1. Introduction
Lesch Nyhan syndrome (LNS) is a rare X-linked
recessive disorder that affects between 1 case per 235,000 births
and 1 case per 380,000 live births (1). It is an inborn error of metabolism
and is associated with a deficiency of the hypoxanthine guanine
phosphoribosyl transferase (HGPRT) enzyme. HGPRT is a purine
salvage enzyme that converts the free purine bases guanine and
hypoxanthine into their utilizable forms, guanosine monophosphate
(GMP) and inosine monophosphate (IMP), respectively. Due to the
X-linked genetic etiology, mostly males develop LNS, with females
exhibiting some mild symptoms (2).
However, females can also develop the disease if the chromosome
containing the wild-type allele for HGPRT undergoes random X
inactivation or lyonization, and the chromosome with the
mutant/defective HGPRT allele is expressed (3). A deficiency in HGPRT is linked to the
following: i) The overproduction of uric acid, resulting in gouty
arthritis and kidney and bladder stones. ii) Hematological
symptoms, such as megaloblastic anemia (4). iii) Neuropathology, causing mental
retardation, spastic cerebral palsy, choreoathetosis and
self-injurious behavior (SIB). SIB includes lip, finger and tongue
biting, and banging head/limbs (5). Patients have been reported to lose
parts or all of their tongue, fingers and toes (6). iv) Abnormal involuntary muscle
movements, such as dystonia, choreoathetosis, opisthotonos, and
ballismus (7,8). Treatment is symptomatic and
supportive, and affected individuals do not survive the first or
second decade of life due to renal failure.
LNS manifests itself severely and has a varying
level of effects on individuals suffering from it due to either the
complete [Lesch-Nyhan disease (LND)] or partial loss of HGPRT
activity [Lesch-Nyhan variants (LNVs)]. These two terms distinguish
spectrum of severity associated with HGPRT deficiency. However, the
severity of neurological and behavioral symptoms may vary depending
on the amount of residual HGPRT activity. One of the most severe
neurological impairments is not being able to walk (9,10).
LNV is divided into two main types of clinical phenotypes: i)
HGPRT-related neurologic dysfunction (HND); and ii) HGPRT-related
hyperuricemia (HRH), also known as Kelley-Seegmiller syndrome,
associated with the marked overproduction of uric acid, resulting
in hyperuricemia, nephrolithiasis and gout (11). LNV is a milder form of the disease
characterized by less severe neurological and motor impairments and
does not include self-injurious behavior.
LNS presents various types of clinical signs, such
as decreased gray and white matter in the brain, hypercoagulability
and intellectual deficit (12,13).
Imaging techniques, such as diffusion tensor imaging reveal the
subtle loss of white matter integrity, particularly in the corpus
callosum, corona radiata, cingulum, internal capsule and superior
longitudinal fasciculus in the brains of patients with LND compared
to controls (14). By contrast,
voxel-based morphometric analyses determine the gray or white
matter volume of the LND. The gray matter volume (GMV) and white
matter volume (WMV) are significantly reduced in the brains of
patients with LND compared to healthy controls and those with LNVs.
For instance, a 20% reduction in the total intracranial volume has
been observed in the brains of patients with LND compared to a 14%
reduction in those with LNVs. Furthermore, reductions in WMV
(26.2%) are more prominent than those in GMV (17%) in the brains of
patients with LND (15). The loss
of gray matter has been shown to be associated with specific sites,
such as the caudate, thalamus and anterior putamen. The putamen and
caudate nucleus together form the corpus striatum, which is part of
the forebrain. However, neurodegeneration was not detected in these
studies, despite the loss of WMV and GMV (14,15).
During the first few months of life, affected
children appear normal. The majority of individuals seek medical
care at a young age, typically before 4 years of age. The main
causes of mortality in these patients are aspiration pneumonitis
and kidney failure (16). Given
that LNS is a genetic condition, there is no known treatment for
it. Treatment with xanthine oxidase inhibitors is generally
effective at reducing the elevated levels of uric acid; however,
there is no specific treatment available for other symptoms. Due to
its low frequency and the incomplete understanding of the
pathophysiological mechanisms, treatments are usually administered
to reduce symptoms. The present review discusses the genomics and
advancements in the pathophysiological mechanisms of the disease,
including promising therapeutics and associated challenges in the
treatment of patients with LNS. While several review articles on
LNS have been published to date (1,17-19),
the present review distinguishes itself by integrating the latest
research up to November 30, 2024, with a focus on genomic
advancements, detailed neuroimaging analysis, pathophysiology and
promising therapeutic interventions. Additionally, the present
review critically evaluates the challenges encountered in the
management of LNS, providing a comprehensive synthesis of recent
findings and their clinical relevance. An extensive search of the
literature was performed using three online databases: PubMed,
Embase and Web of Science. The search was carried out from the
beginning of the databases up until November 30, 2024. A
combination of the following key words was used: LNS, LND, SIB,
mutations in LNS, therapeutic approaches in LNS, therapeutic drugs,
inhibitors, drugs, neurological disorders and clinical trials, case
studies of LNS, case reports of LNS. The inclusion criteria
required studies on participants with confirmed LNS. Studies not
containing sufficient data for analysis, commentaries, or
editorials without primary data, animal or in vitro studies
that lacked direct clinical relevance, studies published in
languages other than English, and duplicate publications or reports
with overlapping data were excluded.
2. Genomics of LNS
Human hgprt1 is a housekeeping gene. It is
present on chromosome Xq26.2-q26.3, and codes for HGPRT. HGPRT,
through its transferase activity, carries out the conversion of
hypoxanthine and guanine to IMP and GMP, respectively by
transferring 5-phosphoribosyl group from 5-phosphoribosyl
1-pyrophosphate (20). The highest
expression of gene has been observed in the testes followed by the
brain tissue. However, the lowest expression has been observed in
the salivary glands, followed by the pancreas. The salivary gland
has only one functional mRNA transcript (21). This gene consists of eight introns
and nine exons. These exons encode 218 amino acids with a protein
size of 24.5 kDa. The majority of mutations are observed within the
intronic and exonic regions of Xq26-q27, which manifests itself as
reduced activity of hypoxanthine guanine phosphoribosyl
transferase, leading to gout and other characteristic abnormalities
(2). Depending on the mutation,
the enzyme exhibits no or residual enzymatic activity (11,22).
Different mutations in hgprt1 lead to a differential
expression and produce different variants designated as LNVs.
Notably, ~68% of the mutations in the LND group are deletion,
insertion, nonsense and splicing mutations, leading to undetectable
enzyme function. In the HND and HRH types of LNVs, the majority of
mutations are missense mutations (88%). Therefore, LNVs
demonstrated residual HGPRT activity (22). Residual activity is associated with
the severity of symptoms, particularly the extent of neurological
disturbances (22). Disease
severity is associated with changes in the purine metabolism rate.
Patients with <2% HGPRT activity demonstrate self-mutilative
behaviors, involuntary movements, intellectual deficits, and
hyperuricemia (23). On the other
hand, the partial deficiency of HGPRT activity (>2%) causes
hyperuricemia with only mild neuropsychiatric symptoms (24). Of note, a previous study
demonstrated there was no significant difference in the levels of
uric acid in the serum of patients with HRD, HRH and LND, ruling
out the possibility of uric acid involvement in developing a
neurobehavioral phenotype in LNS (25). By contrast, mutations are not
always deleterious and may occasionally improve enzyme activity.
However, activity never returns to normal levels (26).
Currently, the etiology of LNS, is caused by
>2,000 identified genetic alterations in the hgprt gene
(http://www.lesch-nyhan.org/en/research/mutations-database/)
(21,27). Mutations are distributed throughout
the gene, and no hotspots or clusters have been identified
(22). This indicates that
multiple mutations in hgprt can cause unique LND. Of note,
>600 pathogenic variations linked to LND have been identified to
date (28). Several novel
mutations have also been identified over the past 5 years (4,29,30).
As per the literature, only 15 cases in females have been shown to
be affected by LNS (23,31-39).
These mutations include: i) Nonsense mutations (p.Arg170*, C151T,
and p.Tyr153*); ii) missense mutations (p.Glu14Lys and p.Tyr72Cys);
iii) splice site mutations (c.609+4A>G and IVS8+4A>G; iv)
translocation severe [46,XX,t(X:2)(q26:p25)]; v) one microdeletion
of HGPRT; and vi) a frameshift mutation (c.539delG). The possible
reason for LNS in females may be the non-random inactivation of the
normal allele; therefore, females have only a mutated copy of the
HGPRT allele. For example, in a previous study, when genomic DNA
from whole blood samples was amplified without HhaI
digestion, two polymorphic alleles at the AR locus were detected.
However, following the HhaI digestion of blood samples from
the affected girl and her mother, only the AR1 allele was
amplified, suggesting the presence of non-random X-inactivation in
the patient (38).
3. Pathophysiology of LNS
Mutations in the HGPRT gene cause enzyme
dysfunction, leading to LND, characterized by gout, self-injurious
behavior and neurological symptoms. HGPRT deficiency disrupts the
purine salvage pathway, causing hypoxanthine accumulation, which is
oxidized to urate, leading to gout, renal dysfunction, and
oxidative stress (40). The
molecular mechanisms behind the neurological and neuropsychiatric
symptoms remain unclear, impeding treatment. The multi-faceted
effects of HGPRT mutations on mitochondrial function, purine
nucleotide metabolism and signaling pathways remain underexplored.
Emerging studies have shown that disruptions in the function of
HGPRT alter exchange protein activated by cAMP (EPAC)/RAP1
signaling, AMP-activated protein kinase activation due to PARP
accumulation, folic acid levels, dopaminergic function, cell
motility and cytoskeletal dynamics, all of which contribute to the
neurodevelopmental and motor deficits observed in LNS (41-43).
Experimental models confirm neurological, renal and metabolic
defects, highlighting potential therapeutics, such as allopurinol,
dopamine receptor agonists and gene editing to manage or treat
HGPRT-related conditions (44).
While attempts have been made to link HGPRT deficiency to these
symptoms, the exact association of HGPRT with LND is not yet fully
understood. It is also not clear how neurons respond to the levels
of HGPRT; hence, treatment is greatly impeded due to the lack of
knowledge about the mechanisms of HGPRT.
Mechanisms of HGPRT-associated
anemia
Macrocytic anemia is another typical feature of LND
and LNV, which is not widely recognized due to insufficient
documentation of its occurrence, intensity, or medical importance
in the literature (45,46). The prevalence of macrocytic
erythrocytes in subjects with LND and LNV with or without anemia is
relatively high. Macrocytic erythrocytes have been shown to occur
in 81-92% of subjects with LND and LNV. This high prevalence
underscores the significance of macrocytic erythrocytes as a common
aspect of the clinical phenotype in individuals with LND and its
variants (12).
Plasma hypoxanthine levels are a sensitive parameter
for hypoxia in fetuses and newborns. Thus, the hypoxanthine level
can be used as a potent indicator of hypoxia. Hypoxia increases
purine nucleotide breakdown, generating high levels of hypoxanthine
as a by-product. In patients with LND, the combination of
hypoxia-induced hypoxanthine generation and HGPRT deficiency
exacerbates the accumulation of hypoxanthine, contributing
biochemical, renal and neurological manifestations (2,47,48).
