Introduction
Pulmonary alveolar proteinosis (PAP), a rare lung
disease of unknown origin, is characterized by the accumulation of
the alveoli with phospholipid, protein and other floccular material
that stain with periodic acid-Schiff. The existence of PAP was only
recognized in 1958 through the seminal report of Rosen et
al(1). Carey and Trapnell
(2) classified PAP into three
groups, i.e., congenital, autoimmune and secondary PAP. Congenital
PAP is a heterogeneous collection of disorders caused by homozygous
mutation of the genes encoding surfactant protein (SP)-B, SP-C and
the ABCA3 transporter or by the absence of the
granulocyte/macrophage colony stimulating factor (GM-CSF) receptor.
Congenital PAP usually occurs in children. Secondary PAP has been
reported in association with various diverse clinical disorders
such as hematological disorders, immunological diseases, lysinuric
protein intolerance and infections, and various toxic inhalation
syndromes, including inhalation of inorganic and organic dusts, and
fumes. Autoimmune PAP is also regarded as a type of idiopathic
PAP.
In this study, idiopathic PAP was employed instead
of autoimmune PAP. Ninety per cent of patients have idiopathic PAP,
with a prevalence of 6–7 individuals per million in the general
population. Additionally, idiopathic PAP occurs in all ethnic
groups, in the third to fourth decades of life and is approximately
twice as common in males. The presence of idiopathic PAP may be
associated with high levels of neutralizing GM-CSF autoantibody
(2,3). High levels of GM-CSF autoantibody are
associated with idiopathic PAP, and these autoantibodies are
assumed to be critical in the pathogenesis of idiopathic PAP, but
are not present in secondary or congenital PAP, other lung diseases
or in healthy donors (4). The
binding affinity of autoantibodies for GM-CSF is higher than that
for the GM-CSF receptor in its low- or high-affinity binding state,
and these autoantibodies eliminate GM-CSF bioactivity in
vivo(5). Subsequently,
idiopathic PAP is a type of autoimmune disorder. However, systemic
studies regarding the immunological function and biochemical index
of idiopathic PAP have yet to be conducted. In the present study,
some pathogenesis of idiopathic PAP was explored, through the
detection of immunocytes, immunoglobulin (Ig), complement (C),
cytokeratin fragment antigen 211 (CYFR211), lactate dehydrogenase
(LDH) and creatine kinase (CK) in the peripheral blood of patients
with idiopathic PAP.
Materials and methods
Subjects
During the 11-year period between 2000 and 2010, a
series of 42 Chinese patients who were diagnosed with PAP in the
Shanghai Pulmonary Hospital Affiliated to Tongji University
(Shanghai, China) were retrospectively analyzed. In this study, the
eligibility criteria used were as proposed by Kavuru et
al(6) and included: i)
histopathological findings of specimens obtained by open lung
biopsy or transbronchial lung biopsy; ii) a milk-like appearance
with typical cytologic findings of bronchoalveolar lavage (BAL);
iii) high-resolution computed tomography (HRCT) scan which showed
ground glass opacity and/or a pattern of crazy paving; iv)
restrictive ventilation and diffusion dysfunction, hypoxemia; with
v) dyspnea and cough being the most common symptoms, although
certain patients were asymptomatic at diagnosis. Exclusion criteria
for this study were: i) PAP resulting from another condition (e.g.,
myeloproliferative disorder or leukemia, occupational exposure to
dust, human immunodeficiency virus disease or respiratory
infections) and ii) age <18 years. The diagnosis of PAP was
based on BAL and the histopathological findings of specimens
obtained by open lung biopsy in eight patients, by transbronchial
lung biopsy in three patients and by only BAL in 31 patients
(6,7). Thirteen patients, including 5
steelworkers, 2 mineworkers, 2 steelworkers and 4 persons with
long-term contact history to oil fume were excluded from the study.
An additional 29 patients had no induction agent, but were
diagnosed with idiopathic PAP and included in this study.
Control group
Between 2000 and 2010, 30 healthy adults who had no
medical histories were randomly selected following examination in
the clinic, and were classified as normal subjects (Table I). All study participants provided
informed consent.
 | Table ICharacteristics of the patients and
normal subjects. |
Table I
Characteristics of the patients and
normal subjects.
| Characteristics | Patients (n=29) | Normal subjects
(n=30) | P-value |
|---|
| Gender
(male/female) | 19/10 | 20/10 | |
| Mean age (years) | 47.8±10.6 | 45.8±14.1 | |
| Smoking status | | | |
| Current smoker | 11 | 13 | |
| Ex-smoker | 3 | 4 | |
| Never-smoker | 15 | 13 | |
| Symptom | | | |
| Dyspnea | 14 | | |
| Dry cough | 11 | | |
| Expectoration | 10 | | |
| Thoracalgia | 5 | | |
| Dyspnea score | 1.17±1.04 | | |
| Score of chest
HRCT | 19.10±9.01 | | |
| FVC% Pred | 72.26±19.12 | 95.97±8.37 | 0.000 |
| FEV1%
Pred | 71.79±18.36 | 95.58±7.83 | 0.000 |
| FEV1/FVC | 83.09±5.70 | 83.47±5.61 | 0.655 |
| DLCO% | 61.56±24.10 | 102.49±11.25 | 0.000 |
| PaO2
(mmHg) | 71.32±14.76 | 94.13±2.99 | 0.000 |
Dyspnea score
Dyspnea was one of the most common symptoms. The
dyspnea score was measured with the Modified British Medical
Research Council (mMRC). The grade of dyspnea was from 0 to 4 (I
only get breathless with strenuous exercise, 0; I get short of
breath when hurrying on the level or walking up a slight hill, 1; I
walk slower than people of the same age on the level due to
breathlessness or I have to stop for breath when walking on my own
pace on the level, 2; I stop for breath after walking ∼100 m or
after a few minutes on the level, 3; I am too breathless to leave
the house or I am breathless when dressing or undressing, 4).
