1. Introduction
Hemophilia is a relatively rare hereditary bleeding
disease characterized by a lack or decreased functionality of
particular coagulation components. The prevailing manifestations of
hemophilia encompass hemophilia A (MIM No 306700), characterized by
a deficiency in factor VIII, and hemophilia B (MIM No 306900),
characterized by a deficiency in factor IX. Factor XI deficiency
sometimes referred to as hemophilia C (MIM No. 612416), is a very
uncommon bleeding ailment characterized by moderate symptoms. This
condition is most frequent among individuals of Ashkenazi Jewish
descent (1).
The global prevalence of hemophilia A is considered
to be ~17.1/100,000 males at birth, whereas the prevalence of
hemophilia B is estimated to be ~3.8/100,000 males at birth. Based
on current estimations, the global population of individuals
affected by hemophilia is ~1,125,000, out of which ~418,000
individuals are diagnosed with severe hemophilia (2).
Clinical manifestations in patients with hemophilia
are solely attributable to the absence of a single protein present
in minute quantities in the circulation; hence, gene therapy has
long been an attractive method for the treatment of hemophilia, as
it provides the possibility of a cure by facilitating the natural
production of the deficient coagulation factor by transferring a
healthy copy of the respective gene (4). It has been demonstrated that a 5%
increase in the levels of the deficient coagulation factor can
substantially alleviate the symptoms of hemorrhage (3). The present review aimed to shed light
on the current status of hemophilia in Iraq. The present review
summarizes the recent advances in gene therapy for hemophilia and
discusses the remaining obstacles to enhancing the availability of
the novel therapy for patients with this disorder.
2. Literature search methods
A literature search was conducted using the PubMed
and Web of Science and Google Scholar platforms for multiple
combinations of different hemophilia types, epidemiology,
management and gene therapy. The present review encompasses
articles produced in the English language, encompassing both
preclinical and clinical studies, up until June, 2023. The search
results were initially screened based on the relevancy of their
titles and abstracts, after which the whole contents of the
relevant articles were examined. In addition, pertinent research of
the reference lists of the relevant documents was performed.
Publications written in languages other than English were
eliminated.
3. Hemophilia status in Iraq
Epidemiological studies on hemophilia in Iraq are
limited in number. According to the World Bleeding Disorder
Registry (WBDR) report in 2021, a total of 2,567 hemophilia cases
have been registered in Iraq. Among these cases, 2,024 were
hemophilia A and 543 were hemophilia B (5).
A recent study including four hemophilia centers in
Baghdad, Iraq indicated that the prevalence of hemophilia has
doubled over a 10-year period, from 7.2 per 100,000 individuals in
2007 to 15.9 per 100,000 population in 2016(6). This rate appears to be greater than
the rates estimated in neighboring countries, including Iran (7.1),
Turkey (7.0), Egypt (6) Jordan
(4.4), Syria (3.5), and Saudi Arabia (1.4) per 100,000 individuals
(6). According to the same study,
hemophilia A comprised the majority, namely 72.9%, of all
documented cases of hemophilia, with a prevalence rate of 5.9 per
100,000 individuals within the population. Conversely, hemophilia B
constituted 24.8% of the cases, with a prevalence rate of 2 per
100,000 individuals within the community and the least prevalent is
hemophilia C with a rate of 0.1 per 100,000 individuals (6). A smaller cohort study conducted at
the National Center of Hematology in Baghdad, Iraq involving 191
children with an average age of 5.3 years revealed that hemophilia
A ranked as the third most registered bleeding disorder among
children with a prevalence of 9.4% following von Willebrand disease
(MIM No 613160) 86.9% and Glanzmann's thrombasthenia (MIM No
273800) 39.8% (7).
The overall survival estimates for patients with
hemophilia fluctuate considerably as a result of treatment
accessibility and advancements in care (8). The life expectancy of individuals
with hemophilia who have access to modern treatment and prophylaxis
approaches is often virtually normal, and the survival rate is
approaching that of the general population (8). However, survival bias is a potential
concern in studies that involve low- and middle-income countries,
which may be a consequence of the fact that neonatal mortality
rates are highest in these countries. The premature fatalities that
disproportionately affect individuals with severe hemophilia in
low- and middle-income countries may be the cause of skewed outcome
comparisons (bleeding events and age of diagnosis) between these
nations (9). Regrettably, the
precise survival rate of hemophilic individuals in Iraq is not
adequately documented in the published data.
Local studies reported that ~40-63% of the
registered hemophilia reported the severe type (6,10);
however, less than half of these patients were on prophylactic
therapy and or on-demand therapy (6).
Current therapeutic approaches in Iraq include
factor concentrate, tranexamic acid and to a lesser extent,
desmopressin (6). However, in a
recently published open-label trial conducted across multiple Iraqi
centers, the efficacy of emicizumab prophylaxis was evaluated
compared to episodic recombinant factor VII treatment in 32
individuals with hemophilia A and high inhibitor titers (7). That study included patients ranging
from 1-46 years of age. The findings revealed a significant
improvement in various outcomes, including bleeding rates, joint
bleeding, hospital admissions, blood transfusions and school or
work absences, when patients switched to emicizumab prophylaxis
from their previous 6 months of episodic recombinant factor VII
treatment. The most commonly observed adverse effect was injection
site reactions, while there was no observed risk of developing
thrombosis (7).
The development of inhibitors in Iraqi patients with
hemophilia was assessed in several small local studies with a
prevalence rate ranging between 5-12%. These studies have
consistently shown that inhibitors are more commonly observed in
individuals with hemophilia A (10-12).
Taresh and Hassan (10) conducted
an evaluation of inhibitor levels in a group of 118 individuals
diagnosed with hemophilia A and 25 individuals diagnosed with
hemophilia B, a total of 22 individuals (constituting 18.6% of the
sample) were found to have inhibitors. Notably, all these patients
were diagnosed with hemophilia A, with a substantial majority (82%)
displaying elevated titers. The likelihood of developing inhibitors
was significantly elevated in patients with severe hemophilia who
had been exposed to factor VIII concentrate at an early age (≤3
months). Furthermore, those who have a familial background of
autoimmune disorders and have immune system challenges had a higher
propensity for the development of inhibitors (10).
4. Current standard management of
hemophilia
The primary purpose of hemophilia therapy is to
effectively manage the frequency and severity of bleeding, apart
from mitigating the risk of long-term joint degeneration and
mortality (13). Depending on the
concentration of defective clotting factors, patients are
classified as having mild, moderate, or severe hemophilia.
Socioeconomic factors greatly affect care standards (4). For severe hemophilia, prophylactic
replacement therapy is the recommended treatment to maintain
clotting factor levels >1% (13). Financial restrictions prevent 75%
of patients with hemophilia in low- and middle-income countries
from receiving regular preventive therapy (13). In such circumstances, the
guidelines established by the World Federation of Hemophilia (WFH)
advocate for the use of low-dose prophylaxis and, in certain cases,
on-demand medication as effective strategies for managing bleeding
episodes (14). Fresh-frozen
plasma or cryoprecipitate, which may cause volume overload and
blood-borne pathogen transmission, makes factor leveling harder
(4). Thus, hemophilia-related
health issues and lower lifespans persist in these countries
Standard recombinant factor VIII and
factor IX factors
These factors have a short half-life in the
bloodstream; thus, lifelong treatment strategy necessitates the
intravenous administration of clotting factor concentrates at least
two to three times per week (15).
Extended half-life factor
prophylaxis
Pegylation (attaching polyethylene glycol) or fusing
clotting factors with proteins such as albumin or Fc have been used
to increase factor VII and factor IX stability and half-lives.
BIVV001, a novel FVIII fusion protein, has been proposed to boost
the half-life of FVIII to 38 h, protecting patients with hemophilia
for longer periods of time (15).
Antifibrinolytic agents
Antifibrinolytic agents are effective in the
management of moderate and mild cases of hemophilia. Aminocaproic
acid and tranexamic acid inhibit the proteolytic activity of
plasmin (14). Desmopressin, on
the other hand, stabilizes the already present factor VIII in the
plasma by releasing the von Willebrand factor from its storage
sites (13).
Targeted therapy
The utilization of immunotherapy, such as
emicizumab, represents a notable advancement in the treatment and
control of hemophilia. Emicizumab, is a recombinant humanized
bispecific IgG antibody that replicates the cofactor activity of
the deficient FVIII in individuals with hemophilia A (16). Emicizumab has a prolonged half-life
of around 4-5 weeks (15).