This highlights the vulnerability of patients with LND to any
additional stressors, such as hypoxia, that may further overload
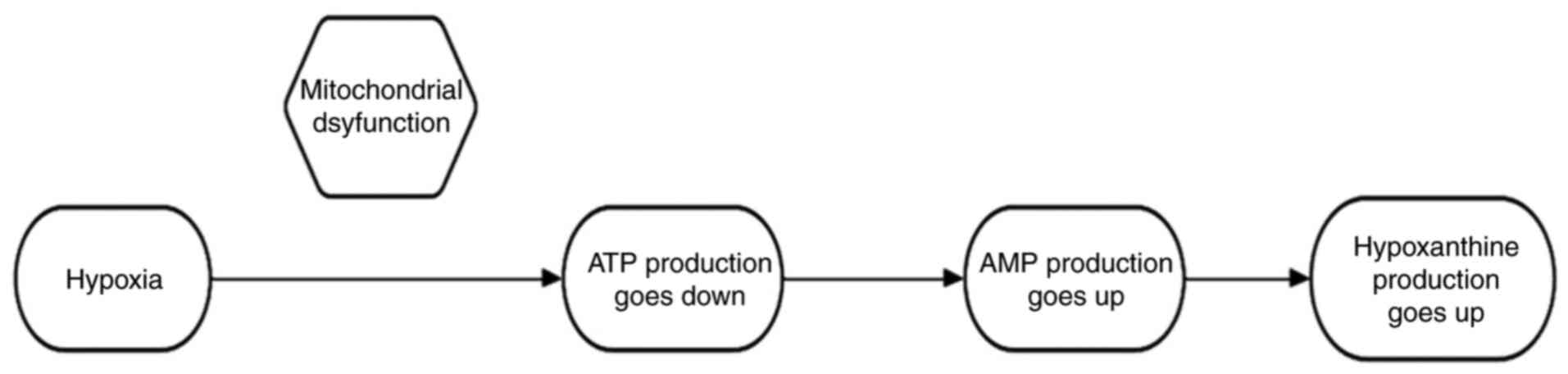
the purine metabolism pathway. In vivo, hypoxanthine is
formed by the dephosphorylation and deamination of ATP and is a
hallmark of hypoxia-induced mitochondrial dysfunction. First,
oxygen is required for the proper functioning of the electron
transport chain and oxidative phosphorylation in the mitochondria,
which ultimately produces ATP. Under low oxygen conditions, these
processes are disrupted, resulting in decreased ATP production and
adenosine monophosphate (AMP) accumulation. To meet these energy
requirements, AMP is degraded to compensate for the loss of ATP.
The increased production and degradation of AMP leads to
hypoxanthine production (Fig. 1).
This is the mechanism through which cells adapt to low oxygen
concentrations by altering their metabolic pathways. In addition,
during hypoxia, xanthine oxidase is inhibited, leading to the
accumulation of hypoxanthine (49,50).
The exposure of red blood cells (RBCs) to oxidative stress, whether
in vivo or ex vivo, enhances purine deamination
through AMP deaminase 3. This process leads to the increased
accumulation of hypoxanthine, a deaminated purine. This increase in
hypoxanthine levels is accompanied by changes in the morphology of
RBCs, followed by increased destruction outside the blood vessels
via splenic sequestration and erythrophagocytosis (51). Additionally, low oxygen levels
trigger erythropoietin (EPO) gene expression, which codes for the
glycoprotein hormone erythropoietin. The kidneys are the main organ
in an adult organism that produces EPO (46). The glycoprotein hormone EPO
increases the ability of the body to carry oxygen by encouraging
the development and differentiation of erythroid precursor cells in
the bone marrow, leading to an increase in red blood cell mass and
macrocytic anemia.
A previous study demonstrated that RBCs from
patients with LNS exhibit an accumulation of glycolytic
intermediates upstream of pyruvate kinase along with elevated
levels of unsaturated fatty acids and long-chain acylcarnitines.
Additionally, there is an increase in highly unsaturated
phosphatidylcholines in the RBCs of these patients, while free
choline levels are decreased. Furthermore, intracellular
concentrations of iron, zinc, selenium and potassium are also
reduced in the RBCs of patients with LNS (2). Global proteomic analyses have
documented alterations in RBC membrane proteins, hemoglobin, redox
homeostasis proteins and the enrichment of coagulation proteins.
These changes are accompanied by increased protein glutamine
deamidation and methylation in both children with LNS and their
carrier mothers. Allopurinol treatment partially reverses these
phenotypes. However, these changes have been specifically noted in
the context of the HGPRT gene mutation c.485 G>A. Ser162Asn
(2). These findings suggest that
complementary treatments, in addition to current regimens, such as
allopurinol, could involve the supplementation of substrates to
activate compensatory regulatory pathways (2). Additionally, the recycling of
hypoxanthine by the X-linked HGPRT plays a crucial role in
maintaining IMP/GMP homeostasis in RBCs. In patients with LNS,
genetic mutations in this enzyme result in various clinical
symptoms, including macrocytic anemia (12).
Mechanisms of HGPRT-associated
neurobehavioral problems
HGPRT deficiency also affects behavioral symptoms,
which are a consequence of disrupted dopamine pathways in the basal
ganglia (52). Previous research
was conducted to elucidate the dysregulation in the development of
dopamine neurons in MN9D derived HGPRT-ve (HGPRT-deficient) cell
lines (52,53). Two mechanisms were proposed based
on the results of that study: i) Microarray analysis of these cell
lines revealed the diminished expression of tyrosine hydroxylase.
This result is in line with previous experiments conducted to
demonstrate the dysregulation of biochemical markers related to the
dopamine phenotype (52). ii)
Another finding was the overexpression of engrailed genes En
1 and En2, which are transcription factors that play
vital roles in neural development, that is, the development and
survival of dopamine neurons. The results revealed that En2
was expressed in all fibroblasts and in higher amounts in patients
with neurobehavioral problems, suggesting an inverse relationship
between HGPRT and En. However, the mechanism by which HGPRT
levels regulate the expression of En genes is yet to be
determined (53).
Similarly, HGPRT deficiency has also been shown to
exhibit a deregulatory effect on guanine metabolism, and hence, it
affects G protein-coupled receptor (GPCR) expression. For instance,
altered and structurally defective expression of P2Y1 (GPCR) is
associated with aberrantly phosphorylated ERK (p-ERK) and cAMP
response element-binding protein (CREB) signaling (54). Notably, p-ERK can be transported
into the nucleus, where it activates various transcription factors,
such as CREB. CREB is crucial for the transcription of numerous
neuronal genes, and is essential for long-term synaptic plasticity.
Therefore, defects in these genes may affect neuronal development.
Compared to LNV, patients with LND exhibit greater reductions in
fractional anisotropy across the brain and specific disruptions in
the corpus callosum, corona radiata, cingulum, internal capsule and
superior longitudinal fasciculus. These deficits in white matter
organization are associated with more severe dystonia and cognitive
impairments, highlighting the need for the further exploration of
the role of white matter in the pathogenesis of LND (14). Emerging studies emphasize that
HGPRT deficiency affects neurodevelopment and neurocognitive
function through metabolic and cellular disruptions. For example,
alterations in white matter integrity, reduced neuronal
connectivity and neurobehavioral symptoms have been linked to
changes in neurexin expression, genes critical for synaptic
function that are associated with autism, schizophrenia, and
Alzheimer's and Parkinson's disease (55). NAV3, another identified gene, has
been implicated in neurodevelopmental disorders and neuromuscular
responses, further suggesting a role in LND-associated neuronal
morphogenesis (56). Additionally,
hypoxia-induced increases in hypoxanthine levels, a characteristic
metabolic disruption in LND, underline the pathophysiological
burden on both the central nervous system and peripheral processes.
This highlights the interplay between metabolic imbalances and
neurobehavioral outcomes in HGPRT deficiency. Stratification
studies from analogous conditions, such as traumatic brain injury,
suggest that white matter disorganization may serve as a prognostic
biomarker for neurocognitive trajectories and may provide a
framework for understanding HGPRT-associated symptoms (57). These findings collectively
underscore the critical need to elucidate the molecular and
structural underpinnings of HGPRT-associated neurobehavioral
deficits, aiming to identify targeted therapeutic interventions and
improve the quality of life of affected individuals. A previous
study revealed that several microRNAs (miRNAs/miRs) from the miR-17
family cluster, along with genes encoding guanine nucleotide
exchange factors, are dysregulated in HGPRT deficiency (41). Notably, the EPAC displays a reduced
expression in HGPRT-deficient human neuron-like cell lines and
fibroblast cells from patients with LNS. Similar alterations have
been observed in the cortex, striatum and midbrain of HGPRT
knockout mice (41). These
dysregulations lead to the impaired activation of the small GTPase
RAP1, which is critical for cytoskeletal dynamics. Consequently,
HGPRT-deficient cells exhibit an altered motility compared to
controls. HGPRT deficiency also results in the dysregulation of
miR-181a (58). The expression of
miR-181a is elevated in HGPRT-deficient human dopaminergic SH-SY5Y
neuroblastoma cells, which in turn leads to the aberrant expression
of target genes involved in mammalian central nervous system (CNS)
development (59). Utilizing
miRNA-based target predictions, researchers have identified
critical signaling pathways for potential therapeutic targeting in
LNS. A hypothetical model further proposes that HGPRT mRNA
transcripts function as competitive endogenous RNAs, engaging in
complex regulatory crosstalk with key neural transcripts and
miRNAs, potentially amplifying the gene's pleiotropic effects on
diverse pathways (60). To further
investigate these mechanisms, fibroblasts derived from patients
with LNS were reprogrammed into induced pluripotent stem cells
(iPSCs) using a combined miRNA/mRNA reprogramming approach
(61). This innovative methodology
facilitated the development of LNS-specific iPSC lines for in-depth
mechanistic and therapeutic research, providing a valuable model
system for dissecting the molecular basis of this rare metabolic
disorder and guiding the identification of novel biological
targets.
Effect of HGPRT deficiency on
mitochondrial function
Another study was conducted to further elucidate the
effects of HGPRT on mitochondrial energy metabolism in the brain
(24). The researchers used a
CRISPR mouse as their model organism, which carried the same
Hgprt1^del8Val mutation as found in humans for LN. This mutation
causes the homodimerization of HGPRT, thereby reducing its
activity. These findings indicate that there is an inhibitory
effect of HGPRT deficiency on complex I, that is, NADH:ubiquinone
oxidoreductase. As a result, the rate of consumption of NADH is
suppressed, which ultimately results in decrease in mitochondrial
membrane potential and increased mitochondrial reactive oxygen
species (ROS) production. The increase in mitochondrial ROS
production may be due to increased levels of xanthine, leading to
the production of superoxide anions by oxidase (62,63).
This diminished membrane potential leads to decreased ATP
production. The low consumption of NADH by complex I results in
NADH accumulation. To compensate for lower levels of ATP, cells
acquire glycolysis (anaerobic metabolism) to fulfil their energy
requirements, particularly when mitochondrial respiration is
compromised owing to the inhibition of complex I. The HGPRT
deficiency inhibits complex I-dependent mitochondrial respiration,
leading to elevated mitochondrial NADH levels, a reduction in
mitochondrial membrane potential, and an increased production rate
of ROS in both the mitochondria and cytosol. Despite the heightened
ROS production, there is no evidence of oxidative stress, as
endogenous glutathione levels remain unaffected. This suggests that
the disruption of mitochondrial energy metabolism, rather than
oxidative stress, may act as a trigger for the brain pathology
characteristic of LNS (24).
