1. Introduction
Trypanosomatids are protozoans that belong to the
order Kinetoplastidae, which includes Trypanosoma cruzi,
T. brucei and different species of Leishmania. These
parasites are transmitted by bite from tsetse fly, bed bug and sand
fly, respectively (1).
Trypanosomatids cause neglected tropical diseases, such as Chagas'
disease, African sleeping sickness and leishmaniasis, which are
debilitating and often fatal (2,3).
These diseases affect >20 million individuals worldwide,
primarily in rural and underdeveloped areas, resulting in
significant suffering in these regions (4,5). For
example, >60 million individuals in sub-Saharan African
countries are at risk of sleeping sickness presenting with fever,
headaches and neurological symptoms, with an annual infection rate
of 300,000-500,000(6).
Furthermore, ~350 million individuals in 92 countries are at risk
of leishmaniasis which causes skin lesions and organ damage,
significantly affecting social structures by exacerbating poverty,
limiting economic productivity and straining healthcare systems in
affected communities, with ~1.2 million new infections each year
(7,8).
Current therapies for trypanosomatids, such as
antimonials, Melarsoprol and pentamidine, have limited efficacy due
to extreme toxicity, resistance, lack of cost efficiency and the
need for parenteral delivery in some cases (6,9-11).
Untreated trypanosomatid infections can be fatal (10). Furthermore, there are no effective
vaccines for the prevention of trypanosomatid diseases, making
rational drug design and the search for excellent therapeutic
targets crucial (4,12). Extensive research has been
conducted on the biochemical characteristics of the life stages of
these parasites, leading to the identification of several potential
targets for anti-trypanosomatid treatment (13,14).
Several potential targets are proposed for anti-trypanosomatid
treatment, such as trypanothione redox, sterol biosynthesis, purine
uptake, folate metabolism, glycosomal enzymes and protein
translocation to the glycosome (15-19).
The present review explored rational drug design strategies
targeting glycosomal enzymes and protein translocation in
trypanosomatids. These targets are crucial because they play
critical roles in the unique metabolic pathways of trypanosomatids,
which are not found in the host organisms. Glycosomal glycolysis is
the only source of energy production, so targeting these pathways
is essential for developing effective and selective therapeutics
against trypanosomatids.
2. Glycosomes
Glycosomes, which are membrane-bound cytoplasmic
peroxisome-like organelles found in the protozoan parasites,
Kinetoplastidae, play a vital role in the metabolism of
trypanosomatids (8). These
organelles have the unique feature of containing numerous enzymes
from the glycolytic and gluconeogenic pathways (20).
Investigating energy metabolism in the amastigote
stage of T. cruzi is challenging due to the complications
arising from host metabolic activity. Nevertheless, studies have
demonstrated that glycolysis serves as the primary energy source
for the amastigote stage (21,22).
Essential proteins and enzymes for parasite survival are
synthesized in the cytosol and must be transported across the
glycosomal membrane into the glycosome. Thus, inhibiting the
transport and activity of these proteins and enzymes can adversely
affect the parasites (23). The
glycosome is a crucial organelle for trypanosomatid metabolism,
containing enzymes essential for glycolysis and researching the
energy metabolism of these parasites and their reliance on
glycosomes is a promising potential therapeutic target.
Protein translocation across the
glycosomal membrane
The formation of the glycosomal membrane, as well as
the synthesis and importation of matrix proteins and certain
membrane proteins, occurs in the cytoplasm. In some cases, membrane
proteins may first be inserted into the endoplasmic reticulum
before being transported via vesicles to developing organelles.
This importation mechanism is conserved (24,25).
Following their synthesis during translation,
glycosomal proteins are produced in the cytosol and then imported
into glycosomes either as fully folded proteins or as complexes
consisting of multiple subunits (26). The protein import mechanisms of
glycosomes and peroxisomes share fundamental similarities and the
process relies on a group of proteins called peroxins (PEX) for
efficient importation of proteins into these organelles (27-29).
In trypanosomatids, more than 10 PEX proteins have been described
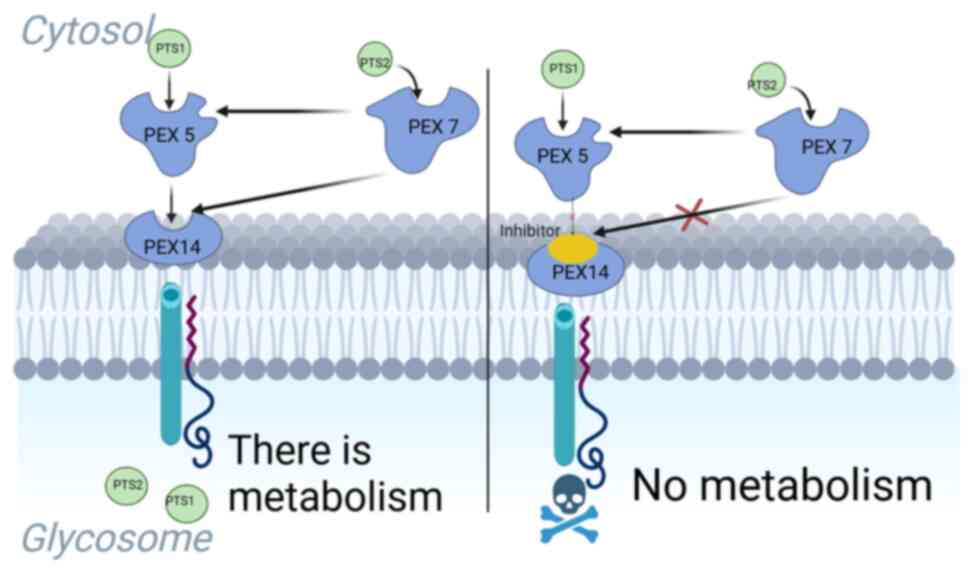
(28,30). PEX5 and PEX7, receptor proteins
that recognize C- or N-terminal import signals in the polypeptide,
are responsible for the importation of most glycosomal proteins
(31).
