
protein kinase regulation, fatty acid oxidation, insulin secretion,
pro-oxidant response, as well as female germ cell preservation
(80-84).
In cancer, TAp63 is known to prevent metastases by
cell apoptosis and senescence (51,78). Previously, the loss of p63 as a
whole was shown to be associated with an accelerated tumour growth
and increased invasiveness (85,86). Current research has even extended
this finding to prove that it is the loss or inactivation of TAp63
coupled with a p53 mutation, that leads to enhanced tumourigenicity
through transforming growth factor (TGF)-β-induced pathways and the
alteration of DNA repair genes (87-89).
These findings regarding the function of TAp63 and the consequences
of its loss or inactivation have also been proven in pancreatic
cancer (88). In fact, TAp63 has
a low expression in T3M4, BxPC3, COLO-357, ASPC-1 and PANC-1
pancreatic cancer cell lines, which supports the anticancer
properties of TAp63(18).
The interaction between p63 and p53 plays an
important role in cancer progression, and both wild-type p53 and
mutant p53 are able to interact with p63 protein (48). Mutant p53 displays a stronger
dominant-negative behaviour against TAp63 in comparison with
wild-type p53, resulting in the impairment of p63 transactivational
target genes (20,35,49). This mechanism has been associated
with enhanced cell invasion and metastases in various types of
cancer, particularly breast cancer (87,90,91).
The role of DNp63 includes maintaining stem and
progenitor cells in stratified and glandular epithelial tissues, as
well as glycolysis and antioxidant defence (92-95).
As the most abundant p63 isoform, the overexpression of DNp63 in
head and neck cancer, non-small cell lung cancer and bladder cancer
suggests its tumour survival properties (96-99).
In spite of this, there are studies that have demonstrated a low
expression of DNp63 in breast and prostate adenocarcinoma, as well
as in urothelial carcinoma (97,100). This could indicate the
suppressive effect of DNp63 in certain types of cancer, alluding to
the paradoxical role of p63. According to Yang et al (2011),
DNp63 overexpression is limited to squamous cell carcinoma in which
it counteracts p53-mediated tumour suppressive activities (101). In pancreatic cancer, DNp63 is
the predominant isoform, with its overexpression being limited to
squamous differentiation (18,102). In BxPC-3, COLO-357 and T3M4
pancreatic cancer cell lines, DNp63 expression is elevated, which
suggests its cancer-enhancing properties (18).
Runt-related transcription factor 2 (RUNX2) is a
nuclear transcription factor generally associated with osteoblast
differentiation and bone formation (103,104). In tumours, RUNX2 overexpression
has been observed in breast, prostate, gastric cancer and melanoma,
as well as in acute myeloid leukaemia (105-110)
through target genes responsible for angiogenesis, invasiveness and
metastasis, such as VEGF, secreted phosphoprotein 1 (Spp1), matrix
metalloproteinase (MMP)9 and MMP13 (107,111). As regards pancreatic cancer,
Kayed et al discovered the pro-oncogenic role of RUNX2
overexpression and its effect on the tumour microenvironment
(109). RUNX2 is responsible for
resistance to gemcitabine (GEM) by attenuating p53-dependent cell
death, and the silencing of RUNX2 using siRNA has been shown to
significantly increase GEM sensitivity, irrespective of the p53
status (20,112-114).
One reason for the strong expression of mutant p53
in pancreatic cancer is the presence of histone deacetylase (HDAC)
1 and 2(115). Therefore, HDAC
inhibitors are under investigation as potential anticancer drugs,
of which SAHA, which also affects RUNX2 levels, has recently
attracted attention (116,117). MiaPaCa-2, a pancreatic cancer
cell line, contains a p53 R248W mutation. As previously
demonstrated, upon treatment with SAHA, although the TAp63, γH2A
histone family member X (γH2AX), p21,
phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, also
known as NOXA) and poly(ADP-ribose) polymerase (PARP) cleavage
levels increased, and mutant p53, RUNX and TAp73 were
downregulated, the response drug response was relatively poor.
However, when p53 was knocked down in MiaPaCa-2 cells, the further
downregulation of RUNX2 and upregulation of TAp63 were found to
lead to an enhanced sensitivity to SAHA. Similarly, the knockdown
of RUNX2 led to the further downregulation of mutant p53 and the
upregulation of TAp63(118).
These findings by Ogata et al (118) provide evidence of a regulatory
axis involving RUNX2, mutant p53 and TAp63.
RUNX2 knockdown in AsPC-1 p53-null pancreatic cancer
cells has been shown to increase GEM sensitivity through
TAp-63-dependent cell death pathway activation (113). In PANC-1 pancreatic cancer cells
with R273H p53 mutation, RUNX2 depletion mediates TAp63 induction
(119). This has been achieved
by the exposure of PANC-1 cells to GEM, after which γH2aX was
increased as a sign of DNA damage, and p73KIP1 and
phosphor-histone H3 at Ser-10 were reduced as a sign of decreased
mitosis. In addition, PARP cleavage was detected at negligible
levels. These data suggest that GEM treatment suppresses the cell
proliferation rate, but does not effectively promote cell death. At
the same time, TAp63 target gene products p21WAF1 and
NOXA are upregulated (119,120). Specific to TAp73, the E2F-1
transcriptional activator is also upregulated (119,121,122). Ozaki et al (119) then examined the effect of GEM
after mutant p53 was knocked down. The depletion of mutant p53 in
pancreatic cancer cell lines with homozygous p53 mutation was not
sufficient to enhance the cytotoxic effect of GEM therapy. However,
when RUNX2 was knocked down, the cytotoxic effect of GEM was
improved in both p53-proficient and deficient pancreatic cancer
cells by enhancing TAp63 target genes (p21WAF1 and
NOXA), but not through TAp73. This was proved by transfecting
PANC-1 cells with TAp63α plasmids, which exhibited an enhanced cell
cycle arrest and/or cell death (119).
miR-301a plays a role in pancreatic cancer
hypoxia-induced chemoresistance by targeting p63 and phosphatase
and tensin homolog (PTEN) in pancreatic cancer cells (123,124). miR-301a has been reported to be
upregulated in pancreatic cancer in comparison with the normal
pancreas and/or pancreatitis (125). Therefore, miR-301a has the
potential to be an independent prognostic marker for pancreatic
cancer (126). In various
tumours, hypoxia or low oxygen tension is associated with
chemoresistance (127) by the
upregulation of hypoxia-inducible factors (HIFs) in tumour cells
(128,129). In pancreatic cancer, a few of
these factors have been identified, including glucose transporter
type 1 (GLUT1), ATP binding cassette subfamily B member 1 (ABCB1)
and ATP binding cassette subfamily G member 2 (ABCG2), all of which
are HIF-1 target genes (130-132).
miR-301a expression is increased in an
NF-κB-independent manner due to hypoxia. The accumulation of
miR-301a leads to a decrease in the TAp63 and PTEN protein levels,
and an increase in the phosphorylation of Akt and HIF-1 factors.
Notably, the overexpression of TAp63 in hypoxic pancreatic cancer
cells leads to reduced cell viability, whereas under normoxic
conditions, this effect is not significant. This finding is
suggestive that a reduction in TAp63 contributes to hypoxia-induced
gemcitabine resistance in pancreatic cancer cells. All in all, Luo
et al suggested that hypoxia reduced TAp63 and PTEN through
the upregulation of miR-301a, which in turn promoted the
accumulation of HIF-1a factors and the phosphorylation of Akt,
leading to gemcitabine resistance (124).