Further research using HGPRT-deficient rat B103
neuroblastoma models has identified a significant reduction in
adenylate cyclase 2 expression, implying a potentially critical
role of adenylate cyclase 2 in LNS-associated neurobehavioral
abnormalities (64). However,
validation in more advanced models, such as in vivo systems,
is necessary to better elucidate the association between adenylate
cyclase 2 and the pathogenesis of LNS (65). These findings highlight the
critical link between HGPRT deficiency, mitochondrial dysfunction,
and the downstream neurological impacts of LNS.
4. Current drugs and promising
therapeutics
To the best of our knowledge, there is no curative
treatment for LNS due to the lack of knowledge about the mechanisms
associated with SIB, low intellect, depression and neurological
dysfunction. Treatments are available on the basis of symptoms.
However, reduction in one symptom will not always necessarily also
lead to the suppression of another symptom. For example, treatments
used to reduce uric acid will not reduce SIB as uric acid levels
are not associated with the neurological manifestations of LNS.
Oral appliances
One strategy for the prevention of SIB is through
tooth removal. However, this method is not ethical and is not
permitted for use by the majority of parents.
Personalized/customized intraoral devices and lip bumpers/lip
guards are alternative options for the removal of teeth and are
used to prevent oral and peri-oral trauma following their
application (66,67).
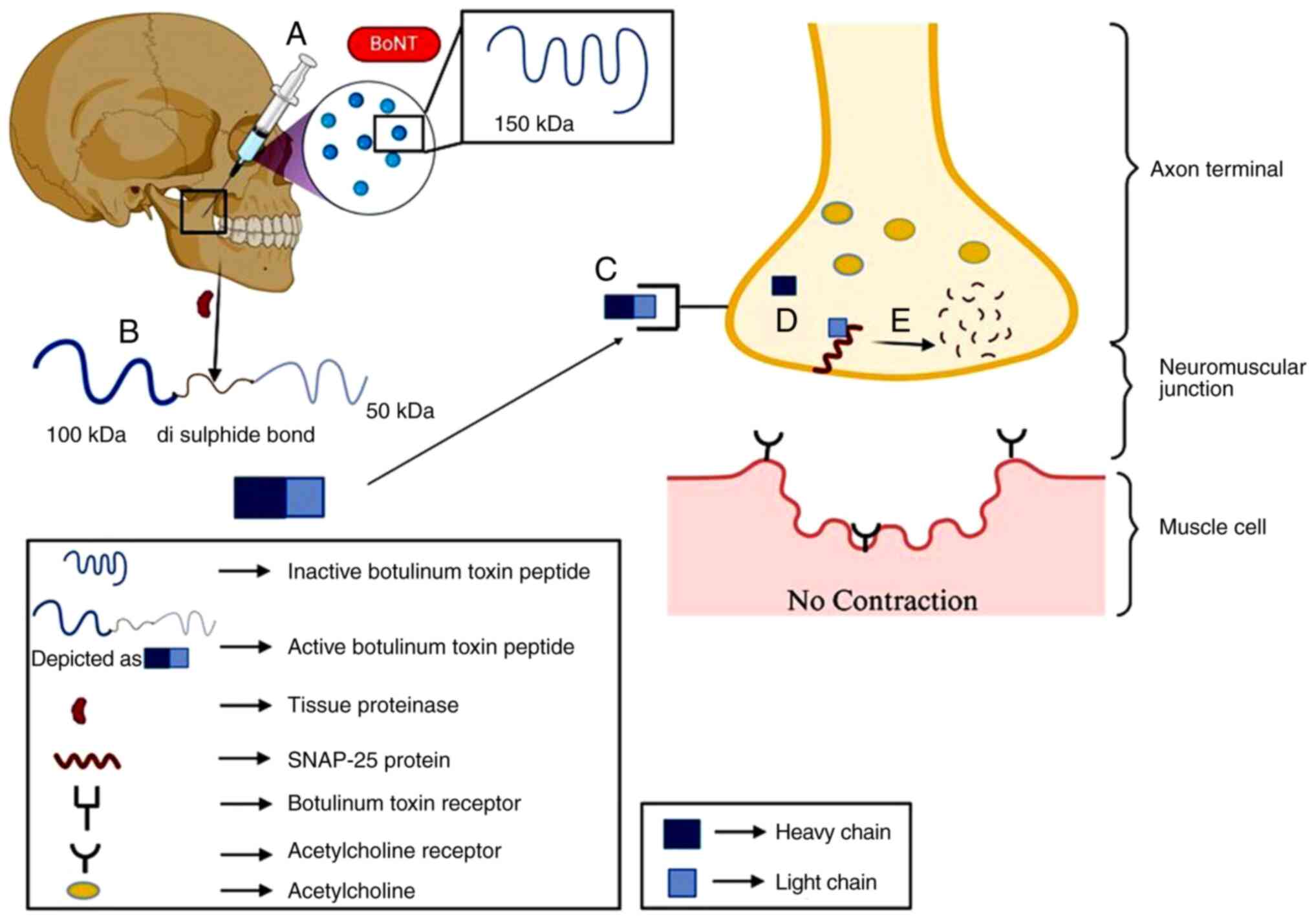
Botulinum toxin A (BTX-A)
Botulinum is a toxic compound produced by
Clostridium botulinum and other related species, such as
Clostridium butyrricum, Clostridium barati, and Clostridium
argentinensis. Botulinum toxin works by blocking the release of
the neurotransmitter acetylcholine, which activates muscle cells
and helps them to contract. Thus, when acetylcholine is not
released, muscles are in a relaxed state. This toxin is potentially
toxic to nerve cells and thus causes paralysis; however, it can be
used in optimal or controlled amounts (Fig. 2). BTX-A is also used to reduce the
need for more invasive interventions, such as tooth extraction in
patients with LNS. To date, there are only a few reported cases of
patients with LNS treated with BTX-A; however, these cases vary in
terms of dose, site and duration of the injection (Table I) (68,69).
 | Table ICase studies on the use of botulinum
toxin A in LNS in the literature. |
Table I
Case studies on the use of botulinum
toxin A in LNS in the literature.
| Age and sex of the
patient(s) | Symptom | Other treatments
given prior to BTX-A | Dose of BTX-A | Outcome | (Refs.) |
|---|
| 10-year-old male
(age at diagnosis, 8 years) | Cerebral palsy,
developmental delay and had cognitive impairments, SIB, spasticity,
dystonic movements, and hyperkinetic dyskinesia | Allopurinol | 20 units of BTX-A
every 12 weeks for three series of visits were given to masseter
muscles. | Overall positive.
i) Sores on hands, lips and tongue healed completely; ii) improved
speech articulation, weight and height; iii) reduced SIB,
aggression and medications. | (68) |
| 30-year-old male
(age at diagnosis, 12 years) | Dystonic posturing,
cognitive disturbances, SIB | Allopurinol,
baclofen, clonazepam, quetiapine, and L-carnitine. | Zygomatic muscles:
12.5 IU each; lower part of the lip orbicularis muscle in 6
injection sites: 2.5 IU each; levator labii inferioris in three
injection sites: 5 IU each. | Overall positive.
i) The muscles became hypotonic and weak; ii) and biting ceased
with progressive healing of the wounds. He is currently on the
waiting list for DBS. | (97) |
| 12-year-old
male | Generalized
dystonia, choreoathetosis, opisthotonus, and spasticity and
SIB | Four-point
restraints to prevent self-harm. | Serial botulinum
toxin type A injections (incobotulinum toxin A (Xeomin) and
onabotulinum toxin A (Botox) to triceps, biceps, gastrocnemius,
hamstring, quadriceps, and extensor hallucis longus at a dose 10
mg/kg every 8-12 weeks. | Overall positive.
i) Dystonia and opisthotonus were significantly improved; ii)
marked decrease in self-injurious behaviors. | (98) |
| 13-year-old
male | SIB, spasticity,
dystonia and anxiety | Carbidopa-levodopa,
fluoxetine, gabapentin, clonazepam, olanzapine, and oral
baclofen. | BTX-A was
administered to bilateral masseters at the dose of 50 units on each
side with the help of ultra sound and electric stimulation for
every 4-6 months. | i) Increased speech
volume and a reduction in articulation errors, ii) decreased SIB
behavior, and increased durations of effects. | (99) |
| 6 patients with LND
Age, 4, 4.5, 6.6, 7.9, 13.9, and 32.3 years | SIB | No information
available. | Botulinum was given
in combination with Dysport, with the highest total dosage of
Dysport being 37.5 units/kg mean, whereas the highest total dose of
Botox was 21.3 units/kg mean. The average number of shots given to
the patients was 20, with a range of 3 to 29. Site of injection:
masticatory muscles (masseter and temporalis), biceps brachii, and
other muscles. Duration of injection: 1.5 to 7.1 years. | 95% success rates.
A total of 119 injections were administered, out of which 113 were
partially or completely effective in ceasing the biting tendencies
of patients, and only 3 produced some adverse effects. | (69) |
Levodopa
Patients with LNS have low levels of dopamine in the
basal ganglia due to the reduced activity of tyrosine hydroxylase,
which is the rate-limiting enzyme in dopamine synthesis (70,71).
Low levels of dopamine are associated with uncontrolled body
movements. Of note, an ~50-63% reduction in the binding of the
WIN-35,428 ligand to dopamine transporter was shown in the caudate
region and a 64-75% reduction in the putamen region in patients
with LNS (72). Therefore, to
compensate for the loss of dopamine, levodopa (L-DOPA) is
administered to treat movement disorders and SIB. Levodopa is a
dopamine precursor, which is metabolized to dopamine in the
periphery and in the CNS. Aromatic-L-amino acid decarboxylase
(AADC) converts levodopa to dopamine. In contrast to dopamine,
levodopa can cross the blood-brain barrier. Therefore, levodopa is
prescribed over the direct injection of dopamine. However, the
bioavailability of levodopa is low in the CNS. To overcome this
issue, levodopa is administered in combination with carbidopa.
Carbidopa (L-alpha-methyldopa hydrazine inhibits AADC by binding
irreversibly to pyridoxal 5'phosphate, thereby blocking the
conversion of levodopa to dopamine in the periphery. However, it
does not block conversion in the CNS. Therefore, carbidopa
increases the bioavailability of dopamine in the CNS. It also
reduces the side-effects associated with the use of levodopa, such
as nausea, vomiting and diarrhea. However, the results have been
inconsistent and not encouraging (Table II) (73).
| Table IICase studies of levodopa A in LNS
identified in the literature. |
Table II
Case studies of levodopa A in LNS
identified in the literature.
| Patient | Age, years | Mutation | Symptoms | Treatment
administered | Dose of L-DOPA | Improvement in
SIB | (Refs.) |
|---|
| 1 | 27 | 428-432del TGCAG,
insAGCAAA | Dystonia, chorea,
ballism, hypotonia, opisthotonus, hyperreflexia, lip and finger
biting | Allopurinol and
chlorothiazide medications | L-DOPA administered
in combination with carbidopa at a 4:1 ratio. L-DOPA/carbidopa for
4 weeks, and the maximum concentration was 600/150 mg daily | No | (73) |
| 2 | 12 | IVS7+5G>A | Dystonia, rigidity,
spasticity, hyperlexia, head banging and biting | Allopurinol,
diazepam, omeprazole, fluticasone, mometasone, cetirizine,
levalbuterol | L-DOPA/carbidopa
for just 1 day, and the maximum concentration he was given was
50/12.5 | Discontinued the
treatment | (73) |
| 3 | 10 | IVS7+5G>A | Dystonia,
hypotonia, and biting of fingers and arms | Allopurinol and
diazepam | L-DOPA/carbidopa
50/12.5 mg | Discontinued after
first dose due to hyperactivity | (73) |
| 4 | 3 | 10delC | Hypotonia,
dystonia, thrashing and mouth biting | Allopurinol,
gabapentin and lorazepam | L-DOPA/carbidopa
for 4 weeks and had the maximum concentration of 150/37.5 mg | Agitated movements
but levodopa treatment had positive effects on his behavioral
aspects. The boy was in A good mood and did not bite his mouth very
often and did not require any restraints | (73) |
Due to these unanticipated complications, which
worsen the condition of patients, none of the patients have
completed the planned titration phase. Levodopa has no effects on
the behavioral aspects, but triggers more adverse motor movements
(73-75).