Proteins designated for transport possess a
peroxisomal targeting signal (PTS), which can be found either at
the far end of the C-terminus (referred to as type-1 or PTS1) or in
proximity to the N-terminus (known as type-2 or PTS2). While a
small number of matrix proteins do not have these PTS signals, most
proteins containing PTS1 are recognized by a cytosolic receptor
called PEX5, while PEX7 recognizes PTS2-containing proteins. PEX5
functions as a co-receptor and also contains a binding box for
PEX7(32) (Fig. 1). The receptors that carry the
cargo bind to a loading complex consisting of PEX14, PEX13.1 and
PEX13.2, facilitating their attachment to the glycosomal membrane
(33,34). The interaction between PEX5 and
PEX14 results in the creation of a transient import pore, allowing
the translocation of the cargo into the glycosomal matrix (35).
Once the cargo protein is internalized into the
glycosomal matrix, the loaded PEX5 and PEX7 receptors are
transported back to the cytosol through interactions with other
peroxins associated with the glycosomal membrane (19,36)
(Fig. 1).
In trypanosomes, the first seven enzymes involved in
glycolysis possess a PTS, which guides them to the glycosomal
matrix (32). These PTS sequences
are specifically present in proteins intended for transport to the
glycosomal matrix (Fig. 1). The
glycolytic enzymes in trypanosomes are equipped with a PTS-1 at the
COOH-terminus or a PTS-2 at the NH2-terminus. Studies conducted on
T. brucei have shown that the cytosolic receptor proteins,
TbPEX5p and TbPEX7p, recognize these newly synthesized enzymes
(32). Subsequently, each complex,
consisting of TbPEX5p with the PTS-1 protein or TbPEX7p with the
PTS-2 protein, interacts with TbPEX14p, a component of the
glycosomal membrane protein import machinery. This interaction
facilitates the translocation of the enzyme complex into the
glycosome. Research has shown that depleting PEX5p, PEX7p, or
PEX14p levels using RNAi leads to mislocalization of the enzymes
into the cytosol in T. brucei. This mislocalization
significantly affects the growth of the parasite and often results
in its death (32,37).
In trypanosomes, the primary pathway for energy
generation is glycolysis, which takes place within the glycosomes
instead of within the cytosol. As a result, the glycolytic enzymes
produced in the cytoplasm exhibit a limited degree of sequence
similarity to their human counterparts, which creates an
opportunity for targeted drug development. The accurate
localization of glycolytic enzymes is of utmost importance, as
misplacement has been demonstrated to lead to the death of the
parasite or hindered growth (32,37,38).
However, this potential of being a target is tested in animal
models (39). Understanding the
mechanisms by which proteins are imported into glycosomes,
especially the role of peroxins, is vital for designing drugs that
disrupt protein importation.
Glycolysis in glycosomes
Glycolysis is vital for trypanosomes, as it is their
main energy source. Any compound that effectively blocks glycolysis
can be considered a potential antiparasitic lead. The damage caused
by glycolysis inhibition will be much greater in trypanosomes than
in the host. Furthermore, trypanosomal glycolysis differs from
mammalian glycolysis in several ways (40,41).
When comparing trypanosomatid glycolytic enzymes with their human
counterparts, trypanosome enzymes exhibit changes in amino acid
sequence, with only 40-50% sequence identity (42).
The significant evolutionary divergence between
trypanosomatids and their mammalian hosts, coupled with the
distinct structure of the glycolytic pathway of parasites, offers
unique glycolytic characteristics that can be harnessed for the
development of selective inhibitor targeting structures or
catalytic mechanisms that do not disrupt host glycolysis (5,9).
Targeting the glycolytic pathway appears promising for intervention
in trypanosomatid. Specialized peroxisome-like organelles called
glycosomes house seven glycolytic enzymes that catalyze the
conversion of glucose to 3-phosphoglycerate (20,43,44).
Metabolic enzymes are attractive therapeutic targets
because the growth and survival of pathogens rely on the
availability of free energy and molecular building blocks provided
by metabolism (45). Despite the
high reliance of parasites on the oxidative degradation of fatty
acids for adenosine triphosphate (ATP) production, glycolysis is an
important mechanism for the stage of mammalian amastigotes of T.
cruzi and Leishmania (46).