5. Factor VIII and IX genotypes
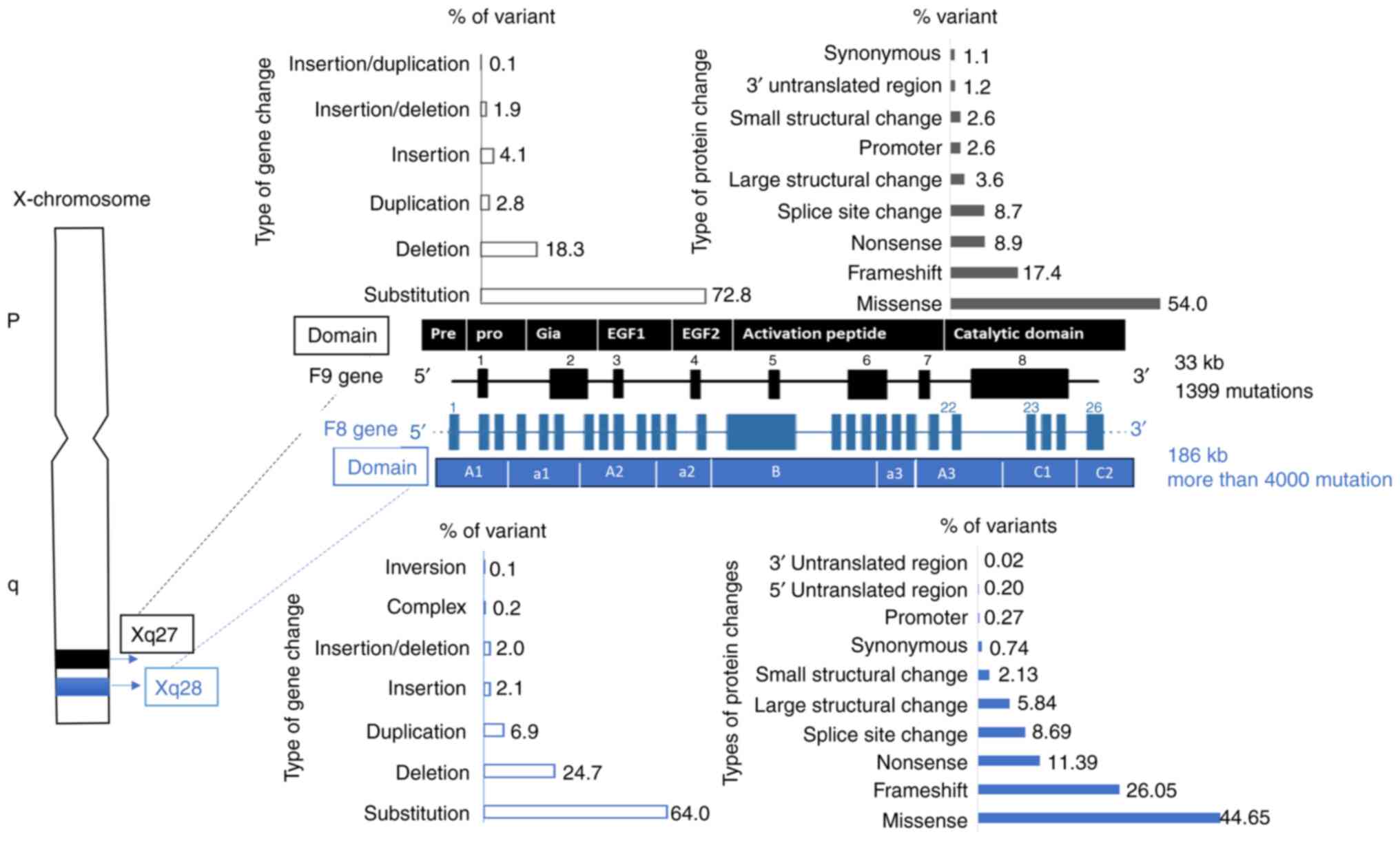
Factor VIII and XI gene characteristics and common
mutations are illustrated in Fig.
1. The most prevalent mutation observed in severe hemophilia A
is the inversion of introns 22 and 1, which accounts for ~55% of
cases and is linked to inhibitor development (17).
In a previous Iraqi study involving 18 patients with
hemophilia A, the most frequent mutations in F8 gene were point
mutations then inversion mutations followed by frameshift
mutations. The severe phenotype was strongly associated with exon
24 mutations with P-value 0.009 and intron 22 with a P-value of
0.036(18). A larger study
included 80 Iraqi Kurdish patients with hemophilia A analyzed the
inversion in intron 22 and intron 1 using inverse
shifting-polymerase chain reaction reported that among patients
with severe hemophilia A, 6.7% had Inv22 and 3.3% (2/60) had
Inv1(19).
Preclinical trials have investigated gene delivery
vehicles whether viral or non-viral to establish the safety and
efficacy of the gene transfer strategy, ascertain an appropriate
starting dose in humans, and evaluate vector biodistribution and
sustainability of delivered transduced gene with therapeutic range.
A gene transfer strategy for the treatment of any disease requires
three components: A therapeutic gene to be transferred, also known
as the transgene; a vector or gene delivery vehicle to facilitate
the transfer; and a physiologically relevant target tissue in the
recipient (20).
In 1989, an early preclinical trial demonstrated
that genetically modified ex vivo human skin fibroblasts
containing human factor IX cDNA could produce factor IX and
discharge it into the bloodstream of laboratory animal (21). However, the level was unstable, and
it was difficult to determine the duration of transduced cell
viability. Subsequent research concentrated on viral vectors, such
as adenovirus, adeno-associated virus (AAV) and retrovirus.
Adenovirus trials were hindered by immune response induction and
hepatotoxicity, whereas AAV and lentivirus exhibited promise in
multiple preclinical scenarios. AVV, a naturally occurring,
non-pathogenic virus, was shown to be capable of attaining the
long-term expression of the desired gene in various tissues
(22).
The sustained repair of the hemophilia phenotype in
inhibitor-prone null mutation hemophilia B dogs was achieved with
direct liver transduction of an AAV2 vector encoding the factor IX
gene in 2009. This approach did not lead to the production of
inhibitors (23). Through the use
of mouse models, scientists have made the noteworthy observation
that hepatic transduction with AAV vectors have the potential to
promote the development of immunological tolerance towards factor
IX. This process is considered to be mediated by the activation of
regulatory T-cells (Tregs) expressing specific markers such as CD4,
CD25 (24,25) and FoxP3. In the same manner,
studies conducted on dogs and non-human primates have shown
sustained transgenic expression over an extended period, with no
discernible CD8+ T-cell reaction to the capsid antigens
of the vector. This observation holds true regardless of the
specific transgene, promoter, or method of delivery (26).
As regards hemophilia A, the capacity of the AAV
vector, which is <4.2 kb was restricted in carrying the full
length of the F8 gene. The F8 cDNA may be truncated by the removal
of the sequence that encodes a nonfunctional domain, known as
B-domain deletion (BDD); this strategy was employed in two distinct
phase I/II clinical trials for transferring a codon-optimized
adeno-associated virus serotype 5 (AAV5) vector in a cohort of 9
patients with severe hemophilia; the results were encouraging with
a sustainable increase in F8 activity over a period of 1 year
(27).
Due to the absence of innate immunity, retroviral
and lentiviral have been found to be attractive vectors for
research (28). Retroviral vectors
that are capable of transducing non-dividing cells were
investigated in pediatric hemophilia, resulting in sustained
therapeutic levels of factor VIII in neonatal mice with hemophilia
A (26). Efficient retroviral
transduction was also achieved in neonatal dogs with hemophilia
(29). Immunosuppression was the
only condition in which liver-directed LV gene therapy attained
therapeutic-range human FVIII activity in non-human primates
(30). Lentiviral vectors could be
administered intravenously to hemophilic adult mice to achieve
therapeutic levels of factor VIII or IX (31,32).
However, these vectors were also adept at transducing
antigen-presenting cells, and antibodies to the transgene product
were detected in the majority of preclinical lentiviral experiments
employing ubiquitous promoters, resulting in unstable levels of the
factor even when using hepatocyte-specific promoters (26). In non-human primates,
liver-directed LV gene therapy achieved therapeutic-range human
FVIII activity in non-human primates, albeit only during
immunosuppression.
The third-generation, self-inactivating lentiviral
vectors further improved safety by splitting the viral genome into
separate plasmids, rendering recombinant virus generation even more
unlikely (33). In their
preclinical study, Wang et al (16) injected mice intraosseously with a
self-inactivating lentivirus vector encoding FVIII under the
control of the Gp1b promoter resulting in partial correction of
hemophilia. This led to platelet-producing cells that persistently
expressed FVIII inside the animals, resulting in sustained FVIII
therapeutic expression levels (16). Additionally, this approach has been
proposed to protect FVIII against inhibitor-induced inactivation
(34). Non-viral gene transfer
includes plasmid and nanoparticles. Dermal fibroblast cells were
used for early non-viral gene transfer trials (35). This was achieved by introducing a
BDD gene-loaded plasmid into the fibroblasts via electropores made
in the cell membranes and then injected into the momentum. After
12-18 months, there were no major side-effects and the FVIII level
had increased (35). Lipid-based
nanoparticles have been also employed. Prophylaxis with PEGylated
liposomes (BAY 79-4980) was found to be safe and effective when
compared to rFVIII-FS in equal doses in a phase I, controlled
crossover design study (36). The
phase II study, on the other hand, was terminated as BAY 79-4980
was unable to demonstrate superior efficacy to rFVIII-FS (37).
6. Clinical trials and FDA approval for gene
therapy in hemophilia
Preclinical trials have provided a rough prediction
of how humans might respond. Animal models typically demonstrate
higher increases in factor VIII and IX levels compared to what has
been observed in human trials (3).