These results suggest that the use of levodopa and carbidopa is not
advantageous for reducing the symptoms of patients with LNS. It is
possible that the mechanism in LND is different than from in
DOPA-responsive dystonia, and the responses to medications may not
be similar. Therefore, it may be interesting to determine the
mechanisms underlying the lack of response to these two drugs in
patients with LNS. These types of studies require replica
HGPRT-deficient models that can help in determining the cause of
drug failure. In addition, instead of combinion therapy,
monotherapies of levodopa and carbidopa should be attempted in LNS
models. It is essential to consider the fact that carbidopa
inhibits AADC activity and, therefore, blocks the conversion of
levodopa to dopamine.
S-adenosyl methionine (SAMe)
SAMe is broken down to yield an adenosyl moiety,
which is then converted into AMP (76). This AMP can be converted into
either ATP or GTP in the brain, thereby replenishing the nucleotide
pool in patients with LND that is otherwise depleted, thereby
improving their condition. SAMe supplementation may help reduce the
extrapyramidal symptoms associated with dopamine hypersensitivity
by boosting the synthesis of GTP, which is crucial for dopamine
production, and by enhancing the function of
catechol-O-methyltransferase, an enzyme that plays a role in
the inactivation of dopamine. Moreover, SAMe is readily taken up
into the bloodstream and can cross the blood brain barrier, thereby
making purine nucleotides available in the brain. Thus, it can also
be used to treat other mental health disorders related to
nucleotide depletion. The results of the use if SAMe have been
encouraging in patients with LNS (Table III) (7,77,78).
From these cases, it is evident that SAMe tends to be more
effective in younger patients with LND. However, further research
in the context of age-related responses to SAMe is required.
| Table IIICase studies of SAMe in LNS
identified in the literature. |
Table III
Case studies of SAMe in LNS
identified in the literature.
| Age and sex of the
patient(s) | Symptoms | Mutation | Other
treatments | Dose | Outcome | (Refs.) |
|---|
| 43 years, male | SIB, dystonic
paralysis, seizures, suspected but ill-defined pain, and renal
stones. | Not available | Fentanyl 25 µg (by
skin patch, every three days) | 800 mg twice daily
(on an empty stomach) | Positive. i)
Delf-abusive behaviors disappearrf in 1.5 years; ii) improved liver
function; iii) mood rating improved from 36 to 96% in 3 years. | (77) |
| 15 years, male (age
at diagnosis, 3.6 years) | SIB, spasticity and
dystonia, severe hyperuricemia and hyperuricosuria | Deletion of exons
1,2 and 3 | Allopurinol,
diazepam and carbamazepine | 22 mg/kg/day | Positive. | (100) |
| 7 years, girl (age
at diagnosis, 6 months) and 3 male cousins (ages: 13, 3.6 years,
and 4 days) | Girl: SIB,
dystonia, medullary nephrocalcinosis Males: Dystonia, limb
hypertonicity, severe hyperuricemia and hyperuricosuria | Girl: Skewed
x-inactivation of >95% of wild-type HPRT1 alleles in peripheral
blood cells Males: HPRT1 intronic splice site mutation
c.610-1G>A | Girl: Allopurinol
(18 mg/kg/day) and oral baclofen (2.5 mg bd) commenced at age 10
months and 15 months, respectively Males: Oral baclofen (1 mg tds),
allopurinol | Girl: 33 mg/kg/day
Males: ~21 mg/kg/day or 26 mg/kg/day | Self-abusive
behaviors and aggression were resolved and it also helped with
dystonia. | (100) |
| 14 patients (age:
11-49 years) | Hyperuricemia, SIB,
anxiety, disturbed sleep pattern. recurrent uncontrolled bodily
movements | Not reported | Allopurinol | Not available | SAMe treatment
worked only for 4 out of 14 patients. P#1 speech and anxiety
resolved, in P#12 and P#14 SIB resolved and P#13 sleep pattern
improved. All others had to drop out of the trial due to SIB,
anxiety, sleep pattern, recurrent uncontrolled bodily movements.
Possible reasons for failure of this conduct could be difference in
individual's age, ethnicity, drug metabolism and tolerance,
severity of genetic mutation, environmental factors. Also, these
patients were previously on other drugs too which weren't stopped
when they received SAMe. This could have interfered with SAMe
action. | (78) |
| 6 months, male | Motor development
delay, poor head control, generalized muscle contractions, SIB,
marked loss of muscle tone hyperuricemia (10.3 mg/dl), and an
increased urinary uric acid/creatinine ratio (5:3), inspiratory
stridor due to laryngeal dystonia and frequently developed
aspiration pneumonitis and bronchitis | g.151C>T (p.
R51X) | Allopurinol
(dose-20 mg/kg/day), potassium citrate, sodium citrate, gabapentin
(40 mg/kg/day) and risperidone (up to 0.05 mg/kg/day at 34 months).
The exact mechanism by risperidone helps/lowers SIB tendencies is
debatable, since it has many targets in the central nervous system.
While SAMe refills the purine pool in brain and hence enhances
brain function. | 20 mg/kg/day | i) At 26 months of
age, the boy's SIB gradually decreased, his head movements became
more stable and he began picking things from both his hands. ii)
Facial, oral and trunk movements improved. iii) The patient's
laryngeal stridor resolved, and he did not experience many episodes
of pneumonia or bronchitis. iv) When he was 26 months old, an MRI
revealed no signs of brain atrophy or aberrant intensity. v)
Uncontrolled bodily movements (dyskinesias) when agitated or
annoyed. | (7) |
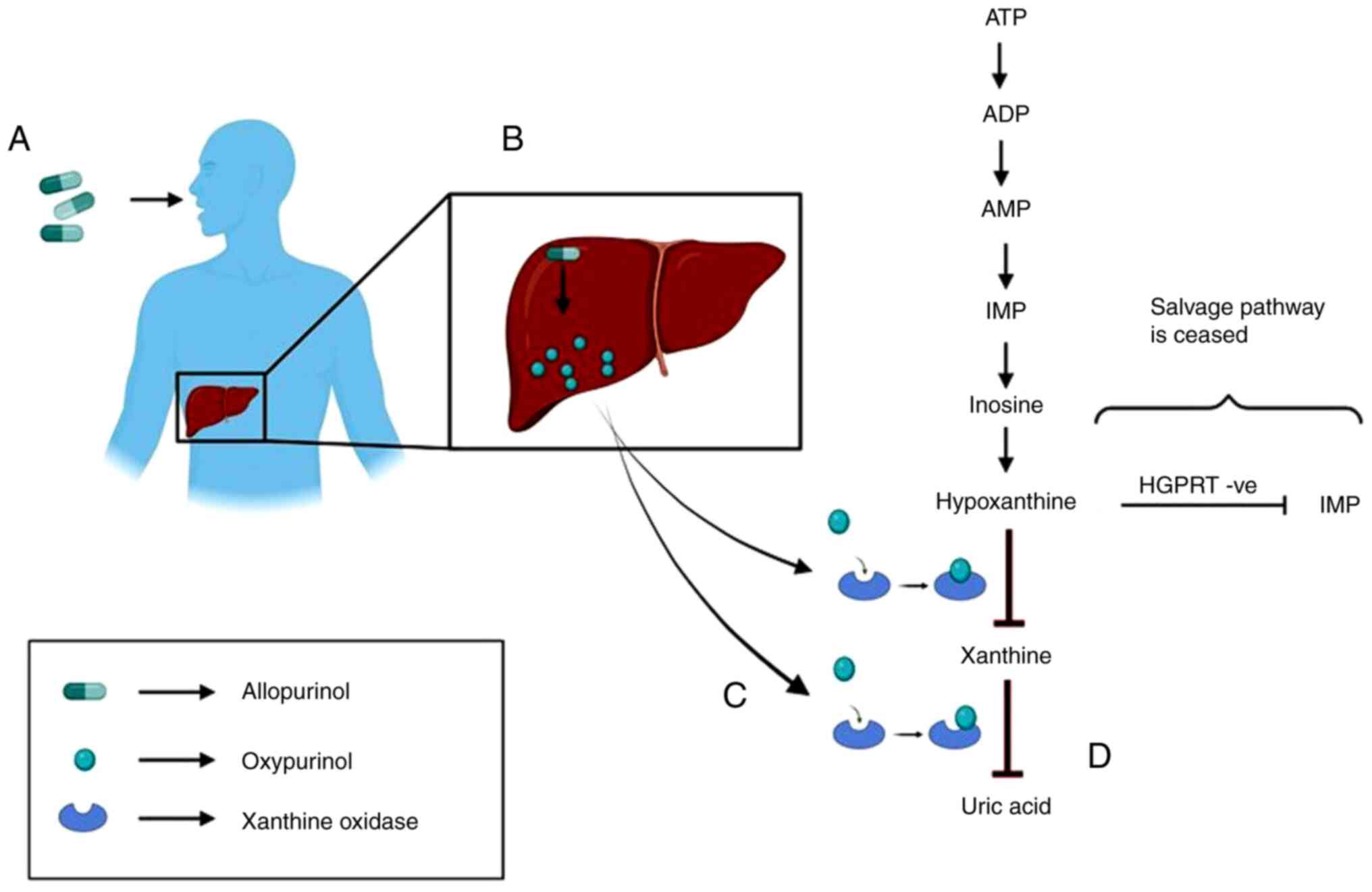
Allopurinol
Allopurinol is converted to its active metabolite,
oxypurinol, which inhibits xanthine oxidase. Xanthine oxidase is
the enzyme that converts hypoxanthine to xanthine and further to
uric acid. Therefore, it first stops the production of uric acid
and then increases the concentration of both hypoxanthine and
xanthine in serum and urine (Fig.