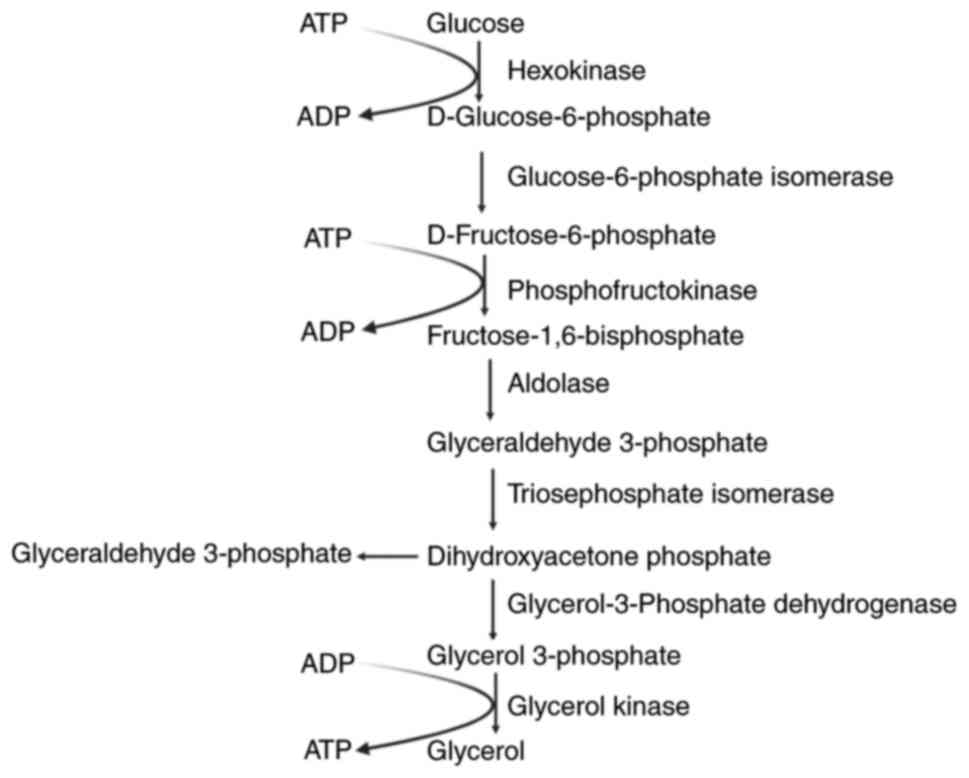
Due to the balance between ATP-consuming processes
mediated by hexokinase and phosphofructokinase and the
ATP-generating phase facilitated by phosphoglycerate kinase, there
is no net ATP production within the glycosome. The glycosome
contains a limited amount of ATP, which restricts the number of
ATP-consuming processes until ATP is replenished by downstream
reactions within the glycosome. Net ATP production occurs in the
cytosol, where pyruvate kinase is located, with the resulting
pyruvate being mostly expelled by the cells (47) (Fig.
2). Given that glycolysis is the primary source of ATP in the
trypanosome lifecycle, it is critical for survival. Therefore,
glycolysis represents a promising target for antitrypanosomal
drugs, as even 50% inhibition can lead to trypanosome death
(48). In summary, glycolysis is
the primary energy source of trypanosomatides, with unique features
that differs it from mammalian glycolysis. Targeting glycolysis
offers a promising avenue for drug development.
Glycolytic enzymes
The bloodstream form of trypanosomatids contains ~14
enzymes involved in glycolysis. Among these enzymes, seven are
located within the glycosome and catalyze the conversion of glucose
to 3-phosphoglycerate, while the remaining enzymes are found in the
cytosol and participate in the final steps of the process (20). The seven enzymes localized in the
glycosome are hexokinase, glucose-6-phosphate isomerase,
phosphofructokinase, aldolase, triosephosphate isomerase,
glycerol-3-phosphate dehydrogenase and glycerol kinase.
Trypanosomatids generate all their ATP by converting glucose to
pyruvate, which is then released into the circulation of the host
(49).
Hexokinase. Hexokinase is a carbohydrate
kinase and it is only 37% identical to the human homolog, implying
that selective inhibitor design is possible, so it has been
targeted for the development of trypanosomatid inhibitor
development (50). Hexokinase
catalyzes glucose conversion to glucose-6-phosphate and various
investigations have found that glucose analogs, such as glucosamine
and 2-C-hydroxymethyl glucose derivatives, block the activity
(51,52). Willson et al (53) tested numerous glucose-6-phosphate
analogs against T. brucei hexokinase since
glucose-6-phosphate possesses an affinity for the active site of
T. brucei hexokinase. The hexokinase of T. brucei is
inhibited by two glucose-6-phosphate derivatives, with a 75%
inhibition at 3 mM for one derivative and a 60% inhibition at 0.2
mM for another derivative. The elimination of Tb HK1 RNA
interference (RNAi) demonstrated that this protein was required for
viability and this enzyme was blocked by lonidamine, a drug that
effectively killed T. brucei parasites (54).