The ability of DNp63 to regulate cell adhesion in
mammary epithelial cells and keratinocytes suggest its role as an
oncogene (133). This was also
shown in pancreatic cancer by a direct association between DNp63α
and β1-integrin, an extracellular matrix component that plays a
critical role in determining the invasive phenotype of PDAC
(134). Upon the upregulation of
DNp63α in PANC-1 cells, increased colony formation and
proliferation was observed through an increase in EGFR signalling
and its downstream kinases, extracellular-signal-regulated kinase
(ERK), Akt and c-Jun N-terminal kinase (JNK) (134). These outcomes, however, have not
been consistently evident in other types of pancreatic cancer
cells.
Similar to p53, p73 induces apoptosis by
transactivating p53-regulated promoters, as well as other p73
target genes, such as p53 upregulated modulator of apoptosis
(PUMA), Bax and GRAM domain containing 4 (GRAMD4), which induce
apoptosis by acting on the cell mitochondria and cytoplasm
(27,47,135-137).
Researchers remain divided as to its role in angiogenesis; some
studies have suggested that TAp73 exerts a suppressive effect
(138,140), whilst others have demonstrated
that it is pro-angiogenic (140-142).
DNp73 has been constantly shown to be pro-angiogenic (139-142).
As the predominant isoform of p73, the loss of TAp73
in various cell lines or mouse models has been associated with
spontaneous tumour development due to an enhanced genomic
instability and the inability of DNA repair mechanisms to be
activated (89,143,144). By contrast, a recent study
suggested the ability of TAp73 to indirectly induce the expression
of interleukin (IL)-1β in lung cancer cell lines, which is
suggestive of its tumour-enhancing properties (145). As regards the tumour-suppressive
properties of TAp73, a mechanism involving miRNA induction has been
identified; these miRNAs, such as miR-3158, inhibit cell migration
through epithelial-mesenchymal transition (EMT) and exhibit
anti-invasive properties in p53-mutant cancer cell lines (146,147).
In the progression of cancer, the interaction
between mutant p53 and p73 plays an important role. Mutant p53 has
the ability to co-precipitate and interact with p73, resulting in a
dominant-negative effect, which inhibits p73 activities (47,48). It is also clear that certain p53
mutations. such as R175H, which have a high frequency in pancreatic
cancer cases (43), exhibit a
stronger binding with p73 in comparison with R273H mutation
(36,48).
GEM is a nucleoside analogue and a standard
chemotherapeutic drug for pancreatic cancer (149). Although the primary action of
GEM is the inhibition of DNA synthesis by the incorporation of
gemcitabine diphosphate into DNA (150), it has a secondary effect of
activating p53 target genes by binding to DNA and terminating DNA
elongation, leading to apoptosis (151-153).
GEM itself requires phosphorylation in order to become active and
cause cytotoxicity (113).
Resistance against GEM is increasing, particularly in pancreatic
cancer cell lines with p53 mutation, such as MiaPacCa-2, or cell
lines that are null-p53, such as AsPC-1 and cell lines, such as
SW1990 that are p53 proficient are yet to exhibit GEM resistance
(20,113,149).
Numerous studies have examined mechanisms through
which to restore GEM sensitivity in pancreatic cancer. In p53-null
pancreatic cancer cell lines, such as AsPC-1, the knockdown and
silencing of RUNX2 has been shown to enhance GEM sensitivity
through TAp73 and TAp63 pathways which activates p21 and NOXA genes
(20,113).
Mouse double minute 2 (MDM2), Itch and neural
precursor cell-expressed developmentally downregulated gene 4
(NEDD4) are ubiquitin ligase proteins that are known to suppress
p73 in pancreatic cancer (149,154,155), and hence, their expression
exhibits enhanced resistance to GEM therapy (156). Targeting these proteins could
enhance GEM sensitivity in pancreatic cancer. MI-319 is an siRNA
that, in combination with cisplatin, is known to inhibit MDM2 and
therefore activate p73(157). In
pancreatic cell lines and xenograft models with p53 mutation, the
knockdown of Itch by anti-Itch shRNA transduction coupled with GEM
therapy has demonstrated improved sensitivity to GEM (149). Similarly, curcumin and curcumin
difluorinated have been identified as an anticancer agents that
inhibit NEDD4 and promote p73 activities, and hence improve the
response to GEM therapy (155,158). Apart from these three proteins,
AKT PI3K protein kinase is known to stabilise mutant p53 protein,
and therefore, its inhibition is related to an enhanced
effectiveness of cancer therapies (159). Although the effect of AKT/PI3K
has not been documented for pancreatic cancer, it is a potential
therapeutic target for future research in this area.
Apart from GEM, imatinib, a chemotherapeutic drug
most commonly used for chronic myeloid leukaemia, is a potential
treatment for pancreatic cancer with mutant p53. It targets the
PDGFRβ pathway, which is constitutively activated by mutant p53
resulting in uncontrollable cell growth (56).
In terms of gene therapy, knocking down the DN
isoform of p63 is promising target for pancreatic cancer therapy
(18). In a study using mouse
models of pancreatic cancer, the shRNA-mediated knockdown of DNp63
was shown to lead to a decrease in tumour volume compared to
identical mice carrying non-targeting shRNA (160). Table II summarises the strategies used
to inhibit various therapeutic targets in pancreatic cancer.
Another promising target for pancreatic cancer is
p53-mediated therapy. MDM2, a feedback regulator of p53, is
upregulated and frequently amplified in cancers, rendering the
MDM2-p53 pathway the optimal target for therapy (161). Several drugs have been
formulated to disrupt the p53-MDM2 pathway such as Nutlin-3a, a
selective inhibitor of MDM2, designed to block the MDM2-p53
interaction, which can induce cell cycle arrest and apoptosis by
blocking the G1 and G2 phases (161-163).
In animal models, Nutlin-3a has been demonstrated to induce the
activation of p53 signalling, as well as the suppression of tumour
growth (161). Another drug,
RITA, is a small molecule that activates the p53 pathway and has
successfully demonstrated suppression of tumour growth in animal
models (164).
Small molecules, including CP-31398, PRIMA-1 and
NSC-319726, can alter mutant p53 to exhibit wild-type p53
functions. PRIMA-1 restores the DNA binding domains by converting
the conformation of mutant p53 (R273H and R175H) to wild-type p53
(165,166). NSC-319726 has been tested to
restore the structure and function of wild type p53 in R175H
mutations, while CP-31398 stabilises the DNA binding domain of p53,
increasing the transcriptional activity (165,167). By restoring the wild-type p53
functions, it is enabled to carry out apoptosis and cell cycle
arrest (168).
Lastly, the degradation of mutant p53 in PDAC using
inhibitory factors, such as HDAC1 and HDAC2 inhibitors, block the
HDAC signalling pathway. HDAC and p53 interaction is responsible
for stabilising the mutant p53, rendering it more stable than the
wild-type p53 (115,169,170). Table III summarises the p53-mediated
therapeutics in pancreatic cancer.
The mortality rate of patients with pancreatic
cancer continues to increase due to the lack of appropriate
screening markers for early detection. As the understanding of
biology surrounding the p53 family grows, their role in
pathogenesis of cancer may be a target for cancer detection or
therapy.