3). The renal clearance of hypoxanthine and xanthine is very
rapid compared to that of uric acid, and its plasma concentration
is only partly elevated (79). The
half-life of allopurinol is ~1-3 h and that of oxypurinol is 12-30
h. When Allopurinol is not used, these oxypurines are secreted
through the urine in the form of uric acid. However, when
allopurinol is administered, the urinary output is composed of
hypoxanthine, xanthine and some amounts of uric acid, thereby
reducing the risk of crystalluria, a condition characterized by the
presence of uric acid crystals in urine, nephrolithiasis,
nephrocalcinosis, and sometimes, acute or chronic kidney impairment
(80). These oxypurines are easily
cleared by kidneys. Advanced stages of gout are characterized by
the formation of tophi, which are yellow-colored lesions that form
around joints. The tophi are composed of a uric acid (monosodium
urate) core and skin that can become stretched and taut, sometimes
to the point of ulceration. Allopurinol aids in the dissolution of
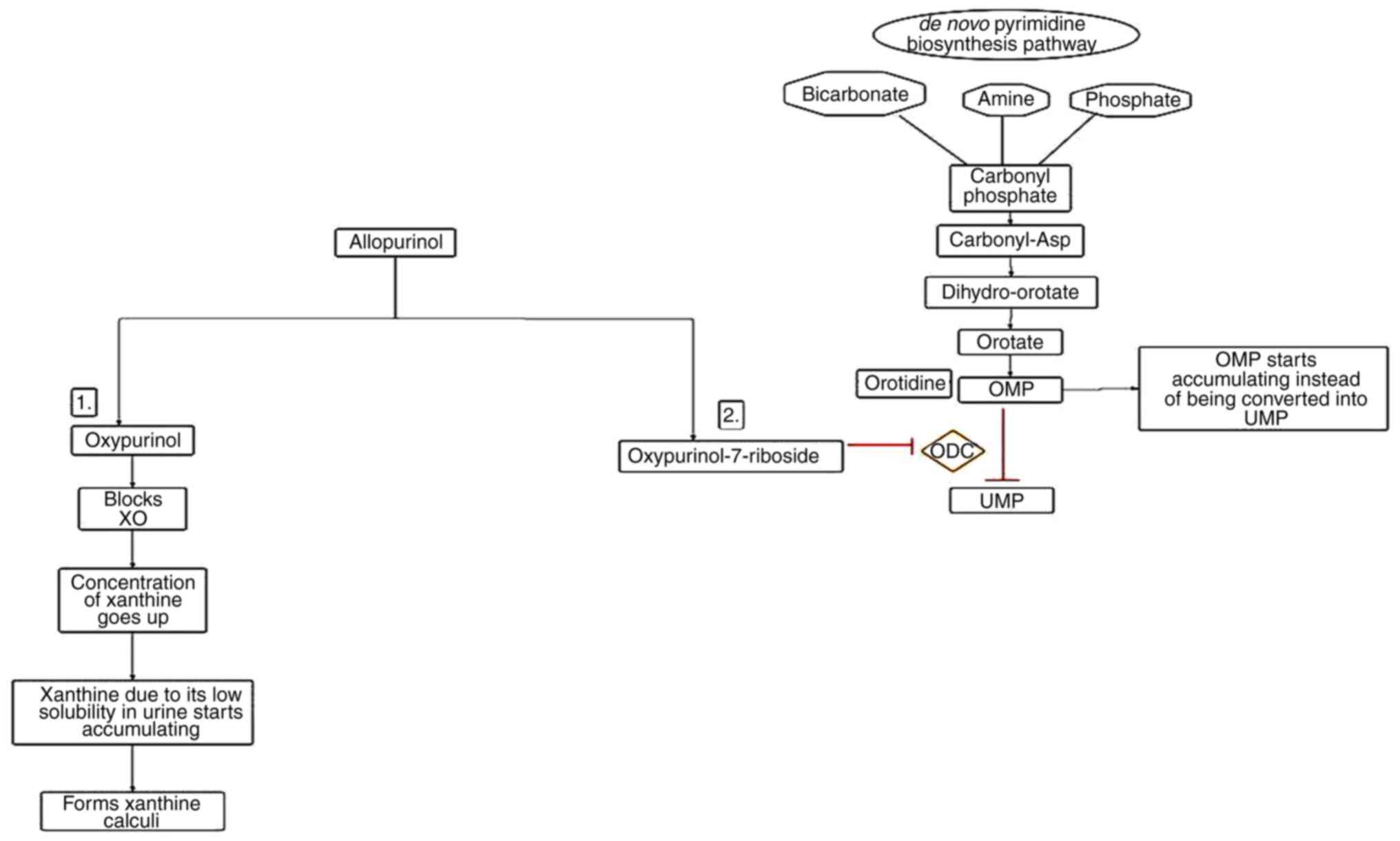
tophi (81). However, higher
concentrations of allopurinol are responsible for the formation of
xanthine stones and oxypurinol-7-riboside, which causes derangement
in the de novo synthetic pathway of pyrimidine by blocking the
enzyme ornithine decarboxylase (82). As a result, orotidine 5'
monophosphate begins accumulating and is rapidly degraded to
orotidine, which cannot be degraded further and thus starts
accumulating in RBCs and urine (Fig.
4) (83). To avoid xanthine
stone deposition, it is advisable to have a high fluid intake to
maintain a neutral or alkaline pH. Treatment with allopurinol did
not correct the movement disorder or SIB. For example, in a study
conducted on two male pediatric patients with a novel LND mutation,
allopurinol normalized urate levels in RBCs and lowered serum uric
acid levels, thereby lowering the risk of kidney stones, but did
not help with movement disorder (2). Although allopurinol is very effective
in maintaining urate levels, its downstream products, namely
5-OHisourate and allantoate, do not respond well to
allopurinol.
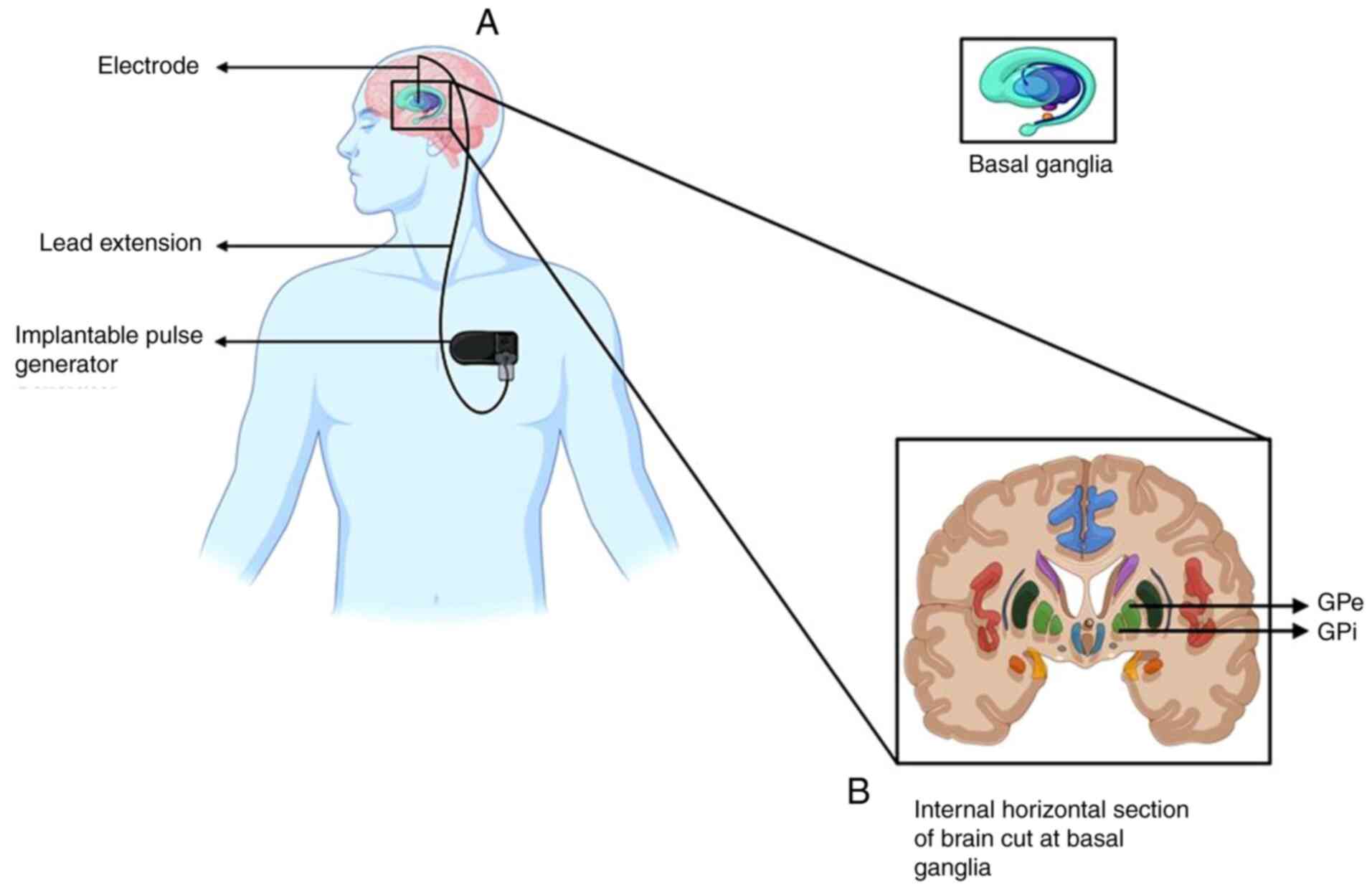
Deep brain stimulation (DBS)
DBS is a neurosurgical procedure that involves
implanting electrodes in specific brain regions to control
activity. The electrical impulses generated by these electrodes are
controlled by an implantable pulse generator, which is fitted to
the chest below the clavicle (beneath the skin) (Fig. 5) (84). DBS is used to treat
neuropsychotic/neurological disorders, where medication has failed
to provide any relief. Depending on the case profile of each
individual, a physician/clinician determines the strength of the
pulse to be delivered, the duration of the pulse, how long it
should last, and how many times it has to be repeated for the
patient to obtain optimal results (85). Achieving optimal effectiveness
requires adjusting stimulation parameters, including voltage, pulse
width and frequency (86). The
exact mechanisms through which DBS functions are not yet fully
understood. It is generally accepted that it functions by
stimulating and/or inhibiting neurons that are in close proximity
to electrodes. Low-frequency stimulation can excite the nearby
cells. However, high-frequency stimulation tends to have an
irreversible effect by lowering local activity (87). Four primary hypotheses have been
proposed to explain the beneficial effects of DBS in movement
disorders: Depolarization blockade, synaptic inhibition, synaptic
depression, and modulation of abnormal oscillations in pathological
networks (88). The globus
pallidus (GP) is a subcortical triangular structure, present below
the cerebral cortex, medial to the putamen. The main function of GP
is to control voluntary movements and consciousness (89). Therefore, DBS of the GP is used to
treat movement disorders (as in Parkinson's disease) and
medication-resistant mental health conditions. Examples of such
case studies are presented in Table
IV (90,91).
| Table IVCase studies of DBS in LNS identified
in the literature. |
Table IV
Case studies of DBS in LNS identified
in the literature.