Glucose-6-phosphate isomerase. In glycolysis
and gluconeogenesis, glucose-6-phosphate isomerase catalyzes the
reversible aldose-ketose isomerization of D-glucose-6-phosphate to
D-fructose-6-phosphate, as well as the recycling of
hexose-6-phosphate in the pentose phosphate pathway, so this enzyme
links glycolysis and the pentose phosphate pathway. When the
crystallographic structure of the parasite glucose-6-phosphate
isomerase is compared to the atomic structure of humans and other
mammalian show that there are unique features of the enzyme of
parasites (55).
Aldolase. The crystal structures of aldolases
in trypanosomatids and mammals have been compared and it is found
that the active site was largely conserved. However, by identifying
minor differences and gaining detailed knowledge of the catalytic
mechanism, the researchers were able to develop a specific
inhibitor that binds quasi-irreversibly to the parasite enzyme. One
such inhibitor is the compound 5-formyl6-hydroxy-2-naphthyl
di-sodium phosphate, which can completely deactivate trypanosomatid
enzymes with a Ki value of 23 µM. By contrast, this compound only
has a weak effect on mammalian aldolase, even at concentrations
that are 1,000 times higher than that used for parasites (56). When formulated as prodrugs to
conceal the charged group, inhibitors derived from
5-formyl6-hydroxy-2-naphthyl disodium phosphate were able to
effectively eliminate cultured trypanosomes with ED50 values in the
low micromolar range. Importantly, these inhibitors did not have
any impact on mammalian cell growth of mammalian cells (57).
Glycerol kinase. Glycerol kinase is
responsible for the ATP-dependent conversion of glycerol into G3P,
which is used for lipid synthesis. Furthermore, G3P can enter the
glycolytic/gluconeogenic pathway by undergoing NAD+-dependent
oxidation to dihydroxyacetone phosphate (DHAP) through the action
of G3P dehydrogenase (G3PDH). However, the reverse reaction, which
converts DHAP back to G3P, would only occur under thermodynamically
favorable conditions characterized by high levels of adenosine
diphosphate (ADP)/ATP and G3P/glycerol ratios. Achieving such
conditions throughout the entire cell would be challenging, but
they can potentially occur within a specialized compartment such as
the glycosome (58). Therefore,
this pathway that consumes glycerol is also important to maintain
the balance of ATP/ADP and NAD+/NADH within the glycosome. In the
bloodstream form of African trypanosomes, the localization of
glycerol kinase (GK) within the glycosome serves as an additional
mechanism to cope with periods of anaerobiosis when G3P cannot be
oxidized through the shuttle mediated by mitochondrial GPO. During
such periods, the inability to further metabolize G3P leads to the
stoppage of glycolysis since glycosomal NADH cannot be oxidized.
However, in trypanosomes where glycerol-3-phosphatase is absent,
this problem is resolved by reversing the GK reaction, despite the
fact that the equilibrium strongly favors phosphorylation (20).
Triosephosphate isomerase (TPI). TPI plays a
crucial role in glycolysis by facilitating the conversion between
dihydroxyacetone phosphate and glyceraldehyde 3-phosphate. This
enzymatic activity ensures complete breakdown of the entire hexose
unit, rather than just half of it. Notably, TPI in glycosomes lacks
the PTS1 and PTS2 motifs, which are established
peroxisome-targeting signals (59). Galland et al (60) conducted a study to identify the
specific region within the TPI polypeptide responsible for its
glycosome targeting. Surprisingly, the glycosomal TPI enzyme does
not contain the recognized peroxisome-targeting signals. However,
the researchers discovered a 22-amino acid fragment located
internally within the polypeptide that can direct a reporter
protein to glycosomes in transfected trypanosomes. This finding was
confirmed by cell fractionation experiments and immunofluorescence
studies. Notably, this internal routing information appears to be
unique to the sequence of the trypanosome enzyme. When a reporter
protein fused with a Saccharomyces cerevisiae peptide
containing the corresponding sequence of the 22 residue fragment of
the enzyme T. brucei was tested, it did not target
glycosomes. In yeast and most other organisms, TPI is found
exclusively found in the cytosol (60).
Phosphofructokinase (PFK). PFK is an enzyme
that exists as a tetramer and uses ATP as a phosphate donor to
facilitate the phosphorylation of fructose 6-phosphate, converting
it into fructose 1,6-bisphosphate. This reaction is considered
practically irreversible under normal physiological conditions.
Notably, trypanosomatid PFKs share 20-30% of their genetic
sequences with humans (61).
However, the structure of PFK in trypanosomatids reveals the
presence of two significant insertions that are unique to these
parasites but absent in mammalian enzymes. These insertions are
situated within and in close proximity to the active site of the
enzyme. Consequently, this structural insight paves the way for
conducting drug discovery experiments based on the structure of the
enzyme, aiming to identify new lead compounds that specifically
target the parasites responsible for diseases caused by the
trypanosomatid family of protozoan parasites (62).
Glycerol-3-phosphate dehydrogenase (GPDH).