Hemophilia B
The very first phase I clinical trial was reported
by Lu et al (38) in 1993
who used the subcutaneous injection of ex vivo skin
fibroblasts in 2 patients with severe B hemophilia and achieved
increased clotting activity from 2.9 to 6.3% for 6 months in 1
patient only. After ~10 years, recombinant AVV vector was first
implicated in factor-IX gene transfer to adult patients diagnosed
with severe hemophilia B. Both intramuscular and transhepatic
routes failed to sustain plasma transgene expression (39).
The first successful trial was reported in 2011. The
administration of codon-optimized Factor IX (co-FIX) contained
inside recombinant AAV8 vectors (specifically, scAAV2/8-LP1-hFIXco)
was performed intravenously in a total of 6 individuals diagnosed
with severe hemophilia B. All individuals expressed 2-11% of normal
Factor IX, which improved bleeding (22).
Several subsequent multicentric open-labelled trials
were conducted, as summarized in Table
I. The preclinical trials used the Padua transgene, which has a
naturally occurring single nucleotide mutation (R338 L) that
results in a gain-of-function. This mutation, known as FIX-Padua,
was applied to enhance the production of FIX by a factor of six to
eight (40). The AAV5-hFIXco-Padua
vector, known as AMT-061 or etranacogene dezaparvovec®,
was used as a replacement for the AMT-060 FIX transgene in order to
enhance the level of expression. A single administration of AMT-061
at a dose of 2x1,013 vg/kg successfully halted bleeding for a
duration of 26 weeks, without the need for FIX replacement
(41). The aforementioned
compelling findings initiated the commencement of the worldwide
HOPE-B (Health Outcomes With Padua Gene; Evaluation in
Hemophilia-B) phase III research (NCT03569891), which aimed to
further assess the efficacy of AMT-061(42). FDA approved AMT-061(CSL Behring's)
in 2022 at a cost of US$3.5 million per dose (43) prescribed as 2x1013
genome copies (gc)/kg IV (2 ml/kg) as a single one-time dose.
 | Table IResults of published clinical trials
that examined the efficacy of gene therapy in hemophilia B and
A. |
Table I
Results of published clinical trials
that examined the efficacy of gene therapy in hemophilia B and
A.
| Sponsor | Authors | Year of publ. | Hemo. type | Type of study | Phase | No. of
patients | Baseline F8
IU/dl | Vector | Transgene |
Mean/mediana follow-up per week | Genome
copies/kg | Mean factor
activity % | Prophylactic factor
replacement status | Trial registration
no. | (Refs.) |
|---|
| - | Lu et
al | 1993 | B | SC | 1 | 2 | | rRV N2CMV-IX | FIX | 24 | - | 92%↑ | - | - | (38) |
| - | Manno et
al | 2006 | B | MC | 1-2 | 7 | <=1 | rAAV-2 | FIX | 111 |
2x1012 | Transient ↑for 8
w | - | - | (39) |
| National Heart,
Lung, and Blood Institute | Nathwani et
al | 2011 | B | SC | 1 | 10 | <=1 | scAAV2/8 | LP1-hFIXco | 165 | 2c1011,
6x1011 2x1012 | 1.8 IU/dl, 2.5
IU/dl, 5.1 IU/dl | 92%↓ 96%↓ | NCT00979238 | (22) |
| Spark Therapeutics
and Pfizer | George et
al | 2017 | B | MC | 1-2a | 14 | ≤2% | AAV-Spark100 | FIX-Padua | 492 |
5x1011 | 33.7% | 98%↓ | NCT02484092 | (90) |
| uniQure B.V | Miesbach et
al | 2018 | B | MC | 1-2 | 10 | ≤2% | AAV5 AMT-060 | FIX-Padua | 54 | 5x1012,
2x1013 | 4.4 IU/dl, 6.9
IU/dl | 81%↓ 37%↓ | NCT02396342 | (91) |
| uniQure and CSL
Behring | Von Drygalski et
al | 2019 | B | SC | 2b | 3 | ≤2% | AAV5 AMT-061 | FIX-Padua | 26 |
2x1013 | ≥40%↑ | - | NCT03489291 | (41) |
| Spark
Therapeutics | George et
al | 2020 | B | MC | 1-2 | 7 | <=1 | AAV2 | hFIX | 780 | 8x1010,
4x1011, 2x1012 | - | 84.5↓ | NCT00515710 | (92) |
| - | Konkle et
al | 2021 | B | MC | 1-2 | 8 | ≤2% | AAV8 (BAX 335) | FIX-Padua | 45.3 | 2.x1011,
1.0x1012, 3.0x1012 | 23-58.5% | N/A | NCT01687608 | (93) |
| Freeline | Chowdary et
al | 2022 | B | SC | 1-2 | 17 | ≤2% | AAVS3
(FLT180a) | FIX-(R338L)
Padua | 108a |
3.84x1011,
6.40x1011, 8.32x1011,
1.28x1012 | 51-78% | | NCT03369444
NCT03641703 | (94) |
| - | Xue et
al | 2022 | B | SC | 1 | 12 | <=1 | rAAV/BBM-H901 | FIX | 58a | 5x1012,
2x1012 | 36·9% | | NCT04135300 | (95) |
| uniQure and CSL
Behring | Pipe et
a. | 2023 | B | SC | 3 | 54 | ≤2% | AAV5 AMT-061 | hFIX-coPadua | 18 |
2x1013 | 39% | 96%↓ | NCT03569891 | (42) |
| - | Roth et
al | 2001 | A | SC | 1 | 6 | <=1 | plasmid | factor VIII | 52 | - | 25%↑ | - | - | (35) |
| - | Powell et
al | 2003 | A | MC | 1 | 13 | <=1 | retroviral
MoMLV | BDD factor
VIII | 13-53 | 2.8,
9.2x107, 2.2, 4.4, 8.8x108 | 2.3-19% | - | - | (96) |
| BioMarin | Rangarajan et
al | 2017 | A | MC | 1 | 9 | <=1 | AAV5 (BMN270) | BDD hFVIII-SQ | 52 | 6x1012,
2x1013, 6x1013 | <1 IU/dl, 1-3
IU/dl, 12-32 IU/dl | 35.1%↓ 88%↓
98%↓ | NCT02576795 | (27) |
| BioMarin | Pasi et
al | 2020 | A | | 1-2 | 15 | <=1 | AAV5 (BMN270) | BDD hFVIII-SQ | 156 | 6x1012
2x1013 6x1013 4x1013 | <1 IU/dl <1
IU/dl 20-60 23 IU/dl | 100↓ 99.6↓ | NCT02576795 | (45) |
| | George et
al | 2021 | A | MC | 1-2 | 18 | ≤2% | AAV3 SPK-8011 | BDD-FVIII | 146.4a | 5x1011,
1x012, 1.5x012, 2x1012 | 12% | 99%↓ | NCT03003533 and
NCT03432520. | (50) |
| Pfizer | Visweshwar et
al | 2021 | | | 1-2 | 11 | | rAAV 2-6 | Modified | 104 |
9x1011 | - | - | NCT03061201 | (48) |
| | | | | | | | | (PF-07055480) | BDD-FVIII | | 2x
1012 | - | | | |
| | | | | | | | | | | |
1x1013 | - | | | |
| | | | | | | | | | | |
3x1013 | 30.9% | | | |
| BioMarin | Ozelo et
al | 2022 | A | MC | 3 | 134 | <=1 | AAV5 (BMN270) | BDD FVIII-SQ | 51 |
6x1013 | 41.9 IU↑ | 98.6%↓ | NCT03370913 | (46) |
| BioMarin | Mahlangu et
al | 2023 | A | MC | 3b | 132 | <1 | AAV5 (BMN270) | BDD-FVIII-SQ | 104-260 |
6x1013 | 22.3 U/dl | 98.2%↓ | NCT03370913 | (97) |
Hemophilia A
The large size and complexity of F8 gene has delayed
the clinical trials on hemophilia A despite its higher prevalence.
Several gene therapies have been developed, and three of them are
approaching the final approval (Table
I).
In the year 2017, a total of 9 patients were
subjected to the administration of a solitary intravenous dosage of
BMN 270 (AAV-hFVIII-SQ), also known as valoctocogene
roxaparvovec® (27).
Over the course of 1 year, the high dosage group demonstrated the
maintenance of normalized FVIII activity, as well as the stability
of hemostasis and a reduction in the occurrence of bleeding events.
A sustained and notable improvement in clinical outcomes was
observed throughout the higher dosage groups over the duration of
the 5-year follow-up period (44).
In addition to the phase 1/2 dose escalation study
and the global phase 3 study GENEr8-1 (45,46),
further investigations, referred to as phase 3b research, were
conducted to assess the efficacy and safety of valoctocogene
roxaparvovec in individuals diagnosed with severe hemophilia A. The
firm is now engaged in a phase 1/2 Study using valoctocogene
roxaparvovec, which aims to include ~10 individuals who had
pre-existing AAV5 antibodies. Additionally, the company is also
undertaking a phase 1/2 study targeting individuals with hemophilia
A who have either active or past FVIII inhibitors (47). FDA approval for valoctocogene
roxaparvovec was deferred until the primary endpoint for ongoing
phase III study, 2-year observation, is completed.