| Age and sex of the
patient(s) | DBS Site | Symptoms | Other
treatments | Outcome | (Refs.) |
|---|
| 15 years/male (age
at diagnosis, 3 years) | Bilateral chronic
stimulation of the middle area of Global Pallidus Internus
(GPI) | Dystonic postures
and sporadic involuntary movements and had trunk opisthotonos | Levodopa with no
benefits | i) Dystonic
involuntary movements vanished and opisthotonus improved. ii) After
3 months follow up, the boy's parents reported that self-mutilative
behaviors have completely disappeared and he no longer needed
physical restraints. iii) Follow up was done again at 24 years,
which revealed that the boy had still not developed self-mutilative
behaviors. These results suggest that the SIB is either due to
dysregulation in basal ganglia pathway or is secondary to the
dystonia. | (90) |
| Two patients; ages,
12 years (age at diagnosis, <1 year | Anterior and
posterior parts of the globus pallidum | Dystonia and
self-mutilative behaviors | Patient 1 was on
risperidone, gabapentin, carbamazepine, amantadine and clonazepam
together with allopurinol, and patient 2 on a combination of
diazepam, clobazam and sodium bicarbonate |
Electrophysiologically, a silent zone was
observed between -4.3 and -3.2 mm from the target, illustrating the
transition between the GPE and the GPI. 56 GPE neurons and 106 GPI
neurons underwent micro-recordings. Within 3 months post-operation,
a number of their self-injurious behaviors vanished and their
limbic dystonic movements significantly diminished, as a result of
which they no longer needed high doses of medications. | (101) |
| 16-year-old, male
(age at diagnosis, 6 months) | DBS of motor and
limbic regions of the GP was suggested. To treat SIB limbic circuit
in the antero-ventral part of the GPI was targeted, and to help
with dystonia and dyskinesia, the sensorimotor part of the GPI was
selected. The stimulation was given at a frequency of 130 Hz and
pulse width of 450 msec. For both limbic and motor leads, voltage
was increased up to 1.6 V. | Slurred speech,
SIB, cerebral palsy, dystonia of the limbs, trunk and head and
hypotonia | Baclofen, diazepam,
clomipramine, and cyamemazine | The changes in
dystonic movements were assessed post and pre-operatively, using
the Burke-Fahn-Marsden Dystonia Rating Scale. Similarly, SIB was
assessed using the Behavior Problems Inventory (BPI-01). i)
Improved aggressive behaviors and dystonic disorders. ii) DBS
efficacy was maintained after 28 months. iii) The quality of life
of the patient and of his family was dramatically improved. iv) At
rest, the four limbs are not tied to the bed and to the wheelchair,
and protection of the fingers by the gloves is not necessary
anymore. Bilateral simple manipulations are possible. | (102) |
| 10-year-old
boy | Single-channel
generators connected to a single electrode on each side of his
brain. | Presented at 8
months of age with poor muscle tone, at 12 months of age cerebral
palsy was detected and at 15 months of age involuntary movements
started. At 22 months of age, he was diagnosed with LNS. His SIB
began at 4 years of cell with biting if fingers and lower lip, that
was so severe that he had to get his primary teeth removed and used
elbow restraints. At age 7, there was a fatal episode of dystonia
that lead to hyperthermia, rhabdomyolysis (the muscle fibers
breakdown, and its components such as myoglobin, creatine kinase
(CK), aldolase, and lactate dehydrogenase, as well as electrolytes,
into the extracellular space and into the blood flow) and transient
renal impairment. | Intrathecal
baclofen pump, baclofen | Barry-Albright
Dystonia Scale was used to assess his progress. Within 3 months of
treatment, he gathered control over his bodily movements and SIB
resolved completely. Physical restraints were no longer needed and
his lips were healed. | (103) |
| 29-year-old male
(age at diagnosis, 14 years) | DBS of the GPI was
suggested | Dystonia,
ambulation disorder and SIB | Allopurinol 400
mg/day; baclofen 40 mg/day; clonazepam 10 mg/day; L-Carnitine 2
tablets/day; clozapine 450 mg/day; carbamazepine 1,600 mg/day;
paroxetine 40 mg/day; gabapentin 1,800 mg/day; amytripline 75
mg/day; risperidone 6 mg/day; olanzapine 20 mg/day; thyoridazine
500 mg/day | His progression was
assessed using the Burke-Fhan-Marsden Dystonia Rating Scale
(BFMDRS) and the Mean Disability Scale (MDS) after the operation at
3, 6, 9 and 12 months, and every 6 months thereafter. i) Right away
after starting the bilateral stimulation, his SIB was under control
and the caretakers no longer needed to tie his hands or put gloves.
ii) After the 2nd month, his dystonic movements markedly improved.
The pulse generator was switched off 10 days after the DBS was
started, and as expected self-harming behavior and dystonic
symptoms returned. On the same day, electrical stimulation was
started again and the benefits resumed. Since then, stimulation was
not stopped. The benefit of stimulation persisted during the
five-year follow-up and is still present now. | (91) |
CRISPR-mediated gene correction
This approach focuses on correcting the mutant HGPRT
gene in individuals with LNS. To check the functionality and
reliability of this procedure, researchers first developed an LNS
disease model using human near-haploid cells (HAP1 cells). Human
near-haploid (HAP1) cells were created from the chronic myelogenous
leukemia cell line, KBM-7, which was obtained from a male patient;
hence, it did not have a Y chromosome; as a result, haploid HAP1
cells have one X chromosome (92).
The researchers opted to work with this particular cell line due to
the following reasons: i) These are of human origin; hence, the
results generated would be much more accurate compared to non-human
cell lines; ii) these are haploid in nature; iii) the HAP1 cell
line is the optimal model for genetic manipulation as these cells
have a high transfection efficiency and responsiveness to
CRISPR-based editing techniques; and iv) this allows researchers to
induce specific mutations and study the underlying pathophysiology
in a controlled environment in vitro.
First, c.430C>T and c.508C>T mutations were
introduced in HAP1 and 293T/17 cell lines using cytosine base
editors (CBE). CBEs have a cytosine deaminase, which is a
catalytically modified cas9 enzyme that can carry out the
transition of C:G to T:A, whereas adenine base editors (ABEs)
induce the transition of A:T to G:C and contain a catalytically
modified cas9 adenine deaminase. These were then successfully
corrected using ABEs, demonstrating the potential of base editing
for gene therapy (93,94). Not only did it correct the
mutation, it also rendered the hgprt gene functional. In
HAP1 cells, mutations can be fixed using ABEs.
According to this study, ABEmax-SpG was able to
repair the c.508C>T mutation by as much as 5.2% without
resulting in bystander mutagenesis (20). ABEmax-xCas9(3.7) also repaired the
c.430C>T mutation by up to 3%. These findings demonstrate the
efficaciousness of ABEs in correcting particular HGPRT1 mutations
in HAP1 cells, while in 293T/17 cells the correction efficiencies
were observed to be as high as 3% for the c.430C>T mutation and
up to 5.2% for the c.508C>T mutation, with only minimal
bystander mutagenesis occurring within the active window of base
editing. highlighting their potential for therapeutic gene editing
applications.
Second, the c.333_334ins(A) mutation was introduced
using prime editors (PEs) in HAP1 cell lines using prime editing
guide RNA (pegRNA) designed for this specific mutation. The same
mutation was also present in patient-derived fibroblasts.
Patient-derived fibroblasts were used to interpret and analyze the
applicability of this approach in a clinically relevant context.
This pegRNA guides the PE to the site of the gene where changes
must be made. Whatever correction/change needs to be induced, the
sequence for that is present in the pegRNA. This specific mutation
was found in a 9-year-old male pediatric Korean patient with LNS,
which mimicked this mutation in the HAP1 cell line and 293T/17
cells, and this was successfully corrected using PE, although the
efficiency varied (20). Mutations
in 293T/17 cells could be corrected only up to an efficiency of
50%, whereas it was much lower in patient-derived fibroblasts (only
14%). Similarly, it was also shown that adenine and cytosine BEs
corrected mutations without DNA cleavage, while improved PEs
achieved up to 14% correction in fibroblasts with minimal
off-target effects, highlighting their potential as therapeutic
tools for this rare genetic disorder (20).
5. Conclusion and future perspectives
It is indispensable to understand the etiology of
the disease to develop effective treatments. Understanding the
mechanisms associated with drugs that have shown positive outcomes
in most patients will also help in the development of new drugs
that can be administered either alone or in combination. For
example, DBS appears to work well for young pediatric patients,
although contradictory results have been observed in adult
patients. Therefore, further intensive research is required to
better understand the exact working mechanisms of these approaches.
Although this is a rare disease, clinical trials need to be
conducted using drugs that have shown effective results in the
majority of patients. For example, two studies demonstrated that a
double-blind placebo-controlled study could be employed to help
generate meaningful data from a small number of subjects (95,96).
Models that can replicate the complete biochemical picture of HGPRT
deficiency in the human brain should also be developed. Stem cells
and HAP1 cells have made it easier to carry out in vitro
investigation/study of the underlying mechanisms in neurons, which
is otherwise not possible. Similarly, to reduce the toxicity of
high allopurinol levels, other xanthine oxidase inhibitors, such as
febuxostat, should be considered. Utilizing new models in
combination with cutting-edge investigative techniques can offer a
greater understanding of the etiology and promising therapeutic
options against LNS.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
DV conceptualized the study and was also involved in
the literature search and the selection of studies for the review,
as well as in the writing and preparation of the original draft of
the manuscript. CJ and MP prepared the tables and were also
involved in the literature search. RG supervised the study and
edited the manuscript. All the authors have read and approved the
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cheppalli V and Nikhilesh A: A review on
lesch-nyhan syndrome: A rare inherited disorder with
hypoxanthine-guanine phosphoribosyltransferase deficiency. Int J
Pharm Sci Rev Res. 78:92–99. 2023.
|
|
2
|
Reisz JA, Dzieciatkowska M, Stephenson D,
Gamboni F, Morton DH and D'Alessandro A: Red blood cells from
individuals with Lesch-Nyhan syndrome: Multi-omics insights into a
novel S162N mutation causing Hypoxanthine-guanine
phosphoribosyltransferase deficiency. Antioxidants (Basel).
12(1699)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nanagiri A and Shabbir N: Lesch-nyhan
syndrome: In: StatPearls. StatPearls Publishing, Treasure Island,
FL, 2024.
|
|
4
|
Torres RJ and Puig JG:
Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency:
Lesch-Nyhan syndrome. Orphanet J Rare Dis. 2(48)2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Baumeister AA and Frye GD: The biochemical
basis of the behavioral disorder in the Lesch-Nyhan syndrome.
Neurosci Biobehav Rev. 9:169–178. 1985.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wurtele SK, King AC and Drabman RS:
Treatment package to reduce SIB in a Lesch-Nyhan patient. J Ment
Defic Res. 28:227–234. 1984.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Momosaki K, Kido J, Matsumoto S, Taniguchi
A, Akiyama T, Sawada T, Ozasa S and Nakamura K: The Effect of
S-Adenosylmethionine treatment on neurobehavioral phenotypes in
Lesch-Nyhan disease: A case report. Case Rep Neurol. 11:256–264.
2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Christie R, Bay C, Kaufman IA, Bakay B,
Borden M and Nyhan WL: Lesch-Nyhan disease: Clinical experience
with nineteen patients. Dev Med Child Neurol. 24:293–306.
1982.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sampat R, Fu R, Larovere LE, Torres RJ,
Ceballos-Picot I, Fischbach M, de Kremer R, Schretlen DJ, Puig JG
and Jinnah HA: Mechanisms for phenotypic variation in Lesch-Nyhan
disease and its variants. Hum Genet. 129:71–78. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Camici M, Micheli V, Ipata PL and Tozzi
MG: Pediatric neurological syndromes and inborn errors of purine
metabolism. Neurochem Int. 56:367–378. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fu R, Sutcliffe D, Zhao H, Huang X,
Schretlen DJ, Benkovic S and Jinnah HA: Clinical severity in
Lesch-Nyhan disease: The role of residual enzyme and compensatory
pathways. Mol Genet Metab. 114:55–61. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cakmakli HF, Torres RJ, Menendez A,
Yalcin-Cakmakli G, Porter CC, Puig JG and Jinnah HA: Macrocytic
anemia in Lesch-Nyhan disease and its variants. Genet Med.