NADH generated by glyceraldehyde-3-phosphate dehydrogenase during
glycolysis is reoxidized by mitochondrial GPO using molecular
oxygen. This electron transfer occurs via a glycosomal
glycerol-3-phosphate dehydrogenase and the
glycerol-3-phosphate:dihydroxyacetone phosphate (DHAP) shuttle. The
resulting G3P is then transported across the glycosomal membrane
and reoxidized through the G3P:DHAP shuttle, connecting with
mitochondrial GPO (20).
Inhibiting the activity of GPDH would hinder the ability of the
trypanosome to produce ATP. Additionally, the inhibition of GPDH
would result in the accumulation of DHAP within the glycosome,
which could be highly detrimental to the trypanosome as DHAP is
naturally converted to methylglyoxal, a toxic compound that
disrupts protein function (63,64).
The unique structure of the glycosome creates an
intraglycosomal environment that maintains the balance of redox and
ATP. In the cytosol, ATP production occurs through the net action
of pyruvate kinase. Under anaerobic conditions, G3P is converted to
glycerol by the reverse action of a glycosomal glycerol kinase,
accompanied by the production of ATP. This allows the glycosome to
maintain the balance of ATP and NAD even in the absence of oxygen.
However, instead of producing two molecules of pyruvate from one
molecule of glucose, the glycosome generates one molecule each of
glycerol and pyruvate, resulting in a reduced net ATP production of
two to one. Due to this compartmentalization, a number of
regulatory mechanisms that function in other cell types do not
operate in trypanosomes. Glycosomal hexokinase and
phosphofructokinase, for example, do not respond to
activity-regulating substances that typically affect these enzymes
in other cell types, reflecting the unique characteristics of
trypanosomes (49,65-67).
In summary, the key glycolytic enzymes highlighted
show promise as targets for selective inhibitors and the
distinctions between trypanosomatid and human enzymes present
opportunities for drug development, however, it is still on animal
models only (68,69).
3. Conclusions and recommendations
Rational drug design targeting glycosomal enzymes
and protein translocation in trypanosomatids offers significant
promise for developing selective and effective treatments. However,
current research faces limitations in elucidating the molecular
interactions within these pathways. The glycosomal enzymes involved
in glycolysis and the translocation of proteins across the
glycosomal membrane are highly specific processes. These
limitations arise mainly due to the complex structures of the
enzymes and the dynamic nature of protein import mechanisms, which
hinder the design of selective inhibitors that can differentiate
between host and parasite proteins. This challenge is further
compounded by the limited availability of high-resolution
structural data, which is essential for understanding these
molecular interactions in greater detail.
Future studies must expand our knowledge of the
molecular mechanisms involved in glycosomal protein translocation
and glycolysis to overcome these challenges. Advanced techniques
such as cryo-electron microscopy, high-throughput screening and
computational modeling should be employed to investigate these
processes at a molecular level. By refining drug design approaches
and conducting in vivo validation studies, the clinical
translation of promising candidates can be ensured, ultimately
leading to more effective treatments for neglected tropical
diseases.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
GA contributed to the conception and design of the
present study, searching, analysis and writing the first draft of
the manuscript. AA contributed to writing the first draft of the
manuscript and YMW, AS and MG contributed to revising the
manuscript. Data authentication is not applicable. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schuster FL and Sullivan JJ: Cultivation
of clinically significant hemoflagellates. Clin Microbiol Rev.
15:374–389. 2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Moreno J and Alvar J: Canine
leishmaniasis: Epidemiological risk and the experimental model.
Trends Parasitol. 18:399–405. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nussbaum K, Honek J, Cadmus CMCVC and
Efferth T: Trypanosomatid parasites causing neglected diseases.
Curr Med Chem. 17:1594–1617. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Robays J, Nyamowala G, Sese C, Betu Ku
Mesu Kande V, Lutumba P, Van der Veken W and Boelaert M: High
failure rates of melarsoprol for sleeping sickness, Democratic
Republic of Congo. Emerg Infect Dis. 14:966–967. 2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
World Health Organization. Tropical
disease research, progress 1995-1996. WHO Tech Rep Ser. 1:108–139.
1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
World Health Organization (WHO): Tropical
disease research. WHO, Geneva, 1997.
|
|
7
|
World Health Organization. Weekly
epidemiological record, 2016, vol. 91, 38 [full issue]. Wkly
Epidemiol Rec. 91:432–440. 2016.
|
|
8
|
Rodrigues JCF, Godinho JLP and de Souza W:
Biology of human pathogenic trypanosomatids: Epidemiology,
lifecycle and ultrastructure. Subcell Biochem. 74:1–42.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Atouguia J and Costa J: Therapy of human
African trypanosomiasis: Current situation. Mem Inst Oswaldo Cruz.
94:221–224. 1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pépin J and Milord F: The treatment of
human African trypanosomiasis. Adv Parasitol. 33:1–47.
1994.PubMed/NCBI View Article : Google Scholar
|
|
11
|
UNICEF/UNDP/World Bank/WHO Special
Programme for Research and Training in Tropical Diseases.