SB-525, also known as PF-07055480 (marketed as
Giroctocogene fitelparvovec®), is a genetically
engineered adeno-associated virus serotype 6 (AAV6) that carries
the complementary DNA sequence for B domain deleted human
coagulation factor VIII (FVIII). The design of the Giroctocogene
fitelparvovec expression cassette aimed at achieving the excellent
expression of the FVIII protein specifically in the liver, while
also enabling high-yield manufacturing of the vector. The
subsequent data obtained from the phase 1 and phase 2 Alta revealed
that the rise in FVIII levels was both dose-dependent and
sustained. Furthermore, there was a notable reduction in the use of
FVIII and an absence of bleeding events seen in the highest dosage
group (48). Of note, 4 patients
in the highest dose cohort maintained modest to normal mean FVIII
activity levels through week 104(48). The clinical suspension of long-term
follow-up research has been implemented by the FDA in response to
the need for a review of a proposed protocol revision to the
current phase III investigation (NCT04370054, AFFINE) (3).
SPK-8011 or (samoparvovec®) is a
recombinant AAV8 that encodes a B-domain-delete FVIII sequence
under the control of a hepatocyte-specific transthyretin promoter
(49). Of note, 12 out of 18
participants in a phase 1-2 trial achieved sustained (>12% of
the normal value level) 2 years after one-stage factor VIII assay
administration with and without glucocorticoids. The annual
hemorrhage rate of the participants decreased by 91.5% prior to
vector administration; however, 8 participants experienced 33
treatment-related adverse events; 17 were vector-related, including
one severe adverse event, and 16 were glucocorticoid-related.
Notably, 2 individuals lost all factor VIII expression as a result
of an anti-AAV capsid cellular immune response that was insensitive
to immune suppression. The phase III trial stopped recruiting in
May, 2023 and the results are to be released soon (50).
7. Other potential pathways for gene therapy
in hemophilia
Restoring functional hemostasis has been
successfully achieved by manipulating natural coagulation
inhibitors via the use of antibodies that neutralize tissue factor
pathway inhibitors, protease nexin 1,25, activated protein
C-specific serpin inhibition and antithrombin inhibition by
nanobodies (51).
Post-transcriptional gene silencing RNA interference
(RNAi) is an additional approach to restore hemostasis equilibrium
in individuals with hemophilia by inhibiting the expression of
coagulation-inhibitory serpins (52). Prophylactic therapy, Fitusiran
(ALN-AT3, Alnylam/Sanofi), is an RNAi-based therapy developed for
patients with hemophilia A and B that targets antithrombin
messenger RNA (53). It is
synthesized by binding siRNA covalently to a triantennary Ga1NAc
ligand which reduces antithrombin production. The safety and
tolerability of fitusiran were assessed in a phase I dose
escalation study in healthy volunteers and adult participants with
moderate-severe non-inhibitor hemophilia A and B. Doses ranging
from 0.015 to 1.8 mg/kg resulted in an ~50% reduction in
antithrombin levels (54). When
antithrombin levels were reduced by >75% from their peak at
baseline, the thrombin generation values were comparable to those
described in mild hemophilia and at the low end of the range
observed in healthy participants (55).
Gene editing is another approach adapted by creating
a targeted DNA double-strand break in genomic DNA near the site of
desired change. The objective of this approach is to maintain
cis-regulatory elements that potentially regulate gene
function, while mitigating any adverse consequences associated with
the mutant gene. This can be achieved using several different
nuclease platforms including, but not limited to, meganucleases,
zinc finger nucleases (ZFNs), transcription activator-like effector
nucleases, and CRISPR-Cas9. Both AAV and non-viral delivery
techniques, including RNPs and lipid nanoparticles have been used
to deliver gene-editing components (56).
Pre-clinical studies on hemophilia A have
demonstrated that an injection of dual AAV vectors containing
Staphylococcus pyogenes Cas 9 (SpCas9) and guided RNA with
human B-domain deleted FVIII integrated human FVIII into the
albumin locus caused the liver in mice to produce FVIII. This
technique improved the hemophilia A phenotype for at least 7 months
without off-target effects or liver damage, suggesting permanent
FVIII substitution (57).
Initial preclinical trials evaluated ZFN, which
aimed at locating the normal copy of the clotting factor gene
within the albumin intron 1, under the control of the endogenous
albumin locus promoter, resulted in high levels of FIX for almost 1
year without any changes in plasma albumin levels, even when only
0.5% of mouse transcripts were modified (58).
Platelet-targeted gene is a promising approach to
gene therapy for hemophilia that involves the targeting of FVIII or
FIX expression and storage on platelets. This strategy holds
promise, not only for enhancing hemostasis, but also for eliciting
immune tolerance. The basic notion is to harvest hematopoietic stem
cells (HSCs) from the peripheral or cord blood of a patient, insert
a platelet-specific promoter into the corrected gene (FVIII or
FIX), and then autologously transplant the cells. The study by Shi
(59) provides a thorough analysis
of this subject.
8. Challenges of gene therapy
Vector related challenges. Viral vectors used
in gene therapy, whether they integrate with the host genome (such
as retroviruses) or remain episomal (such as adeno-associated
viruses or AAVs), carry potential risks (28), including the following:
i) Host immunity. Every vector system used in
gene therapy faces its own unique immune response challenges. These
challenges can be divided into two categories: Those related to
innate immunity and those related to adaptive immunity, which
includes memory and preexisting immunity (60).
Immunological evidence of prior exposure to AAV is
detectable in a substantial proportion of individuals (30-80%),
even in the absence of clinically detectable infection. Even at
modest concentrations, anti-AAV antibodies can neutralize the viral
vector and limit the viability of gene therapy (3). This poses a significant barrier to
the widespread use of AAV-mediated gene transfer, and as a
consequence, a number of clinical trials have precluded patients
with pre-existing anti-AAV antibodies. In a preclinical study, AAV5
transgene expression was successful even with antibodies targeting
AAV5. Primates received rAAV5 with the human secretory embryonic
alkaline phosphatase (hSEAP) gene intravenously 6 weeks prior to
the first rAAV treatment carrying factor IX had therapeutic levels
of factor IX (hFIX) expression, and the researchers determined a
threshold of anti-AAV5 antibodies that allowed for the effective
re-administration of rAAV5. Anti-AAV5 antibodies did not appear to
hinder further AAV5-based therapies (61). The seroprevalence of AAV in
patients with hemophilia in developing countries, including Iraq is
limited (3). Turkey, for instance,
a neighboring country, reported a seroprevalence of pre-existing
AAV8 antibodies of (67%), which is relatively high when compared to
The Netherlands (27%) and Italy (14%) (62). Hence, epidemiological studies are
required to determine the local prevalence.
Further studies involved the use of an endopeptidase
that can degrade circulating anti-rAAV-neutralizing antibodies
prior to the administration of rAAV gene therapy. Imlifidase, an
enzyme derived from Streptococcus pyogenes known for its
ability to degrade immunoglobulin G (IgG), is currently being
studied in transplant recipients (63). In a mouse model of rAAV8-mediated
gene therapy, treatment with imlifidase led to a reduction in
anti-AAV antibodies and improved the expression of the therapeutic
transgene in the liver. This indicates that imlifidase
administration shows promise in addressing the challenge of
pre-existing antibodies in AAV-based gene therapy (20).
Preclinical trials have found that the ability of
host APC transduction varies between retroviruses or lentiviruses
vectors (64). Cytotoxic
T-lymphocytes and antibodies against transgenic products may result
after transduction (26). T-cell
activation against the transgene and pseudo-typed lentiviral
vectors with diverse glycoproteins has lowered transgene expression
over time (64).
Vector design, route and dose affect the gene
expression lifespan. Nasal or intratracheal delivery may activate
the immune system and induce tolerance (65). Liver-directed gene transfer using a
hepatocyte-restricted promoter via hematopoietic lineage-specific
microRNA target sequences limits transgene expression in
professional antigen-presenting cells and reduces immunological
responses (60). The
post-transcriptional microRNA-mediated gene suppression has been
implicated in confining transgene activity to a particular tissue.
This approach involves introducing a specific target sequence into
the messenger RNA of the transgene, which corresponds to a microRNA
found exclusively in the desired cell type (66). In an alternative methodology, the
2bF9/methylguanine-DNA-methyltransferase (MGMT) LV vector was
utilized. This vector incorporates the alpha-2b promoter, FIX and
MGMT 140K genes. Following the transduction of HSCs, this vector
resulted in a 2.9-fold increase in FIX expression and a 3.7-fold
increase in FIX activity within platelets (67,68).
ii) Liver toxicity. The incidence of acute
adverse events subsequent to the administration of AAV vectors is
relatively infrequent. However, it has been shown that liver
toxicity manifested in ~60% of individuals within a timeframe of 4
to 12 weeks subsequent to the therapeutic intervention (27). The observed complication is
distinguished by a range of liver enzyme level elevations, as well
as a reduction or absence of the transgenic clotting proteins in
the circulatory system, primarily caused by the demise of
transduced hepatic cells (27).