In this review, the structure of p53 family
isoforms, and the role of wild-type p53, mutant p53, TAp63, DNp63,
TAp73 and DNp73 were discussed. Particularly in pancreatic cancer,
it is apparent that in addition to the loss of the apoptotic
ability of p53, mutations in this gene leads to gain of
cancer-promoting properties through various pathways, such as the
inhibition of regulatory genes, promoting growth through the PDGFRβ
pathway, as well as the manipulation of autophagy in cells. As for
p63 and p73, the function of each isoform forms a paradox as they
have contradictory properties in cancer. In actuality, the function
of each isoform varies based on the origin of cancer and seems to
be tissue-specific.
There is still much to learn about the exact role of
p53 family isoforms in cancer. In fact, due to their functional
similarity and tissue specificity, how each gene and their isoform
interact with each other is particularly attractive for future
research. In addition, future research should shift its focus to
clinical trials for therapeutic targets such as RUNX2, Itch, MDM2
and DNp63 in order to elucidate more effective strategies for the
treatment of pancreatic cancer.
Not applicable.
This study was supported by the Fundamental Research
Grant Scheme (FRGS/1/2016/SKK08/IMU/03/2) from the Ministry of
Education, Malaysia.
Not applicable.
CLL conceived the study; HJ and ALF acquired and
analysed the information and drafted the review; CLL revised it
critically for important intellectual content; all authors approved
of the version to be published, and are accountable for all aspects
of the work. All authors have read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the united states. Cancer Res. 74:2913–2921.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Siegel R, Miller K and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
What is pancreatic cancer? The American
Cancer Society Atlanta GA 2016. https://www.cancer.org/cancer/pancreatic-cancer/about/what-is-pancreatic-cancer.html.
Accessed February 11, 2019.
|
|
5
|
Kleeff J, Korc M, Apte M, La Vecchia C,
Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH
and Neoptolemos JP: Pancreatic cancer. Nat Rev Dis Primers.
2(16022)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Survival Rates for Pancreatic Cancer. The
American Cancer Society, Atlanta, GA, 2016. https://www.cancer.org/cancer/pancreatic-cancer/detection-diagnosis-staging/survival-rates.html.
Accessed March 14, 2016.
|
|
7
|
Can pancreatic cancer be found early? The
American Cancer Society Atlanta GA , 2016. https://www.cancer.org/cancer/pancreatic-cancer/detection-diagnosis-staging/detection.html.
Accessed February 11, 2019.
|
|
8
|
Yang A, Kaghad M, Wang Y, Gillett E,
Fleming MD, Dötsch V, Andrews NC, Caput D and McKeon F: P63, a P53
homolog at 3Q27-29, encodes multiple products with transactivating,
death-inducing, and dominant-negative activities. Mol Cell.
2:305–316. 1998.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hanel W, Marchenko N, Xu S, Yu SX, Weng W
and Moll U: Two hot spot mutant p53 mouse models display
differential gain of function in tumorigenesis. Cell Death Differ.
20:898–909. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ferraiuolo M, Di Agostino S, Blandino G
and Strano S: Oncogenic intra-p53 family member interactions in
human cancers. Front Oncol. 6(77)2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jost C, Marin M and Kaelin W Jr: p73 is a
human p53-related protein that can induce apoptosis. Nature.
389:191–194. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Kaghad M, Bonnet H, Yang A, Creancier L,
Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, et al:
Monoallelically expressed gene related to p53 at 1p36, a region
frequently deleted in neuroblastoma and other human cancers. Cell.
90:809–819. 1997.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Vanbokhoven H, Melino G, Candi E and
Declercq W: P63, a story of mice and men. J Invest Dermatol.
131:1196–1207. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Monti P, Russo D, Bocciardi R, Foggetti G,
Menichini P, Divizia MT, Lerone M, Graziano C, Wischmeijer A,
Viadiu H, et al: EEC- and ADULT-associated TP63 mutations exhibit
functional heterogeneity toward P63 responsive sequences. Hum
Mutat. 34:894–904. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Allocati N, Di Ilio C and De Laurenzi V:
p63/p73 in the control of cell cycle and cell death. Exp Cell Res.
318:1285–1290. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Flores ER, Tsai KY, Crowley D, Sengupta S,
Yang A, McKeon F and Jacks T: p63 and p73 are required for
p53-dependent apoptosis in response to DNA damage. Nature.
416:560–564. 2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Danilov AV, Neupane D, Nagaraja AS,
Feofanova EV, Humphries LA, DiRenzo J and Korc M:
DeltaNp63alpha-mediated induction of epidermal growth factor
receptor promotes pancreatic cancer cell growth and
chemoresistance. PLoS One. 6(e26815)2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Thakur AK, Nigri J, Lac S, Leca J, Bressy
C, Berthezene P, Bartholin L, Chan P, Calvo E, Iovanna JL, et al:
TAp73 loss favors Smad-independent TGF-β signaling that drives EMT
in pancreatic ductal adenocarcinoma. Cell Death Differ.
23:1358–1370. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Nakamura M, Sugimoto H, Ogata T, Hiraoka
K, Yoda H, Sang M, Sang M, Zhu Y, Yu M, Shimozato O and Ozaki T:
Improvement of gemcitabine sensitivity of p53-mutated pancreatic
cancer MiaPaCa-2 cells by RUNX2 depletion-mediated augmentation of
TAp73-dependent cell death. Oncogenesis. 5:e233. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Levrero M, De Laurenzi V, Costanzo A, Gong
J, Wang JY and Melino G: The p53/p63/p73 family of transcription
factors: Overlapping and distinct functions. J Cell Sci.
113:1661–1670. 2000.PubMed/NCBI
|
|
22
|
Murray-Zmijewski F, Lane DP and Bourdon
JC: p53/p63/p73 isoforms: An orchestra of isoforms to harmonise
cell differentiation and response to stress. Cell Death Differ.
13:962–972. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Enthart A, Klein C, Dehner A, Coles M,
Gemmecker G, Kessler H and Hagn F: Solution structure and binding
specificity of the p63 DNA binding domain. Sci Rep.
6(26707)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen TH, Wu YJ, Hou JN, Chiu CH and Chen
WJ: The p53 gene with emphasis on its paralogues in mosquitoes. J
Microbiol Immunol Infect. 50:747–754. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Heering J, Jonker HR, Löhr F, Schwalbe H
and Dötsch V: Structural investigations of the p53/p73 homologs
from the tunicate species Ciona intestinalis reveal the sequence
requirements for the formation of a tetramerization domain. Protein
Sci. 25:410–422. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dos Santos HG, Nunez-Castilla J and
Siltberg-Liberles J: Functional diversification after gene
duplication: Paralog specific regions of structural disorder and
phosphorylation in p53, p63, and p73. PLoS One.
11(e0151961)2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yoon MK, Ha JH, Lee MS and Chi SW:
Structure and apoptotic function of p73. BMB Rep. 48:81–90.
2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Shin JS, Ha JH, Lee DH, Ryu KS, Bae KH,
Park BC, Park SG, Yi GS and Chi SW: Structural convergence of
unstructured p53 family transactivation domains in MDM2
recognition. Cell Cycle. 14:533–543. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Walker CW, Van Beneden RJ, Muttray AF,
Böttger SA, Kelley ML, Tucker AE and Thomas WK: P53 superfamily
proteins in marine bivalve cancer and stress biology. Adv Mar Biol.
59:1–36. 2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Neira JL and Cámara-Artigas A:
Trifluoroethanol-induced conformational transition of the
C-terminal sterile alpha motif (SAM) of human p73. Arch Biochem
Biophys. 619:1–9. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Brandt T, Kaar JL, Fersht AR and
Veprintsev DB: Stability of p53 homologs. PLoS One.