Operational guidelines for the establishment and functioning of
data and safety monitoring boards: special programme for research
and training in tropical diseases. Int J Pharm Med. 20:25–36.
2006.
|
|
12
|
Legros D, Olilvier G, Gastellu-Etchegorry
M, Paquet C, Burri C, Jannin J and Büscher P: Treatment of human
African trypanosomiasis-present situation and needs for research
and development. Lancet Infect Dis. 2:437–440. 2002.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang CC: Molecular mechanisms and
therapeutic approaches to the treatment of African trypanosomiasis.
Annu Rev Pharmacol Toxicol. 35:93–127. 1995.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Barrett MP, Coombs GH and Mottram JC:
Recent advances in identifying and validating drug targets in
trypanosomes and leishmanias. Trends Microbiol. 7:82–88.
1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Patterson S, Alphey MS, Jones DC, Shanks
EJ, Street IP, Frearson JA, Wyatt PG, Gilbert IH and Fairlamb AH:
Dihydroquinazolines as a novel class of Trypanosoma brucei
trypanothione reductase inhibitors: Discovery, synthesis, and
characterization of their binding mode by protein crystallography.
J Med Chem. 54:6514–6530. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Urbina JA, Concepcion JL, Rangel S, Visbal
G and Lira R: Squalene synthase as a chemotherapeutic target in
Trypanosoma cruzi and Leishmania mexicana. Mol
Biochem Parasitol. 125:35–45. 2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Looker DL, Marr JJ and Berens RL:
Mechanisms of action of pyrazolopyrimidines in Leishmania
donovani. J Biol Chem. 261:9412–9415. 1986.PubMed/NCBI
|
|
18
|
Sienkiewicz N, Jarosławski S, Wyllie S and
Fairlamb AH: Chemical and genetic validation of dihydrofolate
reductase-thymidylate synthase as a drug target in African
trypanosomes. Mol Microbiol. 69:520–533. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kalel VC, Emmanouilidis L, Dawidowski M,
Schliebs W, Sattler M, Popowicz GM and Erdmann R: Inhibitors of
glycosomal protein import provide new leads against
trypanosomiasis. Microb Cell. 4:229–232. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Opperdoes FR and Borst P: Localization of
nine glycolytic enzymes in a microbody-like organelle in
Trypanosoma brucei: The glycosome. FEBS Lett. 80:360–364.
1977.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Clayton CE and Michels P: Metabolic
compartmentation in African trypanosomes. Parasitol Today.
12:465–471. 1996.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Engel JC, de Cazzulo BMF, Stoppani AO,
Cannata JJ and Cazzulo JJ: Aerobic glucose fermentation by
Trypanosoma cruzi axenic culture amastigote-like forms
during growth and differentiation to epimastigotes. Mol Biochem
Parasitol. 26:1–10. 1987.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Moyersoen J, Choe J, Fan E, Hol WGJ and
Michels PAM: Biogenesis of peroxisomes and glycosomes:
Trypanosomatid glycosome assembly is a promising new drug target.
FEMS Microbiol Rev. 28:603–643. 2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gualdrón-López M, Brennand A, Avilán L and
Michels PAM: Translocation of solutes and proteins across the
glycosomal membrane of trypanosomes; possibilities and limitations
for targeting with trypanocidal drugs. Parasitology. 140:1–20.
2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sommer JM, Bradley PJ, Wang C and Johnson
PJ: Biogenesis of specialized organelles: Glycosomes and
hydrogenosomes. In: Smith DF, Parsons M (eds), Molecular biology of
parasitic protozoa, Vol. 13. Oxford Press, New York, N.Y,
pp159-180, 1996.
|
|
26
|
Titorenko VI, Nicaud JM, Wang H, Chan H
and Rachubinski RA: Acyl-CoA oxidase is imported as a
heteropentameric, cofactor-containing complex into peroxisomes of
Yarrowia lipolytica. J Cell Biol. 156:481–494. 2002.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Heiland I and Erdmann R: Biogenesis of
peroxisomes. Topogenesis of the peroxisomal membrane and matrix
proteins. FEBS J. 272:2362–2372. 2005.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Verplaetse E, Rigden DJ and Michels PAM:
Identification, characterization and essentiality of the unusual
peroxin 13 from Trypanosoma brucei. Biochim Biophys Acta.
1793:516–527. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lametschwandtner G, Brocard C, Fransen M,
Van Veldhoven P, Berger J and Hartig A: The difference in
recognition of terminal tripeptides as peroxisomal targeting signal
1 between yeast and human is due to different affinities of their
receptor Pex5p to the cognate signal and to residues adjacent to
it. J Biol Chem. 273:33635–33643. 1998.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cyr N, Madrid KP, Strasser R, Aurousseau
M, Finn R, Ausio J and Jardim A: Leishmania donovani peroxin
14 undergoes a marked conformational change following association
with peroxin 5. J Biol Chem. 283:31488–31499. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Brocard C and Hartig A: Peroxisome
targeting signal 1: Is it really a simple tripeptide? Biochim
Biophys Acta. 1763:1565–1573. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Galland N, Demeure F, Hannaert V,
Verplaetse E, Vertommen D, Van der Smissen P, Courtoy PJ and
Michels PA: Characterization of the role of the receptors PEX5 and
PEX7 in the import of proteins into glycosomes of
Trypanosoma brucei. Biochim Biophys Acta. 1773:521–535.