The exact cause of this liver toxicity remains unclear and may vary
among patients. Some researchers have related this to viral vector
particle load (69). Cytotoxic
T-cells targeting AAV capsid peptides have been reported in a
number of preclinical trials (70). Others have suggested that a high
FVIII transgenes expression increases endoplasmic reticulum stress,
which induces hepatocyte apoptosis. While the underlying mechanisms
require further investigation, current empirical therapies that aim
to manage this issue include oral corticosteroid therapy,
commencing at ~1 mg/kg and then gradually reducing the dose over a
period of 2-3 months (70).
Nevertheless, there are instances where oral steroids prove
ineffective in treating liver toxicity, hence requiring the use of
intravenous methylprednisolone or other immune regulatory drugs
such as tacrolimus or azathioprine (71). In situations where liver toxicity
is more prevalent, prophylactic oral steroid schedules have been
implemented (71).
iii) Genotoxicity. Long-term mutagenic
changes in gene therapy remain a concerning issue. One of the
notable benefits associated with AAV vector administration is its
tendency to avoid the integration of vector sequences into the host
genome. This characteristic significantly reduces the potential
danger of long-term oncogenic consequences (71). The thorough genomic analysis of
liver biopsies from hemophilic dogs that underwent >10 years of
AAV treatment has not revealed any signs of cancerous tumors in the
liver (40). Previous studies,
however, have demonstrated that hepatocellular carcinoma can
develop in neonatal mouse models following recombinant AAV gene
transfer (72). Additionally,
fragmented wild-type AAV genomes have been detected in liver
cancers. In a recently published study, liver biopsy samples from
patients who received AAV5-hFVIII-SQ treatment at a dose of
6x1012 and 4x1013 vg/kg were collected and
analyzed to determine the presence and genomic forms of AAV
persistence. The percentage of hepatocytes exhibiting positive
staining for vector genomes was 1.3 and 32%, respectively. This
staining was observed throughout the liver lobes, resembling
findings seen in earlier studies involving non-human primates
(73). Additionally, both
treatment groups displayed circularized full-length vector genomes
and ITR-fusions, appearing both as individual units and linked
together in chains, in line with observations from previous
research (73).
Insertional mutagenesis is a potential safety
concern of lentivirus, which is more likely to occur when dividing
cells are transduced. However, newer-generation lentivirus designs
have significantly reduced this risk, and there have been no
reported cases of leukemic transformation in human gene therapy
trials (74). The genotoxicity
observed in previous cases involving retroviral vectors could
potentially be attributed to the activation of oncogenes by the
vector's intrinsic promoter. Consequently, the genetic makeup of
the lentivirus has been altered to enable the elimination of the
viral promoter during the reverse-transcription mechanism, commonly
referred to as self-inactivating lentivirus. The aforementioned
alteration has significantly reduced the likelihood of genotoxicity
(20).
iv) Germline transmission. Germline viral
vector transmission is a serious safety issue. Preclinical studies
have not demonstrated AAV in rabbit or dog semen after
intramuscular or portal vein rAAV injections (75). In the rAAV2 experiments, semen
vector sequences were detected transiently. Subsequent rabbit
investigations employing intravascular rAAV vector administration
revealed dose-dependent transitory viral detection in semen
(76). However, detecting the
vector sequences in vasectomy rabbits' semen indicates that viral
shedding into the semen did not need germ cells (76). Therefore, regulatory bodies
recommend barrier contraception for rAAV-containing semen (77).
Gene related challenges
Therapy durability is the main concern of patients.
Uncertainty persists as to whether these levels will stabilize or
decline further (71).
Investigations on the treatment of the FIX gene in adult patients
have demonstrated a minimal decrease in plasma FIX levels for up to
5 years following a single intravenous treatment (78). By contrast, the canine model of
hemophilia A maintained therapeutic expression of FVIII for >10
years after AAV vector infusions (79). A human study utilizing an AAV5
vector for FVIII gene transfer revealed a significant decrease in
FVIII levels during the first 4 years following vector
administration (45). Recently,
FVIII levels have reached ~0.20 IU/ml, which continues to provide
substantial protection against hemorrhage (45).
It is worth noting that in the general population,
natural levels of FVIII and FIX can differ as much as 4-fold among
individuals. The intricate balance and complexity involved in the
production, secretion and clearance of these proteins is only
partially understood at this time (71). Further studies addressing the
mechanisms of vector hepatocyte entry, intracellular trafficking,
nuclear import efficacy and the behavior of the transgene in terms
of remaining episomal or integrating into the host-cell genome are
required in order to be able to predict specific coagulation factor
levels; these are currently extremely difficult to predict
(71).
Ethical concerns. In children
With the continuous advancement of gene therapy, it
is anticipated that it will gain even more appeal as a therapeutic
alternative for children. However, ‘children are not just small
adults’, and customizing gene therapy to hemophilia younger
children will requires special considerations (80). Ethical issues arise when delivering
life-altering medications to young children, given the extended
lifespan of hemophilia patients, with emerging long-term safety
evidence while current standard managing options are available
(80). In addition, the strong
humoral immune response following first vector delivery hinders
successful vector reinfusion. Furthermore, the non-integrating
characteristic of AAV vectors renders them difficult to be
administered to children, since proliferating cells lose a
considerable portion of the vector during liver enlargement
(3).
In Muslim-based societies. Islamic ethics are
interconnected with science, religion and law. Qur'anic exogenesis,
scholastic theology, morality and knowledge acquisition are all
sources from which Islamic ethics is derived. They advocate for the
prevention of corruption and damage, as well as the acquisition of
an advantage or interest. Perhaps the most frequently employed
jurisprudential premise for medical treatment, including somatic
gene therapy, is the theory of advantage or interest. As long as
the concept of fundamental Islamic pillars of public benefit are
prioritized in somatic gene therapy, the technique is ethical
(81).
Cost-related challenges
The issue of gene therapy expenses is a key
obstacle, not only in developing countries, but also in developed
nations (71). The authorized gene
therapy product AMT-061 for hemophilia B by CSL Behring's put
forward a cost of US$3.5 million per dose (82).
A cost analysis conducted prior to the launch of the
first gene therapy product found that the total cost per person for
gene therapy was $1.0 million, resulting in 8.33 quality-adjusted
life years (QALYs), while prophylactic treatment cost $1.7 million
and yielded 6.62 QALYs (82). Over
a lifetime, the average total cost per patient was estimated to be
$16.7 million (with a range of $823,000 to $73.9 million) for those
receiving valoctocogene roxaparvovec, with a gain of 18.07 QALYs
(ranging from 0.06 to 23.38 QALYs). Patients using prophylactic
exogenous FVIII protein had an average total cost of $23.5 million
(with a range of $75,000 to $69.6 million) and gained 17.32 QALYs
(ranging from 0.05 to 22.54 QALYs). These results confirmed that
valoctocogene roxaparvovec was associated with a cost reduction of
$6.8 million per patient (4).
The restricted availability of options and financial
constraints in lower- and middle-income nations, such as Iraq, are
influenced by the high cost of therapy, which is determined by the
expenses associated with alternative treatments in high-income
countries. The potential for substantial improvement in patient
outcomes exists via the use of subsidized, single-dose gene
therapy, contingent upon the facilitation of talks and
international partnerships that would establish a fair and
acceptable price structure (83).
Outcome-based and finance-based payment methods are
two different approaches that have been suggested (84). The previous approach links
compensation to a mutually agreed-upon clinical outcome.
Compensation may be disbursed based on attainable accomplishments,
either on an annual basis if the desired outcome endures, or by
partial or whole reimbursement of the initial payment if the
outcome diminishes after the initial full payment (84). In this strategy, the absence of
agreement on the precise definition of a cure and quality of life
for hemophilia presents a notable obstacle in addition to the lack
of a control group in all gene therapy clinical trials in
hemophilia (85). To overcome
these difficulties, certain approaches have been suggested. Several
strategies may be used to enhance the validity and reliability of
clinical research. These strategies include the utilization of data
derived from prior clinical trials exploring alternative therapies
or real-world evidence to construct synthetic historical cohorts
for comparative analysis. Additionally, forecasts about the
potential outcomes of clinical trials can be made, and data can be
collected from real-world registries after approval (86).
Financial schemes that are based on financial
considerations have fewer practical factors to consider (84). These interventions are particularly
appropriate in cases when the projected patient outcomes are
foreseeable; however, the population of possible patients is either
vast or unclear. In a number of instances, a capitation model is
used, wherein a predetermined sum is levied irrespective of the
number of recipients. This approach may be used in a
subscription-based model, whereby a fixed amount of money provides
access to an infinite number of patients for a duration of 1 year
(84). Alternatively, it is
possible to employ a volume-based method in which the cost per
patient lowers after a specified threshold is reached. In some
cases, the duration of payment is prolonged across multiple years
rather than being disbursed as a single amount at the beginning. In
order to mitigate the potential risk, one possible approach is to
include a number of individuals or corporations that may act as
payers or to transfer a part of the responsibility to reinsurers.
The users have provided a D to support their statement (4).
Patient- and community-related
challenges. Social awareness
The introduction of new healthcare advancements
requires sufficient healthcare education and population awareness.