7(e47889)2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Swiatkowska A, Żydowicz P, Sroka J and
Ciesiołka J: The role of the 5' terminal region of p53 mRNA in the
p53 gene expression. Acta Biochim Pol. 63:645–651. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Vousden KH and Prives C: Blinded by the
light: The growing complexity of p53. Cell. 137:413–431.
2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Luh LM, Kehrloesser S, Deutsch GB, Gebel
J, Coutandin D, Schäfer B, Agostini M, Melino G and Dötsch V:
Analysis of the oligomeric state and transactivation potential of
TAp73α. Cell Death Differ. 20:1008–1016. 2013. View Article : Google Scholar
|
|
35
|
Billant O, Léon A, Le Guellec S, Friocourt
G, Blondel M and Voisset C: The dominant-negative interplay between
p53, p63 and p73: A family affair. Oncotarget. 7:69549–69564.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Muller PA and Vousden KH: P53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 503:333–339. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Joerger AC and Fersht AR: Structural
biology of the tumor suppressor p53 and cancer-associated mutants.
Adv Cancer Res. 97:1–23. 2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Leroy B, Fournier JL, Ishioka C, Monti P,
Inga A, Fronza G and Soussi T: The TP53 website: An integrative
resource centre for the TP53 mutation database and TP53 mutant
analysis. Nucleic Acids Res. 41 (Database issue):D962–D969.
2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zerdoumi Y, Aury-Landas J, Bonaïti-Pellié
C, Derambure C, Sesboüé R, Renaux-Petel M, Frebourg T, Bougeard G
and Flaman JM: Drastic effect of germline TP53 missense mutations
in Li-Fraumeni patients. Hum Mutat. 34:453–461. 2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2(a001008)2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lehmann BD, Ding Y, Viox DJ, Jiang M,
Zheng Y, Liao W, Chen X, Xiang W and Yi Y: Evaluation of public
cancer datasets and signatures identifies TP53 mutant signatures
with robust prognostic and predictive value. BMC Cancer.
15(179)2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Inga A, Cresta S, Monti P, Aprile A, Scott
G, Abbondandolo A, Iggo R and Fronza G: Simple identification of
dominant p53 mutants by a yeast functional assay. Carcinogenesis.
18:2019–2021. 1997. View Article : Google Scholar
|
|
45
|
Monti P, Campomenosi P, Ciribilli Y,
Iannone R, Inga A, Abbondandolo A, Resnick MA and Fronza G: Tumour
p53 mutations exhibit promoter selective dominance over wild type
p53. Oncogene. 21:1641–1648. 2002.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Monti P, Perfumo C, Bisio A, Ciribilli Y,
Menichini P, Russo D, Umbach DM, Resnick MA, Inga A and Fronza G:
Dominant-negative features of mutant p53 in germline carriers have
limited impact on cancer outcomes. Mol Cancer Res. 9:271–279.
2011.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Di Como CJ, Gaiddon C and Prives C: p73
function is inhibited by tumor- derived p53 mutants in mammalian
cells. Mol Cell Biol. 19:1438–1449. 1999.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Gaiddon C, Lokshin M, Ahn J, Zhang T and
Prives C: A subset of tumor-derived mutant forms of p53
down-regulate p63 and p73 through a direct interaction with the p53
core domain. Mol Cell Biol. 21:1874–1887. 2001.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Strano S, Fontemaggi G, Costanzo A, Rizzo
MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M
and Blandino G: Physical interaction with human tumor-derived p53
mutants inhibits p63 activities. J Biol Chem. 277:18817–18826.
2002.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Monti P, Campomenosi P, Ciribilli Y,
Iannone R, Aprile A, Inga A, Tada M, Menichini P, Abbondandolo A
and Fronza G: Characterization of the p53 mutants ability to
inhibit p73 beta transactivation using a yeast-based functional
assay. Oncogene. 22:5252–5260. 2003.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Melino G: P63 is a suppressor of
tumorigenesis and metastasis interacting with mutant P53. Cell
Death Differ. 18:1487–1499. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Oren M and Rotter V: Mutant p53
gain-of-function in cancer. Cold Spring Harbor perspectives in
biology. 2(a001107)2010.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Li DH, Xie KP, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Brody JR, Costantino CL, Potoczek M,
Cozzitorto J, McCue P, Yeo CJ, Hruban RH and Witkiewicz AK:
Adenosquamous carcinoma of the pancreas harbors KRAS2, DPC4 and
TP53 molecular alterations similar to pancreatic ductal
adenocarcinoma. Mod Pathol. 22:651–659. 2009.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Simtniece Z, Vanags A, Strumfa I, Sperga
M, Vasko E, Prieditis P, Trapencieris P and Gardovskis J:
Morphological and immunohistochemical profile of pancreatic
neuroendocrine neoplasms. Pol J Pathol. 66:176–194. 2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Weissmueller S, Manchado E, Saborowski M,
Morris JP IV, Wagenblast E, Davis CA, Moon SH, Pfister NT,
Tschaharganeh DF, Kitzing T, et al: Mutant p53 drives pancreatic
cancer metastasis through cell-autonomous PDGF receptor β
signaling. Cell. 157:382–394. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Morton JP, Timpson P, Karim SA, Ridgway
RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG,
et al: Mutant p53 drives metastasis and overcomes growth
arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA.
107:246–251. 2010.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Wolf D, Harris N and Rotter V:
Reconstitution of p53 expression in a nonproducer
Ab-MuLV-transformed cell line by transfection of a functional p53
gene. Cell. 38:119–126. 1984.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Muller PA, Vousden KH and Norman JC: p53
and its mutants in tumor cell migration and invasion. J Cell Biol.
192:209–218. 2011.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–1286. 2012.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Brosh R and Rotter V: When mutants gain
new powers: News from the mutant p53 field. Nat Rev Cancer.
9:701–713. 2009.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Zhang C, Liu J, Liang Y, Wu R, Zhao Y,
Hong X, Lin M, Yu H, Liu L, Levine AJ, et al: Tumour-associated
mutant p53 drives the Warburg effect. Nat Commun.
4(2935)2013.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Yan W, Liu G, Scoumanne A and Chen X:
Suppression of inhibitor of differentiation 2, a target of mutant
p53, is required for gain-of-function mutations. Cancer Res.
68:6789–6796. 2008.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Do PM, Varanasi L, Fan S, Li C, Kubacka I,
Newman V, Chauhan K, Daniels SR, Boccetta M, Garrett MR, et al:
Mutant p53 cooperates with ETS2 to promote etoposide resistance.
Genes Dev. 26:830–845. 2012.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Yan W and Chen X: Identification of GRO1
as a critical determinant for mutant p53 gain of function. J Biol
Chem. 284:12178–12187. 2009.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Rosenfeldt MT, O'Prey J, Morton JP, Nixon
C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al:
P53 status determines the role of autophagy in pancreatic tumour
development. Nature. 504:296–300. 2013.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Li Y and Prives C: Are interactions with
p63 and p73 involved in mutant p53 gain of oncogenic function?
Oncogene. 26:2220–2225. 2007.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Di Agostino S, Strano S, Emiliozzi V,
Zerbini V, Mottolese M, Sacchi A, Blandino G and Piaggio G: Gain of
function of mutant p53: The mutant p53/NF-Y protein complex reveals
an aberrant transcriptional mechanism of cell cycle regulation.