21:353–360. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Nyhan WL: Clinical features of the
Lesch-nyhan syndrome. Arch Intern Med. 130:186–192. 1972.PubMed/NCBI
|
|
14
|
Del Bene VA, Crawford JL, Gomez-Gastiasoro
A, Vannorsdall TD, Buchholz A, Ojeda N, Harris JC, Jinnah HA and
Schretlen DJ: Microstructural white matter abnormalities in
Lesch-Nyhan disease. Eur J Neurosci. 55:264–276. 2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Schretlen DJ, Varvaris M, Ho TE,
Vannorsdall TD, Gordon B, Harris JC and Jinnah HA: Regional brain
volume abnormalities in Lesch-Nyhan disease and its variants: A
cross-sectional study. Lancet Neurol. 12:1151–1158. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fasullo M and Endres L: Nucleotide salvage
deficiencies, DNA damage and neurodegeneration. Int J Mol Sci.
16:9431–9449. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Krajewski O, Opielka M, Urbanowicz K,
Chojnowski K, Kochany P, Pawlowski K, Tomaszewska J, Peters GJ,
Smoleński RT and Bełdzińska MM: Management of neurological symptoms
in Lesch-Nyhan disease: A systematic review. Neurosci Biobehav Rev.
165(105847)2024.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Visser JE, Bar PR and Jinnah HA:
Lesch-Nyhan disease and the basal ganglia. Brain Res Brain Res Rev.
32:449–475. 2000.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Olson L and Houlihan D: A review of
behavioral treatments used for Lesch-Nyhan syndrome. Behav Modif.
24:202–222. 2000.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jang G, Shin HR, Do HS, Kweon J, Hwang S,
Kim S, Heo SH, Kim Y and Lee BH: Therapeutic gene correction for
Lesch-Nyhan syndrome using CRISPR-mediated base and prime editing.
Mol Ther Nucleic Acids. 31:586–595. 2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Fu R, Ceballos-Picot I, Torres RJ,
Larovere LE, Yamada Y, Nguyen KV, Hegde M, Visser JE, Schretlen DJ,
Nyhan WL, et al: Genotype-phenotype correlations in neurogenetics:
Lesch-Nyhan disease as a model disorder. Brain. 137:1282–1303.
2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ceballos-Picot I, Le Dantec A, Brassier A,
Jais JP, Ledroit M, Cahu J, Ea HK, Daignan-Fornier B and Pinson B:
New biomarkers for early diagnosis of Lesch-Nyhan disease revealed
by metabolic analysis on a large cohort of patients. Orphanet J
Rare Dis. 10(7)2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jinnah HA, Visser JE, Harris JC, Verdu A,
Larovere L, Ceballos-Picot I, Gonzalez-Alegre P, Neychev V, Torres
RJ, Dulac O, et al: Delineation of the motor disorder of
Lesch-Nyhan disease. Brain. 129:1201–1217. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vinokurov AY, Soldatov VO, Seregina ES,
Dolgikh AI, Tagunov PA, Dunaev AV, Skorkina MY, Deykin AV and
Abramov AY: HPRT1 deficiency induces alteration of mitochondrial
energy metabolism in the brain. Mol Neurobiol. 60:3147–3157.
2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jinnah HA, Ceballos-Picot I, Torres RJ,
Visser JE, Schretlen DJ, Verdu A, Laróvere LE, Chen CJ, Cossu A, Wu
CH, et al: Attenuated variants of Lesch-Nyhan disease. Brain.
133:671–689. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
ThinkGenetic Foundation: Lesch-nyhan
syndrome. https://thinkgenetic.org/diseases/lesch-nyhan-syndrome/.
Accessed July 17, 2024.
|
|
27
|
Mak BS, Chi CS, Tsai CR, Lee WJ and Lin
HY: New mutations of the HPRT gene in Lesch-Nyhan syndrome. Pediatr
Neurol. 23:332–335. 2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nguyen KV, Naviaux RK and Nyhan WL: Novel
mutation in the human HPRT1 gene and the Lesch-Nyhan disease.
Nucleosides Nucleotides Nucleic Acids. 36:704–711. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang D, Zhao J, Teng J, Li W, Zhao X and
Li L: Genetic analysis of a Chinese pedigree with Lesch-Nyhan
syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 40:723–726.
2023.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
30
|
Li L, Qiao X, Liu F, Wang J, Shen H, Fu H
and Mao JH: Description of the molecular and phenotypic spectrum of
Lesch-nyhan disease in eight chinese patients. Front Genet.
13(868942)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ogasawara N, Stout JT, Goto H, Sonta S,
Matsumoto A and Caskey CT: Molecular analysis of a female
Lesch-Nyhan patient. J Clin Invest. 84:1024–1027. 1989.PubMed/NCBI View Article : Google Scholar
|
|
32
|
van Bogaert P, Ceballos I, Desguerre I,
Telvi L, Kamoun P and Ponsot G: Lesch-Nyhan syndrome in a girl. J
Inherit Metab Dis. 15:790–791. 1992.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yukawa T, Akazawa H, Miyake Y, Takahashi
Y, Nagao H and Takeda E: A female patient with Lesch-Nyhan
syndrome. Dev Med Child Neurol. 34:543–546. 1992.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yamada Y, Goto H, Yukawa T, Akazawa H and
Ogasawara N: Molecular mechanisms of the second female Lesch-Nyhan
patient. Adv Exp Med Biol. 370:337–340. 1994.PubMed/NCBI View Article : Google Scholar
|
|
35
|
De Gregorio L, Nyhan WL, Serafin E and
Chamoles NA: An unexpected affected female patient in a classical
Lesch-Nyhan family. Mol Genet Metab. 69:263–268. 2000.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Sebesta I, Stiburkova B, Dvorakova L,
Hrebicek M, Minks J, Stolnaja L, Vernerova Z and Rychlik I: Unusual
presentation of Kelley-seegmiller syndrome. Nucleosides Nucleotides
Nucleic Acids. 27:648–655. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Aral B, de Saint Basile G, Al-Garawi S,
Kamoun P and Ceballos-Picot I: Novel nonsense mutation in the
hypoxanthine guanine phosphoribosyltransferase gene and nonrandom
X-inactivation causing Lesch-Nyhan syndrome in a female patient.
Hum Mutat. 7:52–58. 1996.PubMed/NCBI View Article : Google Scholar
|
|
38
|
AlBakheet A, AlQudairy H, Alkhalifah J,
Almoaily S, Kaya N and Rahbeeni Z: Detailed genetic and clinical
analysis of a novel de novo variant in HPRT1: Case report of a
female patient from Saudi Arabia with Lesch-Nyhan syndrome. Front
Genet. 13(1044936)2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hooft C, Van Nevel C and De Schaepdryver
AF: Hyperuricosuric encephalopathy without hyperuricaemia. Arch Dis
Child. 43:734–737. 1968.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sekine M, Fujiwara M, Okamoto K, Ichida K,
Nagata K, Hille R and Nishino T: Significance and amplification
methods of the purine salvage pathway in human brain cells. J Biol
Chem. 300(107524)2024.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Guibinga GH, Murray F, Barron N, Pandori W
and Hrustanovic G: Deficiency of the purine metabolic gene HPRT
dysregulates microRNA-17 family cluster and guanine-based cellular
functions: A role for EPAC in Lesch-Nyhan syndrome. Hum Mol Genet.
22:4502–4515. 2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Lopez JM, Outtrim EL, Fu R, Sutcliffe DJ,
Torres RJ and Jinnah HA: Physiological levels of folic acid reveal
purine alterations in Lesch-Nyhan disease. Proc Natl Acad Sci USA.
117:12071–12079. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Saito Y and Takashima S: Neurotransmitter
changes in the pathophysiology of Lesch-Nyhan syndrome. Brain Dev.
22 (Suppl 1):S122–S131. 2000.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bell S, Kolobova I, Crapper L and Ernst C:
Lesch-Nyhan syndrome: Models, theories, and therapies. Mol
Syndromol. 7:302–311. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
van der Zee SP, Schretlen ED and Monnens
LA: Megaloblastic anaemia in the Lesch-Nyhan syndrome. Lancet.
1(1427)1968.PubMed/NCBI View Article : Google Scholar
|
|
46
|
McKeran RO: Factors in the pathogenesis of
the brain damage and anaemia in the Lesch-Nyhan syndrome. Ciba
Found Symp. 83–96. 1977.PubMed/NCBI View Article : Google Scholar : doi:
10.1002/9780470720301.ch6.
|
|
47
|
Nemkov T, Sun K, Reisz JA, Song A, Yoshida
T, Dunham A, Wither MJ, Francis RO, Roach RC, Dzieciatkowska M, et
al: Hypoxia modulates the purine salvage pathway and decreases red
blood cell and supernatant levels of hypoxanthine during
refrigerated storage. Haematologica. 103:361–372. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Laróvere LE, Escuredo E, Becerra A and
Dodelson de Kremer R: Lesch-nyhan disease and its variants:
Phenotypic and mutation spectrum of hypoxanthine-guanine
phosphoribosyltransferase deficiency in argentine patients. J
Inborn Errors Metab: 9, 2021
doi:10.1590/2326-4594-jiems-2020-0027.
|
|
49
|
Saugstad OD: Hypoxanthine as a measurement
of hypoxia. Pediatr Res. 9:158–161. 1975.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Saugstad OD: Hypoxanthine as an indicator
of hypoxia: Its role in health and disease through free radical
production. Pediatr Res. 23:143–150. 1988.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Roussel C, Morel A, Dussiot M, Marin M,
Colard M, Fricot-Monsinjon A, Martinez A, Chambrion C, Henry B,
Casimir M, et al: Rapid clearance of storage-induced
microerythrocytes alters transfusion recovery. Blood.
137:2285–2298. 2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ceballos-Picot I, Mockel L, Potier MC,
Dauphinot L, Shirley TL, Torero-Ibad R, Fuchs J and Jinnah HA:
Hypoxanthine-guanine phosphoribosyl transferase regulates early
developmental programming of dopamine neurons: Implications for
Lesch-Nyhan disease pathogenesis. Hum Mol Genet. 18:2317–2327.
2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Lewers JC, Ceballos-Picot I, Shirley TL,
Mockel L, Egami K and Jinnah HA: Consequences of impaired purine
recycling in dopaminergic neurons. Neuroscience. 152:761–772.
2008.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Mastrangelo L, Kim JE, Miyanohara A, Kang
TH and Friedmann T: Purinergic signaling in human pluripotent stem
cells is regulated by the housekeeping gene encoding hypoxanthine
guanine phosphoribosyltransferase. Proc Natl Acad Sci USA.
109:3377–3382. 2012.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Cuttler K, Hassan M, Carr J, Cloete R and
Bardien S: Emerging evidence implicating a role for neurexins in
neurodegenerative and neuropsychiatric disorders. Open Biol.
11(210091)2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Ghaffar A, Akhter T, Stromme P, Misceo D,
Khan A, Frengen E, Umair M, Isidor B, Cogné B, Khan AA, et al:
Variants of NAV3, a neuronal morphogenesis protein, cause
intellectual disability, developmental delay, and microcephaly.