2007.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Verplaetse E, Gualdrón-López M, Chevalier
N and Michels PAM: Studies on the organization of the docking
complex involved in matrix protein import into glycosomes of
Trypanosoma brucei. Biochem Biophys Res Commun. 424:781–785.
2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Brennand A, Rigden DJ and Michels PAM:
Trypanosomes contain two highly different isoforms of peroxin PEX13
involved in glycosome biogenesis. FEBS Lett. 586:1765–1771.
2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Meinecke M, Cizmowski C, Schliebs W,
Krüger V, Beck S, Wagner R and Erdmann R: The peroxisomal
importomer constitutes a large and highly dynamic pore. Nat Cell
Boil. 12:273–277. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Francisco T, Rodrigues TA, Freitas MO,
Grou CP, Carvalho AF, Sá-Miranda C, Pinto MP and Azevedo JE: A
cargo-centered perspective on the PEX5 receptor-mediated
peroxisomal protein import pathway. J Biol Chem. 288:29151–29159.
2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Moyersoen J, Choe J, Kumar A, Voncken FG,
Hol WG and Michels PA: Characterization of Trypanosoma
brucei PEX14 and its role in the import of glycosomal matrix
proteins. Eur J Biochem. 270:2059–2067. 2003.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Krazy H and Michels PAM: Identification
and characterization of three peroxins-PEX6, PEX10 and
PEX12-involved in glycosome biogenesis in Trypanosoma
brucei. Biochim Biophys Acta. 1763:6–17. 2006.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Dawidowski M, Emmanouilidis L, Kalel VC,
Tripsianes K, Schorpp K, Hadian K, Kaiser M, Mäser P, Kolonko M,
Tanghe S, et al: Inhibitors of PEX14 disrupt protein import into
glycosomes and kill Trypanosoma parasites. Science.
355:1416–1420. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Opperdoes FR, Baudhuin P, Coppens I, De
Roe C, Edwards SW, Weijers PJ and Misset O: Purification,
morphometric analysis, and characterization of the glycosomes
(microbodies) of the protozoan hemoflagellate Trypanosoma
brucei. J Cell Biol. 98:1178–1184. 1984.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Visser N and Opperdoes FR: Glycolysis in
Trypanosoma brucei. Eur J Biochem. 103:623–632.
1980.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Michels PA: Evolutionary aspects of
trypanosomes: Analysis of genes. J Mol Evol. 24:45–52.
1986.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Fernandes AP, Nelson K and Beverley SM:
Evolution of nuclear ribosomal RNAs in kinetoplastid protozoa:
perspectives on the age and origins of parasitism. Proc Natl Acad
Sci USA. 90:11608–11612. 1993.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Opperdoes FR: Compartmentation of
carbohydrate metabolism in trypanosomes. Annu Rev Microbiol.
41:127–151. 1987.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Ruppin E, Papin JA, de Figueiredo LF and
Schuster S: Metabolic reconstruction, constraint-based analysis and
game theory to probe genome-scale metabolic networks. Curr Opin
Biotechnol. 21:502–510. 2010.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hart D and Coombs GH: Leishmania
mexicana: Energy metabolism of amastigotes and promastigotes. Exp
Parasitol. 54:397–409. 1982.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Creek DJ, Mazet M, Achcar F, Anderson J,
Kim DH, Kamour R, Morand P, Millerioux Y, Biran M, Kerkhoven EJ, et
al: Probing the metabolic network in bloodstream-form
Trypanosoma brucei using untargeted metabolomics with stable
isotope labelled glucose. PLoS Pathog. 11(e1004689)2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Haanstra JR, Kerkhoven EJ, van Tuijl A,
Blits M, Wurst M, van Nuland R, Albert MA, Michels PA, Bouwman J,
Clayton C, et al: A domino effect in drug action: From metabolic
assault towards parasite differentiation. Mol Microbiol. 79:94–108.
2011.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Michels P, Hannaert V and Bringaud F:
Metabolic aspects of glycosomes in trypanosomatidae-new data and
views. Parasitol Today. 16:482–489. 2000.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Verlinde CL, Hannaert V, Blonski C,
Willson M, Périé JJ, Fothergill-Gilmore LA, Opperdoes FR, Gelb MH,
Hol WG and Michels PA: Glycolysis as a target for the design of new
anti-trypanosome drugs. Drug Resist Updat. 4:50–65. 2001.PubMed/NCBI View Article : Google Scholar
|
|
51
|
D'Antonio EL, Deinema MS, Kearns SP, Frey
TA, Tanghe S, Perry K, Roy TA, Gracz HS, Rodriguez A and D'Antonio
J: Structure-based approach to the identification of a novel group
of selective glucosamine analogue inhibitors of Trypanosoma
cruzi glucokinase. Mol Biochem Parasitol. 204:64–76.