Cancer Cell. 10:191–202. 2006.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Fiorini C, Cordani M, Padroni C, Blandino
G, Di Agostino S and Donadelli M: Mutant p53 stimulates
chemoresistance of pancreatic adenocarcinoma cells to gemcitabine.
Biochim Biophys Acta. 1853:89–100. 2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Song H, Hollstein M and Xu Y: p53
gain-of-function cancer mutants induce genetic instability by
inactivating ATM. Nat Cell Biol. 9:573–580. 2007.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Restle A, Färber M, Baumann C, Böhringer
M, Scheidtmann KH, Müller-Tidow C and Wiesmüller L: Dissecting the
role of p53 phosphorylation in homologous recombination provides
new clues for gain-of-function mutants. Nucleic Acids Res.
36:5362–5375. 2008.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Liu DP, Song H and Xu Y: A common gain of
function of p53 cancer mutants in inducing genetic instability.
Oncogene. 29:949–956. 2010.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Müller BF, Paulsen D and Deppert W:
Specific binding of MAR/SAR DNA-elements by mutant p53. Oncogene.
12:1941–1952. 1996.PubMed/NCBI
|
|
74
|
Will K, Warnecke G, Wiesmüller L and
Deppert W: Specific interaction of mutant p53 with regions of
matrix attachment region DNA elements (MARs) with a high potential
for base-unpairing. Proc Natl Acad Sci USA. 95:13681–13686. 1998.
View Article : Google Scholar
|
|
75
|
Neilsen PM, Noll JE, Mattiske S, Bracken
CP, Gregory PA, Schulz RB, Lim SP, Kumar R, Suetani RJ, Goodall GJ
and Callen DF: Mutant p53 drives invasion in breast tumors through
up-regulation of miR-155. Oncogene. 32:2992–3000. 2013.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Wang W, Cheng B, Miao L, Mei Y and Wu M:
Mutant p53-R273H gains new function in sustained activation of EGFR
signaling via suppressing miR-27a expression. Cell Death Dis.
4(e574)2013.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Gonfloni S, Caputo V and Iannizzotto V:
P63 in health and cancer. Int J Dev Biol. 59:87–93. 2015.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Flores ER, Sengupta S, Miller JB, Newman
JJ, Bronson R, Crowley D, Yang A, McKeon F and Jacks T: Tumor
predisposition in mice mutant for p63 and p73: Evidence for broader
tumor suppressor functions for the p53 family. Cancer Cell.
7:363–373. 2005.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Keyes WM, Vogel H, Koster MI, Guo X, Qi Y,
Petherbridge KM, Roop DR, Bradley A and Mills AA: P63 heterozygous
mutant mice are not prone to spontaneous or chemically induced
tumors. Proc Natl Acad Sci USA. 103:8435–8440. 2006.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Su X, Gi YJ, Chakravarti D, Chan IL, Zhang
A, Xia X, Tsai KY and Flores ER: TAp63 is a master transcriptional
regulator of lipid and glucose metabolism. Cell Metab. 16:511–525.
2012.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Giacobbe A, Bongiorno-Borbone L,
Bernassola F, Terrinoni A, Markert EK, Levine AJ, Feng Z, Agostini
M, Zolla L, Agrò AF, et al: P63 regulates glutaminase 2 expression.
Cell Cycle. 12:1395–1405. 2013.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Liu G and Chen X: The ferredoxin reductase
gene is regulated by the p53 family and sensitizes cells to
oxidative stress-induced apoptosis. Oncogene. 21:7195–7204.
2002.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Suh EK, Yang A, Kettenbach A, Bamberger C,
Michaelis AH, Zhu Z, Elvin JA, Bronson RT, Crum CP and McKeon F:
P63 protects the female germ line during meiotic arrest. Nature.
444:624–628. 2006.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Su X, Napoli M, Abbas HA, Venkatanarayan
A, Bui NHB, Coarfa C, Gi YJ, Kittrell F, Gunaratne PH, Medina D, et
al: TAp63 suppresses mammary tumorigenesis through regulation of
the Hippo pathway. Oncogene. 36:2377–2393. 2017.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Urist MJ, Di Como CJ, Lu M-L,
Charytonowicz E, Verbel D, Crum CP, Ince TA, McKeon FD and
Cordon-Cardo C: Loss of p63 expression is associated with tumor
progression in bladder cancer. Am J Pathol. 161:1199–1206.
2002.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Barbieri CE, Tang LJ, Brown KA and
Pietenpol JA: Loss of p63 leads to increased cell migration and
up-regulation of genes involved in invasion and metastasis. Cancer
Res. 66:7589–7597. 2006.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Adorno M, Cordenonsi M, Montagner M,
Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo
V, et al: A mutant-p53/Smad complex opposes p63 to empower
TGFbeta-induced metastasis. Cell. 137:87–98. 2009.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Tan EH, Morton JP, Timpson P, Tucci P,
Melino G, Flores ER, Sansom OJ, Vousden KH and Muller PA: Functions
of TAp63 and p53 in restraining the development of metastatic
cancer. Oncogene. 33:3325–3333. 2014.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Lin YL, Sengupta S, Gurdziel K, Bell GW,
Jacks T and Flores ER: p63 and p73 transcriptionally regulate genes
involved in DNA repair. PLoS Genet. 5(e1000680)2009.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Marine JC and Berx G: Transforming growth
factor-beta and mutant p53 conspire to induce metastasis by
antagonizing p63: A (ternary) complex affair. Breast Cancer Res.
11(304)2009.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Neilsen PM, Noll JE, Suetani RJ, Schulz
RB, Al-ejeh F, Evdokiou A, Lane DP and Callen DF: Mutant p53 uses
p63 as a molecular chaperone to alter gene expression and induce a
pro-invasive secretome. Oncotarget. 2:1203–1217. 2011.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Viticchiè G, Agostini M, Lena AM, Mancini
M, Zhou H, Zolla L, Dinsdale D, Saintigny G, Melino G and Candi E:
p63 supports aerobic respiration through hexokinase II. Proc Natl
Acad Sci USA. 112:11577–11582. 2015.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Yan W and Chen X: GPX2, a direct target of
p63, inhibits oxidative stress-induced apoptosis in a p53-dependent
manner. J Biol Chem. 281:7856–7862. 2006.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Senoo M, Pinto F, Crum CP and McKeon F:
p63 is essential for the proliferative potential of stem cells in
stratified epithelia. Cell. 129:523–536. 2007.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Pignon JC, Grisanzio C, Geng Y, Song J,
Shivdasani RA and Signoretti S: P63-expressing cells are the stem
cells of developing prostate, bladder, and colorectal epithelia.
Proc Natl Acad Sci USA. 110:8105–8110. 2013.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Rocco JW, Leong CO, Kuperwasser N, DeYoung
MP and Ellisen LW: P63 mediates survival in squamous cell carcinoma
by suppression of p73-dependent apoptosis. Cancer Cell. 9:45–56.
2006.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Di Como CJ, Urist MJ, Babayan I, Drobnjak
M, Hedvat CV, Teruya-Feldstein J, Pohar K, Hoos A and Cordon-Cardo
C: P63 expression profiles in human normal and tumor tissues. Clin
Cancer Res. 8:494–501. 2002.PubMed/NCBI
|
|
98
|
Deyoung MP and Ellisen LW: P63 and P73 in
human cancer: Defining the network. Oncogene. 26:5169–5183.