Commun Biol. 7(831)2024.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ignacio DA, Babikian T, Dennis EL, Bickart
KC, Choe M, Snyder AR, Brown A, Giza CC and Asarnow RF: The
neurocognitive correlates of DTI indicators of white matter
disorganization in pediatric moderate-to-severe traumatic brain
injury. Front Hum Neurosci. 18(1470710)2024.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Guibinga GH, Hrustanovic G, Bouic K,
Jinnah HA and Friedmann T: MicroRNA-mediated dysregulation of
neural developmental genes in HPRT deficiency: Clues for
Lesch-Nyhan disease? Hum Mol Genet. 21:609–622. 2012.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Kang TH, Park Y, Bader JS and Friedmann T:
The housekeeping gene hypoxanthine guanine
phosphoribosyltransferase (HPRT) regulates multiple developmental
and metabolic pathways of murine embryonic stem cell neuronal
differentiation. PLoS One. 8(e74967)2013.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Guibinga GH: MicroRNAs: Tools of
mechanistic insights and biological therapeutics discovery for the
rare neurogenetic syndrome Lesch-Nyhan disease (LND). Adv Genet.
90:103–131. 2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Sutcliffe DJ, Dinasarapu AR, Visser JE,
Hoed JD, Seifar F, Joshi P, Ceballos-Picot I, Sardar T, Hess EJ,
Sun YV, et al: Induced pluripotent stem cells from subjects with
Lesch-Nyhan disease. Sci Rep. 11(8523)2021.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Bortolotti M, Polito L, Battelli MG and
Bolognesi A: Xanthine oxidoreductase: One enzyme for multiple
physiological tasks. Redox Biol. 41(101882)2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Abramov AY, Scorziello A and Duchen MR:
Three distinct mechanisms generate oxygen free radicals in neurons
and contribute to cell death during anoxia and reoxygenation. J
Neurosci. 27:1129–1138. 2007.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Pinto CS and Seifert R: Decreased
GTP-stimulated adenylyl cyclase activity in HPRT-deficient human
and mouse fibroblast and rat B103 neuroblastoma cell membranes. J
Neurochem. 96:454–459. 2006.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Gray M, Nash KR and Yao Y: Adenylyl
cyclase 2 expression and function in neurological diseases. CNS
Neurosci Ther. 30(e14880)2024.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Arhakis A, Topouzelis N, Kotsiomiti E and
Kotsanos N: Effective treatment of self-injurious oral trauma in
Lesch-Nyhan syndrome: A case report. Dent Traumatol. 26:496–500.
2010.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Lee J, Lee E, Shin J, Kim S and Jeong T:
Semi-fixed lip bumper in lesch-nyhan syndrome: An interim treatment
modality. J Korean Acad Pediatr Dent. 47:93–98. 2020.
|
|
68
|
Dabrowski E, Smathers SA, Ralstrom CS,
Nigro MA and Leleszi JP: Botulinum toxin as a novel treatment for
self-mutilation in Lesch-Nyhan syndrome. Dev Med Child Neurol.
47:636–639. 2005.PubMed/NCBI
|
|
69
|
Garcia-Romero MDM, Torres RJ, Garcia-Puig
J and Pascual-Pascual SI: Safety and efficacy of botulinum toxin in
the treatment of Self-biting behavior in Lesch-nyhan disease.
Pediatr Neurol. 127:6–10. 2022.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Gottle M, Prudente CN, Fu R, Sutcliffe D,
Pang H, Cooper D, Veledar E, Glass JD, Gearing M, Visser JE and
Jinnah HA: Loss of dopamine phenotype among midbrain neurons in
Lesch-Nyhan disease. Ann Neurol. 76:95–107. 2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Lloyd KG, Hornykiewicz O, Davidson L,
Shannak K, Farley I, Goldstein M, Shibuya M, Kelley WN and Fox IH:
Biochemical evidence of dysfunction of brain neurotransmitters in
the Lesch-Nyhan syndrome. N Engl J Med. 305:1106–1111.
1981.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Wong DF, Harris JC, Naidu S, Yokoi F,
Marenco S, Dannals RF, Ravert HT, Yaster M, Evans A, Rousset O, et
al: Dopamine transporters are markedly reduced in Lesch-Nyhan
disease in vivo. Proc Natl Acad Sci USA. 93:5539–5543.
1996.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Visser JE, Schretlen DJ, Bloem BR and
Jinnah HA: Levodopa is not a useful treatment for Lesch-Nyhan
disease. Mov Disord. 26:746–749. 2011.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Jankovic J, Caskey TC, Stout JT and Butler
IJ: Lesch-Nyhan syndrome: A study of motor behavior and
cerebrospinal fluid neurotransmitters. Ann Neurol. 23:466–469.
1988.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Watts RW, Spellacy E, Gibbs DA, Allsop J,
McKeran RO and Slavin GE: Clinical, post-mortem, biochemical and
therapeutic observations on the Lesch-Nyhan syndrome with
particular reference to the Neurological manifestations. Q J Med.
51:43–78. 1982.PubMed/NCBI
|
|
76
|
Montero C, Smolenski RT, Duley JA and
Simmonds HA: S-adenosylmethionine increases erythrocyte ATP in
vitro by a route independent of adenosine kinase. Biochem
Pharmacol. 40:2617–2623. 1990.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Glick N: Dramatic reduction in self-injury
in Lesch-Nyhan disease following S-adenosylmethionine
administration. J Inherit Metab Dis. 29(687)2006.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Dolcetta D, Parmigiani P, Salmaso L,
Bernardelle R, Cesari U, Andrighetto G, Baschirotto G, Nyhan WL and
Hladnik U: Quantitative evaluation of the clinical effects of
S-adenosylmethionine on mood and behavior in Lesch-Nyhan patients.
Nucleosides Nucleotides Nucleic Acids. 32:174–188. 2013.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Elion GB, Benezra FM, Beardmore TD and
Kelley WN: Studies with allopurinol in patients with impaired renal
function. Adv Exp Med Biol. 122A:263–267. 1980.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Daudon M: Crystalluria. Nephrol Ther:
174-190, 2015 (In French).
|
|
81
|
Sriranganathan MK, Vinik O, Pardo Pardo J,
Bombardier C and Edwards CJ: Interventions for tophi in gout.
Cochrane Database Syst Rev. 8(CD010069)2021.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Greene ML, Fujimoto WY and Seegmiller JE:
Urinary xanthine stones-a rare complication of allopurinol therapy.
N Engl J Med. 280:426–427. 1969.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Simmonds HA, Reiter S, Davies PM and
Cameron JS: Orotidine accumulation in human erythrocytes during
allopurinol therapy: Association with high urinary
oxypurinol-7-riboside concentrations in renal failure and in the
Lesch-Nyhan syndrome. Clin Sci (Lond). 80:191–197. 1991.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Krauss JK, Lipsman N, Aziz T, Boutet A,
Brown P, Chang JW, Davidson B, Grill WM, Hariz MI, Horn A, et al:
Technology of deep brain stimulation: Current status and future
directions. Nat Rev Neurol. 17:75–87. 2021.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Lozano AM, Lipsman N, Bergman H, Brown P,
Chabardes S, Chang JW, Matthews K, McIntyre CC, Schlaepfer TE,
Schulder M, et al: Deep brain stimulation: Current challenges and
future directions. Nat Rev Neurol. 15:148–160. 2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
van Westen M, Rietveld E, Bergfeld IO, de
Koning P, Vullink N, Ooms P, Graat I, Liebrand L, van den Munckhof
P, Schuurman R and Denys D: Optimizing deep brain stimulation
parameters in Obsessive-compulsive disorder. Neuromodulation.
24:307–315. 2021.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Pycroft L, Stein J and Aziz T: Deep brain
stimulation: An overview of history, methods, and future
developments. Brain Neurosci Adv.
2(2398212818816017)2018.PubMed/NCBI View Article : Google Scholar
|
|
88
|
McIntyre CC, Savasta M, Kerkerian-Le Goff
L and Vitek JL: Uncovering the mechanism(s) of action of deep brain
stimulation: Activation, inhibition, or both. Clin Neurophysiol.
115:1239–1248. 2004.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Javed N and Cascella M: Neuroanatomy,
Globus Pallidus. Publishing ISITIFS, editor, 2023.
|
|
90
|
Taira T, Kobayashi T and Hori T:
Disappearance of self-mutilating behavior in a patient with
lesch-nyhan syndrome after bilateral chronic stimulation of the
globus pallidus internus. Case report. J Neurosurg. 98:414–416.
2003.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Piedimonte F, Andreani JC, Piedimonte L,
Micheli F, Graff P and Bacaro V: Remarkable clinical improvement
with bilateral globus pallidus internus deep brain stimulation in a
case of Lesch-Nyhan disease: Five-year follow-up. Neuromodulation.
18:118–122. 2015.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Llargues-Sistac G, Bonjoch L and
Castellvi-Bel S: HAP1, a new revolutionary cell model for gene
editing using CRISPR-Cas9. Front Cell Dev Biol.
11(1111488)2023.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Anzalone AV, Koblan LW and Liu DR: Genome
editing with CRISPR-Cas nucleases, base editors, transposases and
prime editors. Nat Biotechnol. 38:824–844. 2020.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Gaudelli NM, Komor AC, Rees HA, Packer MS,
Badran AH, Bryson DI and Liu DR: Programmable base editing of A•T
to G•C in genomic DNA without DNA cleavage. Nature. 551:464–471.
2017.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Khasnavis T, Torres RJ, Sommerfeld B, Puig
JG, Chipkin R and Jinnah HA: A double-blind, placebo-controlled,
crossover trial of the selective dopamine D1 receptor antagonist
ecopipam in patients with Lesch-Nyhan disease. Mol Genet Metab.
118:160–166. 2016.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Lauber M, Plecko B, Pfiffner M, Nuoffer JM
and Haberle J: The Effect of S-Adenosylmethionine on
Self-mutilation in a patient with Lesch-nyhan disease. JIMD Rep.
32:51–57. 2017.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Gutierrez C, Pellene A and Micheli F:
Botulinum toxin: Treatment of self-mutilation in patients with
Lesch-Nyhan syndrome. Clin Neuropharmacol. 31:180–183.
2008.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Zecavati N, Santos C and Oyegbile T:
Botulinum toxin for the treatment of Extra-pyramidal symptoms of
Lesch-nyhan syndrome (P6.252). Neurology. 84(252)2015.
|
|
99
|
Gilbert C, Sauer M and Cheng J: Reduction
of self-mutilating behavior and improved oromotor function in a
patient with Lesch-Nyhan syndrome following botulinum toxin
injection: A case report. J Pediatr Rehabil Med. 14:133–136.
2021.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Chen BC, Balasubramaniam S, McGown IN,
O'Neill JP, Chng GS, Keng WT, Ngu LH and Duley JA: Treatment of
Lesch-Nyhan disease with S-adenosylmethionine: Experience with five
young Malaysians, including a girl. Brain Dev. 36:593–600.
2014.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Pralong E, Pollo C, Coubes P, Bloch J,
Roulet E, Tetreault MH, Debatisse D and Villemure JG:
Electrophysiological characteristics of limbic and motor globus
pallidus internus (GPI) neurons in two cases of Lesch-Nyhan
syndrome. Neurophysiol Clin. 35:168–173. 2005.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Cif L, Biolsi B, Gavarini S, Saux A,
Robles SG, Tancu C, Vasques X and Coubes P: Antero-ventral internal
pallidum stimulation improves behavioral disorders in Lesch-Nyhan
disease. Mov Disord. 22:2126–2129. 2007.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Deon LL, Kalichman MA, Booth CL, Slavin KV
and Gaebler-Spira DJ: Pallidal deep-brain stimulation associated
with complete remission of self-injurious behaviors in a patient
with Lesch-Nyhan syndrome: A case report. J Child Neurol.
27:117–120. 2012.PubMed/NCBI View Article : Google Scholar
|