2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Merritt C, Silva LE, Tanner AL, Stuart K
and Pollastri MP: Kinases as druggable targets in trypanosomatid
protozoan parasites. Chem Rev. 114:11280–11304. 2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Willson M, Alric I, Perie J and Sanejouand
YH: Yeast hexokinase inhibitors designed from the 3-D enzyme
structure rebuilding. J Enzyme Inhib. 12:101–121. 1997.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Chambers JW, Fowler ML, Morris MT and
Morris JC: The anti-trypanosomal agent lonidamine inhibits
Trypanosoma brucei hexokinase 1. Mol Biochem Parasitol.
158:202–207. 2008.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Cordeiro AT, Michels PAM, Delboni LF and
Thiemann OH: The crystal structure of glucose-6-phosphate isomerase
from Leishmania mexicana reveals novel active site features.
Eur J Biochem. 271:2765–2772. 2004.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Dax C, Duffieux F, Chabot N, Coincon M,
Sygusch J, Michels PAM and Blonski C: Selective irreversible
inhibition of fructose 1,6-bisphosphate aldolase from
Trypanosoma brucei. J Med Chem. 49:1499–1502.
2006.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Azéma L, Lherbet C, Baudoin C and Blonski
C: Cell permeation of a Trypanosoma brucei aldolase
inhibitor: Evaluation of different enzyme-labile phosphate
protecting groups. Bioorg Med Chem Lett. 16:3440–3443.
2006.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Bakker BM, Michels PA, Opperdoes FR and
Westerhoff HV: Glycolysis in bloodstream form Trypanosoma
brucei can be understood in terms of the kinetics of the glycolytic
enzymes. J Biol Chem. 272:3207–3215. 1997.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wierenga R, Noble M, Vriend G, Nauche S
and Hol W: Refined 1.83 A structure of trypanosomal triosephosphate
isomerase crystallized in the presence of 2.4 M-ammonium sulphate:
A comparison with the structure of the trypanosomal triosephosphate
isomerase-glycerol-3-phosphate complex. J Mol Biol. 220:995–1015.
1991.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Galland N, de Walque S, Voncken FGJ,
Verlinde CLMJ and Michels PAM: An internal sequence targets
Trypanosoma brucei triosephosphate isomerase to glycosomes.
Mol Biochem Parasitol. 171:45–49. 2010.PubMed/NCBI View Article : Google Scholar
|
|
61
|
López C, Chevalier N, Hannaert V, Rigden
DJ, Michels PAM and Ramirez JL: Leishmania donovani
phosphofructokinase. Gene characterization, biochemical properties
and structure-modeling studies. Eur J Biochem. 269:3978–3989.
2002.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Martinez-Oyanedel J, McNae IW, Nowicki MW,
Keillor JW, Michels PA, Fothergill-Gilmore LA and Walkinshaw MD:
The first crystal structure of phosphofructokinase from a
eukaryote: Trypanosoma brucei. J Mol Biol. 366:1185–1198.
2007.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lo T, Westwood ME, McLellan AC, Selwood T
and Thornalley PJ: Binding and modification of proteins by
methylglyoxal under physiological conditions. A kinetic and
mechanistic study with N alpha-acetylarginine, N
alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum
albumin. J Biol Chem. 269:32299–32305. 1994.PubMed/NCBI
|
|
64
|
Denise H, Giroud C, Barrett MP and Baltz
T: Affinity chromatography using trypanocidal arsenical drugs
identifies a specific interaction between glycerol-3-phosphate
dehydrogenase from Trypanosoma brucei and Cymelarsan. Eur J
Biochem. 259:339–346. 1999.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Nwagwu M and Opperdoes FR: Regulation of
glycolysis in Trypanosoma brucei: Hexokinase and
phosphofructokinase activity. Acta Trop. 39:61–72. 1982.PubMed/NCBI
|
|
66
|
Cronin CN and Tipton KF: Purification and
regulatory properties of phosphofructokinase from
Trypanosoma (Trypanozoon) brucei brucei. Biochem J.
227:113–124. 1985.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Bakker BM, Mensonides FI, Teusink B, van
Hoek P, Michels PA and Westerhoff HV: Compartmentation protects
trypanosomes from the dangerous design of glycolysis. Proc Natl
Acad Sci USA. 97:2087–2092. 2000.PubMed/NCBI View Article : Google Scholar
|
|
68
|
McNae IW, Kinkead J, Malik D, Yen LH,
Walker MK, Swain C, Webster SP, Gray N, Fernandes PM, Myburgh E, et
al: Fast acting allosteric phosphofructokinase inhibitors block
trypanosome glycolysis and cure acute African trypanosomiasis in
mice. Nat Commun. 12(1052)2021.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Cortés-Figueroa AA, Pérez-Torres A,
Salaiza N, Cabrera N, Escalona-Montaño A, Rondán A, Aguirre-García
M, Gómez-Puyou A, Pérez-Montfort R and Becker I: A monoclonal
antibody that inhibits Trypanosoma cruzi growth in vitro and
its reaction with intracellular triosephosphate isomerase.
Parasitol Res. 102:635–643. 2008.PubMed/NCBI View Article : Google Scholar
|