2007.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Leong CO, Vidnovic N, DeYoung MP, Sgroi D
and Ellisen LW: The p63/p73 network mediates chemosensitivity to
cisplatin in a biologically defined subset of primary breast
cancers. J Clin Invest. 117:1370–1380. 2007.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Fukushima H, Koga F, Kawakami S, Fujii Y,
Yoshida S, Ratovitski E, Trink B and Kihara K: Loss of
DeltaNp63alpha promotes invasion of urothelial carcinomas via
N-cadherin/Src homology and collagen/extracellular signal-regulated
kinase pathway. Cancer Res. 69:9263–9270. 2009.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Yang X, Lu H, Yan B, Romano RA, Bian Y,
Friedman J, Duggal P, Allen C, Chuang R, Ehsanian R, et al: ΔNp63
versatilely regulates a broad NF-κB gene program and promotes
squamous epithelial proliferation, migration, and inflammation.
Cancer Res. 71:3688–3700. 2011.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Basturk O, Khanani F, Sarkar F, Levi E,
Cheng JD and Adsay NV: DeltaNp63 expression in pancreas and
pancreatic neoplasia. Mod Pathol. 18:1193–1198. 2005.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Komori T, Yagi H, Nomura S, Yamaguchi A,
Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al:
Targeted disruption of Cbfa1 results in a complete lack of bone
formation owing to maturational arrest of osteoblasts. Cell.
89:755–764. 1997.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Otto F, Thornell AP, Crompton T, Denzel A,
Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen
BR, et al: Cbfa1, a candidate gene for cleidocranial dysplasia
syndrome, is essential for osteoblast differentiation and bone
development. Cell. 89:765–771. 1997.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Barnes GL, Javed A, Waller SM, Kamal MH,
Hebert KE, Hassan MQ, Bellahcene A, Van Wijnen AJ, Young MF, Lian
JB, et al: Osteoblast-related transcription factors Runx2
(Cbfa1/AML3) and MSX2 mediate the expression of bone sialoprotein
in human metastatic breast cancer cells. Cancer Res. 63:2631–2637.
2003.PubMed/NCBI
|
|
106
|
Akech J, Wixted JJ, Bedard K, van der Deen
M, Hussain S, Guise TA, van Wijnen AJ, Stein JL, Languino LR,
Altieri DC, et al: Runx2 association with progression of prostate
cancer in patients: Mechanisms mediating bone osteolysis and
osteoblastic metastatic lesions. Oncogene. 29:811–821.
2010.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Pratap J, Javed A, Languino LR, van Wijnen
AJ, Stein JL, Stein GS and Lian JB: The Runx2 osteogenic
transcription factor regulates matrix metalloproteinase 9 in bone
metastatic cancer cells and controls cell invasion. Mol Cell Biol.
25:8581–8591. 2005.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Kuo Y, Zaidi SK, Gornostaeva S, Komori T,
Stein GS and Castilla LH: Runx2 induces acute myeloid leukemia in
cooperation with Cbfbeta-SMMHC in mice. Blood. 113:3323–3333.
2019.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Kayed H, Jiang X, Keleg S, Jesnowski R,
Giese T, Berger M, Esposito I, Löhr M, Friess H and Kleeff J:
Regulation and functional role of the Runt-related transcription
factor-2 in pancreatic cancer. Br J Cancer. 97:1106–1115.
2007.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Boregowda R, Olabisi O, Abushahba W, Jeong
B, Haenssen K, Chen W, Chekmareva M, Lasfar A, Foran DJ, Goydos JS
and Cohen-Solal KA: RUNX2 is overexpressed in melanoma cells and
mediates their migration and invasion. Cancer Lett. 348:61–70.
2014.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Zelzer E, Glotzer DJ, Hartmann C, Thomas
D, Fukai N, Soker S and Olsen BR: Tissue specific regulation of
VEGF expression during bone development requires Cbfa1/Runx2. Mech
Dev. 106:97–106. 2001.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Ozaki T, Wu D, Sugimoto H, Nagase H and
Nakagawara A: Runt-related transcription factor 2 (RUNX2) inhibits
p53-dependent apoptosis through the collaboration with HDAC6 in
response to DNA damage. Cell Death Dis. 4(e610)2013.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Sugimoto H, Nakamura M, Yoda H, Hiraoka K,
Shinohara K, Sang M, Fujiwara K, Shimozato O, Nagase H and Ozaki T:
Silencing of RUNX2 enhances gemcitabine sensitivity of
p53-deficient human pancreatic cancer AsPC-1 cells through the
stimulation of TAp63-mediated cell death. Cell Death Dis.
6(e1914)2015.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Ozaki T, Sugimoto H, Nakamura M, Hiraoka
K, Yoda H, Sang M, Fujiwara K and Nagase H: Runt-related
transcription factor 2 attenuates the transcriptional activity as
well as DNA damage-mediated induction of pro-apoptotic TAp73 to
regulate chemosensitivity. FEBS J. 282:114–128. 2015.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Stojanovic N, Hassan Z, Wirth M, Wenzel P,
Beyer M, Schäfer C, Brand P, Kroemer A, Stauber RH, Schmid RM, et
al: HDAC1 and HDAC2 integrate the expression of p53 mutants in
pancreatic cancer. Oncogene. 36:1804–1815. 2017.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Grant S, Easley C and Kirkpatrick P:
Vorinostat. Nat Rev Drug Discov. 6:21–22. 2007.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Gryder B, Sodji Q and Oyelere A: Targeted
cancer therapy: Giving histone deacetylase inhibitors all they need
to succeed. Futur Med Chem. 4:505–524. 2012.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Ogata T, Nakamura M, Sang M, Yoda H,
Hiraoka K, Yin D, Sang M, Shimozato O and Ozaki T: Depletion of
runt-related transcription factor 2 (RUNX2) enhances SAHA
sensitivity of p53-mutated pancreatic cancer cells through the
regulation of mutant p53 and TAp63. PLoS One.
12(e0179884)2017.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Ozaki T, Nakamura M, Ogata T, Sang M, Yoda
H, Hiraoka K, Sang M and Shimozato O: Depletion of pro-oncogenic
RUNX2 enhances gemcitabine (GEM) sensitivity of p53-mutated
pancreatic cancer Panc-1 cells through the induction of
pro-apoptotic TAp63. Oncotarget. 7:71937–71950. 2016.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Nakaya N, Ishigaki Y, Nakajima H, Murakami
M, Shimasaki T, Takata T, Ozaki M, Dusetti NJ, Iovanna JL and Motoo
Y: Meaning of tumor protein 53-induced nuclear protein 1 in the
molecular mechanism of gemcitabine sensitivity. Mol Clin Oncol.
1:100–104. 2013.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Irwin M, Marin MC, Phillips AC, Seelan RS,
Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden KH and Kaelin
WG Jr: Role for the p53 homologue p73 in E2F-1-induced apoptosis.
Nature. 407:645–648. 2000.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Stiewe T and Putzer BM: Role of the
p53-homologue p73 in E2F1-induced apoptosis. Nat Genet. 26:464–469.
2000.PubMed/NCBI View
Article : Google Scholar
|
|
123
|
Xia X, Zhang K, Luo G, Cen G, Cao J, Huang
K and Qiu Z: Downregulation of miR-301a-3p sensitizes pancreatic
cancer cells to gemcitabine treatment via PTEN. Am J Transl Res.