A survey conducted by the International Society on Thrombosis and
Hemostasis focused on healthcare teams and scientists to assess
their understanding and awareness of gene therapy, particularly as
regards hemophilia (87). The
survey included responses from physicians (66% of respondents),
with 59% directly involved in caring for hemophilia patients. The
results of that study indicated that ~33% of doctors had challenges
when attempting to articulate the core scientific concepts behind
AAV gene therapy (87).
Furthermore, a significant proportion of doctors, namely 40%,
acknowledged their lack of confidence in effectively addressing
patient queries pertaining to gene therapy. A survey was undertaken
by the WFH, which included the participation of 103 national member
organizations and 109 clinicians from 76 countries (88). The findings of that study revealed
that a notable proportion of patients (68%) exhibited a main level
of comprehension about gene therapy. By contrast, a notable
percentage of medical professionals (44%) exhibited only a
rudimentary or moderate grasp of the topic (88).
Health status of patients. A considerable
number of patients with hemophilia have a history of HIV, hepatitis
B, or hepatitis C virus infection. All these patients, in addition
to those with other inflammation, cardiovascular and neoplastic
diseases,were excluded from clinical trials (89). A study conducted in Iraq revealed
that the prevalence rates of hepatitis C virus, hepatitis B virus
and HIV infections among individuals with hemophilia were 22.9, 0.9
and 0.2%, respectively (6).
A number of trials excluded patients who had factor inhibitors and
AAV-neutralizing antibodies, as well as pediatric or elderly
populations (4).
Post-marketing trials have the potential to
accommodate these patients and loosen the stringent eligibility
criteria commonly employed in premarketing trials, although the
cost remains a prohibiting factor.
9. Conclusion and future perspectives
The approval of gene therapy for patients with
hemophilia B is expected to be followed shortly by the approval of
another agent for hemophilia A. As we enter the era of gene
therapy, it is crucial to prioritize education for healthcare
providers and the community to ensure they are well-prepared to
embrace these advancements. Regional collaboration and the
establishment of multinational-sponsored centers can aid in the
development of the necessary infrastructure, while ensuring
hematologists receive adequate training to effectively deliver this
cutting-edge treatment.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All the authors contributed equally in the
preparation and design of the study. MAA and IMAB contributed to
the preparation and design of the manuscript. EAKDAS was involved
in drafting and editing the manuscript. IMAB and MAA contributed to
the design and preparation of the table and figure. All authors
have read and approved the final manuscript. Data authentication is
not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bolton-Maggs PH and Pasi KJ: Haemophilias
A and B. Lancet. 361:1801–1809. 2003.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Iorio A, Stonebraker JS, Chambost H,
Makris M, Coffin D, Herr C and Germini F: Data and Demographics
Committee of the World Federation of Hemophilia. Establishing the
prevalence and prevalence at birth of hemophilia in males: A
meta-analytic approach using national registries. Ann Intern Med.
171:540–546. 2019.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Kavaklı K, Antmen B, Okan V, Şahin F,
Aytaç S, Balkan C, Berber E, Kaya Z, Küpesiz A and Zülfikar B: Gene
therapy in haemophilia: Literature review and regional perspectives
for Turkey. Ther Adv Hematol. 13(20406207221104591)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bolous NS, Bhatt N, Bhakta N, Neufeld EJ,
Davidoff AM and Reiss UM: Gene therapy and hemophilia: Where do we
go from here? J Blood Med. 13:559–580. 2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
World Federation of Hemophilia: The report
on the WFH annual global survey 2020: World Federation of
Hemophilia, 2021. Available from: https://www1.wfh.org/publications/files/pdf-2045.pdf.
|
|
6
|
Kadhim KAR, Al-Lami FH and Baldawi KH:
Epidemiological profile of hemophilia in Baghdad-Iraq. Inquiry.
56(46958019845280)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Abdulsalam AH, Al-Rahal NK and Ghiath Y:
Inherited bleeding disorders in pediatric patients; experience of
the national referral center in Iraq. Indian J Hematol Blood
Transfus. 37:96–100. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hassan S, Monahan RC, Mauser-Bunschoten
EP, van Vulpen LFD, Eikenboom J, Beckers EAM, Hooimeijer L, Ypma
PF, Nieuwenhuizen L, Coppens M, et al: Mortality, life expectancy,
and causes of death of persons with hemophilia in the Netherlands
2001-2018. J Thromb Haemost. 19:645–653. 2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Coffin D, Gouider E, Konkle B, Hermans C,
Lambert C, Diop S, Ayoub E, Tootoonchian E, Youttananukorn T, Dakik
P, et al: The world federation of hemophilia world bleeding
disorders registry: Insights from the first 10,000 patients. Res
Pract Thromb Haemost. 7(102264)2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Taresh AK and Hassan MK: Inhibitors among
patients with hemophilia in Basra, Iraq-A single center experience.
Niger J Clin Pract. 22:416–421. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lateef IA: Evaluation of the clinical
status of patients with inherited bleeding disorders in
Diyala-Iraq. Diyala J Medicine. 9:23–29. 2015.
|
|
12
|
Lateef IA, Hamood HJ and Khaleel OA:
Spectrum of hemophilia in Diyala-Iraq. Diyala J Medicine. 10:53–58.
2016.
|
|
13
|
Sharma A, Mathew ME, Sriganesh V and Reiss
UM: Gene therapy for haemophilia. Cochrane Database Syst Rev.
4(CD010822)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Srivastava A, Santagostino E, Dougall A,
Kitchen S, Sutherland M, Pipe SW, Carcao M, Mahlangu J, Ragni MV,
Windyga J, et al: WFH guidelines for the management of hemophilia.
Haemophilia. 26:1–158. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Nathwani AC: Gene therapy for hemophilia.
Hematology Am Soc Hematol Educ Program. 2019:1–8. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang X, Shin SC, Chiang AF, Khan I, Pan D,
Rawlings DJ and Miao CH: Intraosseous delivery of lentiviral
vectors targeting factor VIII expression in platelets corrects
murine hemophilia A. Mol Ther. 23:617–626. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Doncel SS, Mosquera GA, Pelaez RG, Cortes
JM, Rico CA, Cadavid FJ, Plazas NR, Amar IAP, Siado JEP, Rey FAP,
et al: Genetic characterization of the factor VIII gene in a cohort
of colombian patients with severe hemophilia A with inhibitors.
Hematol Rep. 14:149–154. 2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hassan MM and Jabber AD: Identification of
factor VIII gene mutations in Iraqi patient with hemophilia A. Int
J Med Res Prof. 2:192–199. 2016.
|
|
19
|
Abdulqader AMR, Mohammed AI, Rachid S,
Ghoraishizadeh P and Mahmood SN: Identification of the intron 22
and intron 1 inversions of the factor VIII gene in Iraqi Kurdish
patients with hemophilia A. Clin Appl Thromb Hemost.
26(1076029619888293)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lisowski L, Staber JM, Wright JF and
Valentino LA: The intersection of vector biology, gene therapy, and
hemophilia. Res Pract Thromb Haemost. 5(e12586)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Palmer TD, Thompson AR and Miller AD:
Production of human factor IX in animals by genetically modified
skin fibroblasts: Potential therapy for hemophilia B. Blood.
73:438–445. 1989.PubMed/NCBI
|
|
22
|
Nathwani AC, Tuddenham EG, Rangarajan S,
Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ,
Harrington C, et al: Adenovirus-associated virus vector-mediated
gene transfer in hemophilia B. N Engl J Med. 365:2357–2365.
2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Niemeyer GP, Herzog RW, Mount J, Arruda
VR, Tillson DM, Hathcock J, van Ginkel FW, High KA and Lothrop CD
Jr: Long-term correction of inhibitor-prone hemophilia B dogs
treated with liver-directed AAV2-mediated factor IX gene therapy.
Blood. 113:797–806. 2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liao G, Nayak S, Regueiro JR, Berger SB,
Detre C, Romero X, de Waal Malefyt R, Chatila TA, Herzog RW and
Terhorst C: GITR engagement preferentially enhances proliferation
of functionally competent CD4+ CD25+ FoxP3+ regulatory T cells. Int
Immunol. 22:259–270. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cooper M, Nayak S, Hoffman BE, Terhorst C,
Cao O and Herzog RW: Improved induction of immune tolerance to
factor IX by hepatic AAV-8 gene transfer. Hum Gene Ther.
20:767–776. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mátrai J, Chuah MKL and VandenDriessche T:
Preclinical and clinical progress in hemophilia gene therapy. Curr
Opin Hematol. 17:387–392. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rangarajan S, Walsh L, Lester W, Perry D,
Madan B, Laffan M, Yu H, Vettermann C, Pierce GF, Wong WY and Pasi
KJ: AAV5-factor VIII gene transfer in severe hemophilia A. N Engl J
Med. 377:2519–2530. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Guo XL, Chung TH, Qin Y, Zheng J, Zheng H,
Sheng L, Wynn T and Chang LJ: Hemophilia gene therapy: New
development from bench to bed side. Curr Gene Ther. 19:264–273.
2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xu L, Nichols TC, Sarkar R, McCorquodale
S, Bellinger DA and Ponder KP: Absence of a desmopressin response
after therapeutic expression of factor VIII in hemophilia A dogs
with liver-directed neonatal gene therapy. Proc Natl Acad Sci USA.