9:1886–1895. 2017.PubMed/NCBI
|
|
124
|
Luo G, Xia X, Wang X, Zhang K, Cao J,
Jiang T, Zhao Q and Qiu Z: miR-301a plays a pivotal role in
hypoxia-induced gemcitabine resistance in pancreatic cancer. Exp
Cell Res. 369:120–128. 2018.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner
MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ and Schmittgen
TD: Expression profiling identifies microRNA signature in
pancreatic cancer. Int J Cancer. 120:1046–1054. 2007.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Xia X, Zhang K, Cen G, Jiang T, Cao J,
Huang K, Huang C, Zhao Q and Qiu Z: MicroRNA-301a-3p promotes
pancreatic cancer progression via negative regulation of SMAD4.
Oncotarget. 6:21046–21063. 2015.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Rohwer N and Cramer T: Hypoxia-mediated
drug resistance: Novel insights on the functional interaction of
HIFs and cell death pathways. Drug Resist Updat. 14:191–201.
2011.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Nakazawa MS, Keith B and Simon MC: Oxygen
availability and metabolic adaptations. Nat Rev Cancer. 16:663–673.
2016.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Rankin EB and Giaccia AJ: Hypoxic control
of metastasis. Science. 352:175–180. 2016.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Shukla SK, Purohit V, Mehla K, Gunda V,
Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A, et
al: MUC1 and HIF-1alpha signaling crosstalk induces anabolic
glucose metabolism to impart gemcitabine resistance to pancreatic
cancer. Cancer Cell. 32:71–87.e7. 2017.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Comerford KM, Wallace TJ, Karhausen J,
Louis NA, Montalto MC and Colgan SP: Hypoxia-inducible
factor-1-dependent regulation of the multidrug resistance (MDR1)
gene. Cancer Res. 62:3387–3394. 2002.PubMed/NCBI
|
|
132
|
He X, Wang J, Wei W, Shi M, Xin B, Zhang T
and Shen X: Hypoxia regulates ABCG 2 activity through the
activivation of ERK1/2/HIF-1α and contributes to chemoresistance in
pancreatic cancer cells. Cancer Biol Ther. 17:188–198.
2016.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Choi W, Shah JB, Tran M, Svatek R, Marquis
L, Lee I, Yu D, Adam L, Wen S, Shen Y, et al: p63 expression
defines a lethal subset of muscle-invasive bladder cancers. PLoS
One. 7(e30206)2012.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Dang TT, Westcott JM, Maine EA, Kanchwala
M, Xing C and Pearson GW: ΔNp63α induces the expression of FAT2 and
slug to promote tumor invasion. Oncotarget. 7:28592–28611.
2016.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Melino G, Bernassola F, Ranalli M, Yee K,
Zong WX, Corazzari M, Knight RA, Green DR, Thompson C and Vousden
KH: p75 induces apoptosis via PUMA transactivation and Bax
mitochondrial translocation. J Biol Chem. 279:8076–8083.
2004.PubMed/NCBI View Article : Google Scholar
|
|
136
|
John K, Alla V, Meier C and Pützer BM:
GRAMD4 mimics p53 and mediates the apoptotic function of p73 at
mitochondria. Cell Death Differ. 18:874–886. 2011.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Deng Y and Wu X: Peg3/Pw1 promotes
p53-mediated apoptosis by inducing Bax translocation from cytosol
to mitochondria. Proc Natl Acad Sci USA. 97:12050–12055.
2000.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Stantic M, Sakil HAM, Zirath H, Fang T,
Sanz G, Fernandez-Woodbridge A, Marin A, Susanto E, Mak TW,
Arsenian Henriksson M and Wilhelm MT: TAp73 suppresses tumor
angiogenesis through repression of proangiogenic cytokines and
HIF-1α activity. Proc Natl Acad Sci USA. 112:220–225.
2015.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Amelio I, Inoue S, Markert EK, Levine AJ,
Knight RA, Mak TW and Melino G: TAp73 opposes tumor angiogenesis by
promoting hypoxia-inducible factor 1α degradation. Proc Natl Acad
Sci USA. 112:226–231. 2015.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Dulloo I, Hooi PB and Sabapathy K:
Hypoxia-induced DNp73 stabilization regulates Vegf-A expression and
tumor angiogenesis similar to TAp73. Cell Cycle. 14:3533–3539.
2015.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Dulloo I, Phang BH, Othman R, Tan SY,
Vijayaraghavan A, Goh LK, Martin-Lopez M, Marques MM, Li CW, Wang
de Y, et al: Hypoxia-inducible TAp73 supports tumorigenesis by
regulating the angiogenic transcriptome. Nat Cell Biol. 17:511–523.
2015.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Fernandez-Alonso R, Martin-Lopez M,
Gonzalez-Cano L, Garcia S, Castrillo F, Diez-Prieto I,
Fernandez-Corona A, Lorenzo-Marcos ME, Li X, Claesson-Welsh L, et
al: p73 is required for endothelial cell differentiation, migration
and the formation of vascular networks regulating VEGF and TGFβ
signaling. Cell Death Differ. 22:1287–1299. 2015.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Tomasini R, Tsuchihara K, Wilhelm M,
Fujitani M, Rufini A, Cheung CC, Khan F, Itie-Youten A, Wakeham A,
Tsao MS, et al: TAp73 knockout shows genomic instability with
infertility and tumor suppressor functions. Genes Dev.
22:2677–2691. 2008.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Tomasini R, Tsuchihara K, Tsuda C, Lau SK,
Wilhelm M, Ruffini A, Tsao MS, Iovanna JL, Jurisicova A, Melino G
and Mak TW: TAp73 regulates the spindle assembly checkpoint by
modulating BubR1 activity. Proc Natl Acad Sci USA. 106:797–802.
2009.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Vikhreva P, Petrova V, Gokbulut T,
Pestlikis I, Mancini M, Di Daniele N, Knight RA and Melino G: TAp73
upregulates IL-1β in cancer cells: Potential biomarker in lung and
breast cancer? Biochem Biophys Res Commun. 482:498–505.
2017.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Galtsidis S, Logotheti S, Pavlopoulou A,
Zampetidis CP, Papachristopoulou G, Scorilas A, Vojtesek B,
Gorgoulis V and Zoumpourlis V: Unravelling a p73-regulated network:
The role of a novel p73-dependent target, MIR3158, in cancer cell
migration and invasiveness. Cancer Lett. 388:96–106.
2017.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Ory B, Ramsey MR, Wilson C, Vadysirisack
DD, Forster N, Rocco JW, Rothenberg SM and Ellisen LW: A
microRNA-dependent program controls p53-independent survival and
chemosensitivity in human and murine squamous cell carcinoma. J
Clin Invest. 121:809–820. 2011.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Bardeesy N, Cheng K, Berger JH, Chu GC,
Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D and
DePinho RA: Smad4 is dispensable for normal pancreas development
yet critical in progression and tumor biology of pancreas cancer.
Genes Dev. 20:3130–3146. 2006.PubMed/NCBI View Article : Google Scholar
|
|
149
|
de la Fuente M, Jones MC, Santander-Ortega
MJ, Mirenska A, Marimuthu P, Uchegbu I and Schätzlein A: A
nano-enabled cancer-specific ITCH RNAi chemotherapy booster for
pancreatic cancer. Nanomedicine. 11:369–377. 2015.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Plunkett W, Huang P, Xu YZ, Heinemann V,
Grunewald R and Gandhi V: Gemcitabine: Metabolism, mechanisms of
action, and self-potentiation. Semin Oncol. 22 (4 Suppl 11):S3–S10.
1995.PubMed/NCBI
|
|
151
|
Huang P, Chubb S, Hertel L, Grindey G and
Plunkett W: Action of 2',2'-difluorodeoxycytidine on DNA synthesis.
Cancer Res. 51:6110–6117. 1991.PubMed/NCBI
|
|
152
|
Achanta G, Pelicano H, Feng L, Plunkett W
and Huang P: Interaction of p53 and DNA-PK in response to
nucleoside analogues: Potential role as a sensor complex for DNA
damage. Cancer Res. 61:8723–8729. 2001.PubMed/NCBI
|
|
153
|
Galmarini CM, Clarke ML, Falette N,
Puisieux A, Mackey JR and Dumontet C: Expression of a
non-functional p53 affects the sensitivity of cancer cells to
gemcitabine. Int J Cancer. 97:439–445. 2002.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Zeng X, Chen L, Jost CA, Maya R, Keller D,
Wang X, Kaelin WG Jr, Oren M, Chen J and Lu H: MDM2 suppresses p73
function without promoting p73 degradation. Mol Cell Biol.
19:3257–3266. 1999.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Su J, Zhou X, Yin X, Wang L, Zhao Z, Hou
Y, Zheng N, Xia J and Wang Z: The effects of curcumin on
proliferation, apoptosis, invasion, and NEDD4 expression in
pancreatic cancer. Biochem Pharmacol. 140:28–40. 2017.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Yang SH, Lee JC, Guo JC, Kuo SH, Tien YW,
Kuo TC, Cheng AL and Yeh KH: Association of MDM2 expression with
shorter progression-free survival and overall survival in patients
with advanced pancreatic cancer treated with gemcitabine-based
chemotherapy. PLoS One. 12(e0180628)2017.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Azmi AS, Aboukameel A, Banerjee S, Wang Z,
Mohammad M, Wu J, Wang S, Yang D, Philip PA, Sarkar FH and Mohammad
RM: MDM2 inhibitor MI-319 in combination with cisplatin is an
effective treatment for pancreatic cancer independent of p53
function. Eur J Cancer. 46:1122–1131. 2010.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Azmi AS, Ali S, Banerjee S, Bao B, Maitah
M, Padhye S, Philip PA, Mohammad RM and Sarkar FH: Network modeling
of CDF treated pancreatic cancer cells reveals a novel c-myc-p73
dependent apoptotic mechanism. Am J Transl Res. 3:374–382.
2011.PubMed/NCBI
|
|
159
|
Hamilton G, Abraham A, Morton J, Sampson
O, Pefani D, Khoronenkova S, Grawenda A, Papaspyropoulos A,
Jamieson N, McKay C, et al: AKT regulates NPM dependent ARF
localization and p53mut stability in tumors. Oncotarget.
5:6142–6167. 2014.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Bid HK, Roberts RD, Cam M, Audino A,
Kurmasheva RT, Lin J, Houghton PJ and Cam H: ΔNp63 promotes
pediatric neuroblastoma and osteosarcoma by regulating tumor
angiogenesis. Cancer Res. 74:320–329. 2014.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Zhang Y, Zeng SX, Hao Q and Lu H:
Monitoring p53 by MDM2 and MDMX is required for endocrine pancreas
development and function in a spatio-temporal manner. Dev Biol.
423:34–45. 2017.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Vassilev LT, Vu BT, Craves B, Carvajal D,
Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et
al: In vivo activation of the p53 pathway by small-molecule
antagonists of MDM2. Science. 303:844–848. 2004.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Grasberger BL, Lu T, Schubert C, Parks DJ,
Carver TE, Koblish HK, Cummings MD, LaFrance LV, Milkiewicz KL,
Calvo RR, et al: Discovery and cocrystal structure of
benzodiazepinedione HDM2 antagonists that activate p53 in cells. J
Med Chem. 48:909–912. 2005.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Issaeva N, Bozko P, Enge M, Protopopova M,
Verhoef LGGC, Masucci M, Pramanik A and Selivanova G: Small
molecule RITA binds to p53, blocks p53-HDM-2 interaction and
activates p53 function in tumors. Nat Med. 10:1321–1328.
2004.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Yu X, Vazquez A, Levine AJ and Carpizo DR:
Allele-specific p53 mutant reactivation. Cancer Cell. 21:614–625.
2012.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Lambert JMR, Moshfegh A, Hainaut P, Wiman
KG and Bykov VJ: Mutant p53 reactivation by PRIMA-1MET induces
multiple signaling pathways converging on apoptosis. Oncogene.
29:1329–1338. 2010.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Tang X, Zhu Y, Han L, Kim AL, Kopelovich
L, Bickers DR and Athar M: CP-31398 restores mutant p53 tumor
suppressor function and inhibits UVB-induced skin carcinogenesis in
mice. J Clin Invest. 117:3753–3764. 2007.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Bykov VJN, Issaeva N, Shilov A, Hultcrantz
M, Pugacheva E, Chumakov P, Bergman J, Wiman KG and Selivanova G:
Restoration of the tumor suppressor function to mutant p53 by a
low-molecular-weight compound. Nat Med. 8:282–288. 2002.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Li D, Marchenko ND, Schulz R, Fischer V,
Velasco-Hernandez T, Talos F and Moll UM: Functional inactivation
of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization
of mutant p53 in human cancer cells. Mol Cancer Res. 9:577–588.
2011.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Li D, Marchenko ND and Moll UM: SAHA shows
preferential cytotoxicity in mutant p53 cancer cells by
destabilizing mutant p53 through inhibition of the HDAC6-Hsp90
chaperone axis. Cell Death Differ. 18:1904–1913. 2011.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Liu J, Zhang C and Feng Z: Tumor
suppressor p53 and its gain-of-function mutants in cancer. Acta
Biochim Biophys Sin (Shanghai). 46:170–179. 2014.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Stindt MH, Muller PAJ, Ludwig RL,
Kehrloesser S, Dötsch V and Vousden KH: Functional interplay
between MDM2, p63/p73 and mutant p53. Oncogene. 34:4300–4310.
2015.PubMed/NCBI View Article : Google Scholar
|
|
173
|
Ludes-Meyers JH, Subler MA, Shivakumar CV,
Munoz RM, Jiang P, Bigger JE, Brown DR, Deb SP and Deb S:
Transcriptional activation of the human epidermal growth factor
receptor promoter by human p53. Mol Cell Biol. 16:6009–6019.
1996.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Weisz L, Zalcenstein A, Stambolsky P,
Cohen Y, Goldfinger N, Oren M and Rotter V: Transactivation of the
EGR1 gene contributes to mutant p53 gain of function. Cancer Res.
64:8318–8327. 2004.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Scian MJ, Stagliano KER, Anderson MAE,
Hassan S, Bowman M, Miles MF, Deb SP and Deb S: Tumor-derived p53
mutants induce NF-kappaB2 gene expression. Mol Cell Biol.
25:10097–10110. 2005.PubMed/NCBI View Article : Google Scholar
|
|
176
|
Strano S, Dell'Orso S, Di Agostino S,
Fontemaggi G, Sacchi A and Blandino G: Mutant p53: An oncogenic
transcription factor. Oncogene. 26:2212–2219. 2007.PubMed/NCBI View Article : Google Scholar
|
|
177
|
Wang Q, Selth LA and Callen DF: MiR-766
induces p53 accumulation and G2/M arrest by directly targeting
MDM4. Oncotarget. 8:29914–29924. 2017.PubMed/NCBI View Article : Google Scholar
|