102:6080–6085. 2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Milani M, Canepari C, Liu T, Biffi M,
Russo F, Plati T, Curto R, Patarroyo-White S, Drager D, Visigalli
I, et al: Liver-directed lentiviral gene therapy corrects
hemophilia A mice and achieves normal-range factor VIII activity in
non-human primates. Nat Commun. 13(2454)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tsui LV, Kelly M, Zayek N, Rojas V, Ho K,
Ge Y, Moskalenko M, Mondesire J, Davis J, Roey MV, et al:
Production of human clotting Factor IX without toxicity in mice
after vascular delivery of a lentiviral vector. Nat Biotechnol.
20:53–57. 2002.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Stein CS, Kang Y, Sauter SL, Townsend K,
Staber P, Derksen TA, Martins I, Qian J, Davidson BL and McCray PB
Jr: In vivo treatment of hemophilia A and mucopolysaccharidosis
type VII using nonprimate lentiviral vectors. Mol Ther. 3:850–856.
2001.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Dull T, Zufferey R, Kelly M, Mandel RJ,
Nguyen M, Trono D and Naldini L: A third-generation lentivirus
vector with a conditional packaging system. J Virol. 72:8463–8471.
1998.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kuether EL, Schroeder JA, Fahs SA, Cooley
BC, Chen Y, Montgomery RR, Wilcox DA and Shi Q: Lentivirus-mediated
platelet gene therapy of murine hemophilia A with pre-existing
anti-factor VIII immunity. J Thromb Haemost. 10:1570–1580.
2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Roth DA, Tawa NE Jr, O'Brien JM, Treco DA
and Selden RF: Factor VIII Transkaryotic Therapy Study Group.
Nonviral transfer of the gene encoding coagulation factor VIII in
patients with severe hemophilia A. N Engl J Med. 344:1735–1742.
2001.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Spira J, Plyushch OP, Andreeva TA and
Andreev Y: Prolonged bleeding-free period following prophylactic
infusion of recombinant factor VIII reconstituted with pegylated
liposomes. Blood. 108:3668–3673. 2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Di Minno G, Cerbone AM, Coppola A, Cimino
E, Di Capua M, Pamparana F, Tufano A and Di Minno MN: Longer-acting
factor VIII to overcome limitations in haemophilia management: The
PEGylated liposomes formulation issue. Haemophilia. 16:2–6.
2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lu DR, Zhou JM, Zheng B, Qiu XF, Xue JL,
Wang JM, Meng PL, Han FL, Ming BH and Wang XP: Stage I clinical
trial of gene therapy for hemophilia B. Sci China B. 36:1342–1351.
1993.PubMed/NCBI
|
|
39
|
Manno CS, Pierce GF, Arruda VR, Glader B,
Ragni M, Rasko JJ, Ozelo MC, Hoots K, Blatt P, Konkle B, et al:
Successful transduction of liver in hemophilia by AAV-factor IX and
limitations imposed by the host immune response. Nat Med.
12:342–347. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
40
|
Crudele JM, Finn JD, Siner JI, Martin NB,
Niemeyer GP, Zhou S, Mingozzi F, Lothrop CD Jr and Arruda VR: AAV
liver expression of FIX-Padua prevents and eradicates FIX inhibitor
without increasing thrombogenicity in hemophilia B dogs and mice.
Blood. 125:1553–1561. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Von Drygalski A, Giermasz A, Castaman G,
Key NS, Lattimore S, Leebeek FWG, Miesbach W, Recht M, Long A, Gut
R, et al: Etranacogene dezaparvovec (AMT-061 phase 2b): Normal/near
normal FIX activity and bleed cessation in hemophilia B. Blood Adv.
3:3241–3247. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pipe SW, Leebeek FWG, Recht M, Key NS,
Castaman G, Miesbach W, Lattimore S, Peerlinck K, Van der Valk P,
Coppens M, et al: Gene therapy with etranacogene dezaparvovec for
hemophilia B. N Engl J Med. 388:706–718. 2023.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pipe SW, Reddy KR and Chowdary P: Gene
therapy: Practical aspects of implementation. Haemophilia. 28
(Suppl 4):S44–S52. 2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Pasi KJ, Laffan M, Rangarajan S, Robinson
TM, Mitchell N, Lester W, Symington E, Madan B, Yang X, Kim B, et
al: Persistence of haemostatic response following gene therapy with
valoctocogene roxaparvovec in severe haemophilia A. Haemophilia.
27:947–956. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Pasi KJ, Rangarajan S, Mitchell N, Lester
W, Symington E, Madan B, Laffan M, Russell CB, Li M, Pierce GF and
Wong WY: Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for
hemophilia A. N Engl J Med. 382:29–40. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ozelo MC, Mahlangu J, Pasi KJ, Giermasz A,
Leavitt AD, Laffan M, Symington E, Quon DV, Wang JD, Peerlinck K,
et al: Valoctocogene roxaparvovec gene therapy for hemophilia A. N
Engl J Med. 386:1013–1025. 2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Long BR, Veron P, Kuranda K, Hardet R,
Mitchell N, Hayes GM, Wong WY, Lau K, Li M, Hock MB, et al: Early
phase clinical immunogenicity of valoctocogene roxaparvovec, an
AAV5-mediated gene therapy for hemophilia A. Mol Ther. 29:597–610.
2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Visweshwar N, Harrington TJ, Leavitt AD,
Konkle BA, Giermasz A, Stine K, et al: Updated results of the alta
study, a phase 1/2 study of giroctocogene fitelparvovec
(PF-07055480/SB-525) gene therapy in adults with severe hemophilia
A. Blood. 138 (Suppl 1)(564)2021.
|
|
49
|
Elkouby L, Armour SM, Toso R, DiPietro M,
Davidson RJ, Nguyen GN, Willet M, Kutza S, Silverberg J, Frick J,
et al: Preclinical assessment of an optimized AAV-FVIII vector in
mice and non-human primates for the treatment of hemophilia A. Mol
Ther Methods Clin Dev. 24:20–29. 2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
George LA, Monahan PE, Eyster ME, Sullivan
SK, Ragni MV, Croteau SE, Rasko JEJ, Recht M, Samelson-Jones BJ,
MacDougall A, et al: Multiyear factor VIII expression after AAV
gene transfer for hemophilia A. N Engl J Med. 385:1961–1973.
2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Barbon E, Ayme G, Mohamadi A, Ottavi JF,
Kawecki C, Casari C, Verhenne S, Marmier S, van Wittenberghe L,
Charles S, et al: Single-domain antibodies targeting antithrombin
reduce bleeding in hemophilic mice with or without inhibitors. EMBO
Mol Med. 12(e11298)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Setten RL, Rossi JJ and Han SP: The
current state and future directions of RNAi-based therapeutics. Nat
Rev Drug Discov. 18:421–446. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Patnaik MM and Moll S: Inherited
antithrombin deficiency: A review. Haemophilia. 14:1229–1239.
2008.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Pasi KJ, Rangarajan S, Georgiev P, Mant T,
Creagh MD, Lissitchkov T, Bevan D, Austin S, Hay CR, Hegemann I, et
al: Targeting of antithrombin in hemophilia A or B with RNAi
therapy. N Engl J Med. 377:819–828. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Dargaud Y, Béguin S, Lienhart A, Al Dieri
R, Trzeciak C, Bordet JC, Hemker HC and Negrier C: Evaluation of
thrombin generating capacity in plasma from patients with
haemophilia A and B. Thromb Haemost. 93:475–480. 2005.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Segurado OG, Jiang R and Pipe SW:
Challenges and opportunities when transitioning from in vivo gene
replacement to in vivo CRISPR/Cas9 therapies-a spotlight on
hemophilia. Expert Opin Biol Ther. 22:1091–1098. 2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Chen H, Shi M, Gilam A, Zheng Q, Zhang Y,
Afrikanova I, Li J, Gluzman Z, Jiang R, Kong LJ, et al: Hemophilia
A ameliorated in mice by CRISPR-based in vivo genome editing of
human factor VIII. Sci Rep. 9(16838)2019.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Anguela XM, Sharma R, Doyon Y, Miller JC,
Li H, Haurigot V, Rohde ME, Wong SY, Davidson RJ, Zhou S, et al:
Robust ZFN-mediated genome editing in adult hemophilic mice. Blood.
122:3283–3287. 2013.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Shi Q: Platelet-targeted gene therapy for
hemophilia. Mol Ther Methods Clin Dev. 9:100–108. 2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Nayak S and Herzog RW: Progress and
prospects: Immune responses to viral vectors. Gene Ther.
17:295–304. 2010.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Salas D, Kwikkers KL, Zabaleta N, Bazo A,
Petry H, van Deventer SJ, Aseguinolaza GG and Ferreira V:
Immunoadsorption enables successful rAAV5-mediated repeated hepatic
gene delivery in nonhuman primates. Blood Adv. 3:2632–2641.
2019.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Ferla R, Claudiani P, Savarese M, Kozarsky
K, Parini R, Scarpa M, Donati MA, Sorge G, Hopwood JJ, Parenti G,
et al: Prevalence of anti-adeno-associated virus serotype 8
neutralizing antibodies and arylsulfatase B cross-reactive
immunologic material in mucopolysaccharidosis VI patient candidates
for a gene therapy trial. Hum Gene Ther. 26:145–152.
2015.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Elmore ZC, Oh DK, Simon KE, Fanous MM and
Asokan A: Rescuing AAV gene transfer from neutralizing antibodies
with an IgG-degrading enzyme. JCI Insight.
5(e139881)2020.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Follenzi A, Santambrogio L and Annoni A:
Immune responses to lentiviral vectors. Curr Gene Ther. 7:306–315.
2007.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Limberis MP, Bell CL, Heath J and Wilson
JM: Activation of transgene-specific T cells following
lentivirus-mediated gene delivery to mouse lung. Mol Ther.
18:143–150. 2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Geisler A and Fechner H:
MicroRNA-regulated viral vectors for gene therapy. World J Exp Med.
6:37–54. 2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Chen Y, Schroeder JA, Gao C, Li J, Hu J
and Shi Q: In vivo enrichment of genetically manipulated platelets
for murine hemophilia B gene therapy. J Cell Physiol. 236:354–365.
2021.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Lundstrom K: Viral vectors in gene
therapy: Where do we stand in 2023? Viruses. 15(698)2023.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Nathwani AC, Rosales C, McIntosh J,
Rastegarlari G, Nathwani D, Raj D, Nawathe S, Waddington SN,
Bronson R, Jackson S, et al: Long-term safety and efficacy
following systemic administration of a self-complementary AAV
vector encoding human FIX pseudotyped with serotype 5 and 8 capsid
proteins. Mol Ther. 19:876–885. 2011.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Nathwani AC, Reiss UM, Tuddenham EG,
Rosales C, Chowdary P, McIntosh J, Peruta MD, Lheriteau E, Patel N,
Raj D, et al: Long-term safety and efficacy of factor IX gene
therapy in hemophilia B. N Engl J Med. 371:1994–2004.
2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Batty P and Lillicrap D: Hemophilia gene
therapy: Approaching the first licensed product. Hemasphere.
5(e540)2021.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Chandler RJ, LaFave MC, Varshney GK,
Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V,
Elkahloun AG, Burgess SM and Venditti CP: Vector design influences
hepatic genotoxicity after adeno-associated virus gene therapy. J
Clin Invest. 125:870–880. 2015.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Fong S, Rangarajan S and Mitchell N: First
in human liver biopsy study following gene therapy for hemophilia
A. Res Pract Thromb Haemost. 4(3)2020.
|
|
74
|
Marcucci KT, Jadlowsky JK, Hwang WT,
Suhoski-Davis M, Gonzalez VE, Kulikovskaya I, Gupta M, Lacey SF,
Plesa G, Chew A, et al: Retroviral and lentiviral safety analysis
of gene-modified T cell products and infused HIV and oncology
patients. Mol Ther. 26:269–279. 2018.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Arruda VR, Fields PA, Milner R, Wainwright
L, De Miguel MP, Donovan PJ, Herzog RW, Nichols TC, Biegel JA,
Razavi M, et al: Lack of germline transmission of vector sequences
following systemic administration of recombinant AAV-2 vector in
males. Mol Ther. 4:586–592. 2001.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Favaro P, Downey HD, Zhou JS, Wright JF,
Hauck B, Mingozzi F, High KA and Arruda VR: Host and
vector-dependent effects on the risk of germline transmission of
AAV vectors. Mol Ther. 17:1022–1030. 2009.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Food and Drug Administration: Guidance for
industry: Gene therapy clinical trials-Observing subjects for
delayed adverse events. 2006.
|
|
78
|
Leebeek FWG, Meijer K, Coppens M, Kampmann
P, Klamroth R, Schutgens R, Castaman G, Seifried E, Schwaeble J,
Bönig H, et al: AMT-060 gene therapy in adults with severe or
moderate-severe hemophilia B confirm stable FIX expression and
durable reductions in bleeding and factor IX consumption for up to
5 years. Blood. 136(26)2020.
|
|
79
|
Sabatino DE, Lange AM, Altynova ES, Sarkar
R, Zhou S, Merricks EP, Franck HG, Nichols TC, Arruda VR and
Kazazian HH Jr: Efficacy and safety of long-term prophylaxis in
severe hemophilia A dogs following liver gene therapy using AAV
vectors. Mol Ther. 19:442–449. 2011.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Gollomp KL, Doshi BS and Arruda VR: Gene
therapy for hemophilia: Progress to date and challenges moving
forward. Transfus Apher Sci. 58:602–612. 2019.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Samori Z and Badran I: Somatic gene
therapy: Ethical consideration and Islamic Fiqhi perspective. J
Engineering and Applied Sciences. 13:4353–4561. 2018.
|
|
82
|
Bolous NS, Chen Y, Wang H, Davidoff AM,
Devidas M, Jacobs TW, Meagher MM, Nathwani AC, Neufeld EJ, Piras
BA, et al: The cost-effectiveness of gene therapy for severe
hemophilia B: A microsimulation study from the United States
perspective. Blood. 138:1677–1690. 2021.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Reiss UM, Zhang L and Ohmori T: Hemophilia
gene therapy-New country initiatives. Haemophilia. 27:132–141.
2021.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Goodman C, Berntorp E and Wong O:
International Haemophilia Access Strategy Council. Alternative
payment models for durable and potentially curative therapies: The
case of gene therapy for haemophilia A. Haemophilia. 28:27–34.
2022.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Sharma A, Mathew ME, Sriganesh V and Reiss
UM: Gene therapy for haemophilia. Cochrane Database Syst Rev.
12(CD010822)2016.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Drummond MF, Neumann PJ, Sullivan SD,
Fricke FU, Tunis S, Dabbous O and Toumi M: Analytic considerations
in applying a general economic evaluation reference case to gene
therapy. Value Health. 22:661–668. 2019.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Peyvandi F, Garagiola I and Young G: The
past and future of haemophilia: Diagnosis, treatments, and its
complications. Lancet. 388:187–197. 2016.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Pierce GF and Coffin D: Members of the WFH
Gene Therapy Round Table Program Committee and Organizing
Committee. The 1st WFH gene therapy round table: Understanding the
landscape and challenges of gene therapy for haemophilia around the
world. Haemophilia. 25:189–194. 2019.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Doshi BS and Arruda VR: Gene therapy for
hemophilia: What does the future hold? Ther Adv Hematol. 9:273–293.
2018.PubMed/NCBI View Article : Google Scholar
|
|
90
|
George LA, Sullivan SK, Giermasz A, Rasko
JEJ, Samelson-Jones BJ, Ducore J, Cuker A, Sullivan LM, Majumdar S,
Teitel J, et al: Hemophilia B gene therapy with a
high-specific-activity factor IX variant. N Engl J Med.
377:2215–2227. 2017.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Miesbach W, Meijer K, Coppens M, Kampmann
P, Klamroth R, Schutgens R, Tangelder M, Castaman G, Schwäble J,
Bonig H, et al: Gene therapy with adeno-associated virus vector
5-human factor IX in adults with hemophilia B. Blood.
131:1022–1031. 2018.PubMed/NCBI View Article : Google Scholar
|
|
92
|
George LA, Ragni MV, Rasko JEJ, Raffini
LJ, Samelson-Jones BJ, Ozelo M, Hazbon M, Runowski AR, Wellman JA,
Wachtel K, et al: Long-Term follow-up of the first in human
intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for
severe hemophilia B. Mol Ther. 28:2073–2082. 2020.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Konkle BA, Walsh CE, Escobar MA, Josephson
NC, Young G, Von Drygalski A, McPhee SWJ, Samulski RJ, Bilic I, de
la Rosa M, et al: BAX 335 hemophilia B gene therapy clinical trial
results: Potential impact of CpG sequences on gene expression.
Blood. 137:763–774. 2021.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Chowdary P, Shapiro S, Makris M, Evans G,
Boyce S, Talks K, Dolan G, Reiss U, Phillips M, Riddell A, et al:
Phase 1-2 trial of AAVS3 gene therapy in patients with hemophilia
B. N Engl J Med. 387:237–247. 2022.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Xue F, Li H, Wu X, Liu W, Zhang F, Tang D,
Chen Y, Wang W, Chi Y, Zheng J, et al: Safety and activity of an
engineered, liver-tropic adeno-associated virus vector expressing a
hyperactive Padua factor IX administered with prophylactic
glucocorticoids in patients with haemophilia B: A single-centre,
single-arm, phase 1, pilot trial. Lancet Haematol. 9:e504–e513.
2022.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Powell JS, Ragni MV, White GC II, Lusher
JM, Hillman-Wiseman C, Moon TE, Cole V, Ramanathan-Girish S, Roehl
H, Sajjadi N, et al: Phase 1 trial of FVIII gene transfer for
severe hemophilia A using a retroviral construct administered by
peripheral intravenous infusion. Blood. 102:2038–2045.
2003.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Mahlangu J, Kaczmarek R, Von Drygalski A,
Shapiro S, Chou SC, Ozelo MC, Kenet G, Peyvandi F, Wang M, Madan B,
et al: Two-Year outcomes of valoctocogene roxaparvovec therapy for
hemophilia A. N Engl J Med. 388:694–705. 2023.PubMed/NCBI View Article : Google Scholar
|