Score of chest HRCT
The high-resolution computed tomography (HRCT) of
chest was analyzed for 21 patients. The chest HRCT of an additional
8 patients had been examined in other hospitals, but records were
inadequate. HRCT scans of the chest were graded according to the
visual scoring methods proposed by Lee et al(8). The chest radiographs were read and
interpreted independently by a radiologist and a respiratory
medicine physician. The mean values obtained from the two readers
were used for analysis. The scores were decided on four
representative layers including aortic arch, tracheal carina,
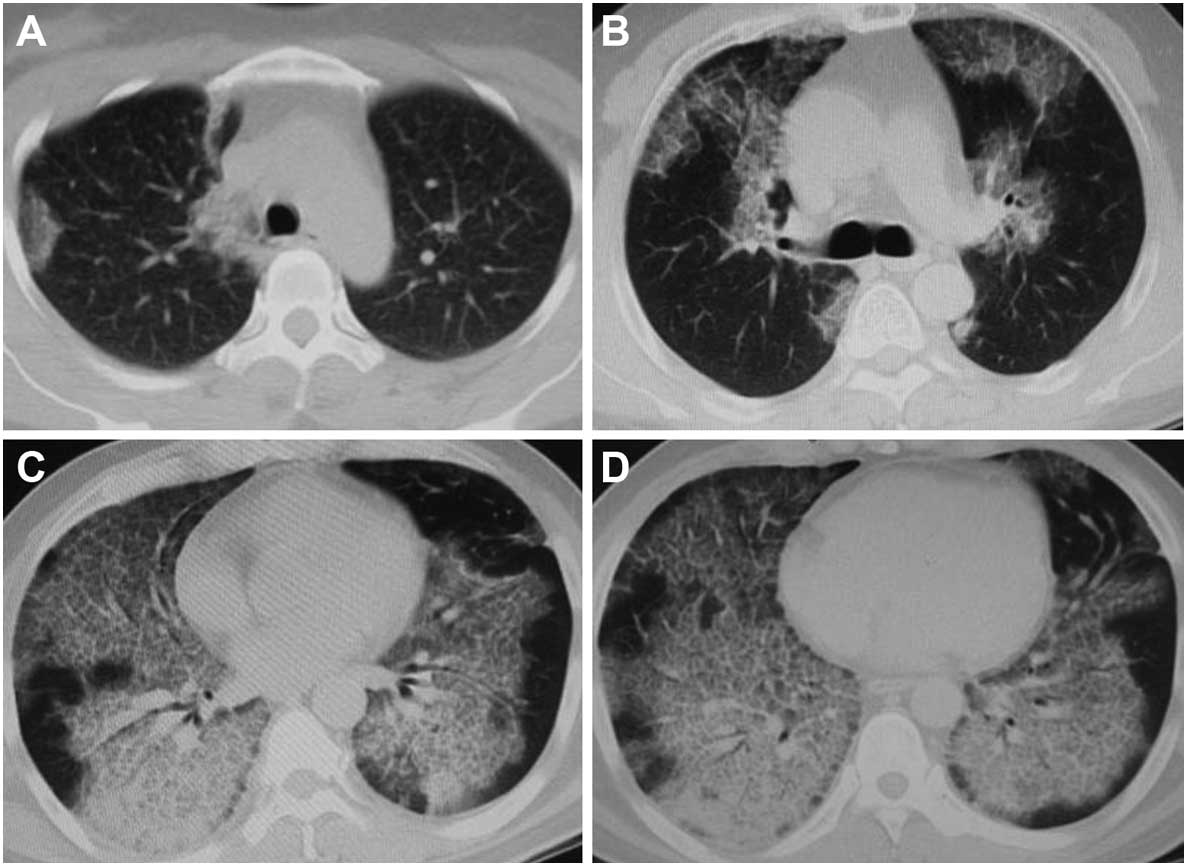
left/right inferior lung vein converging and above of diaphragm
layer score (Fig. 1). The ‘ground
glass opacity’ referred to the presence of increased lung opacity
associated with partial obscuring of normal vascular structures.
The extent of lung opacity was estimated by means of a five-point
scale as: no opacity, 0; opacity involving <25% of a layer of
hemithorax, 1; ≥25 – <50%, 2; ≥50 – <75%, 3; ≥75%, 4. The
score of chest HRCT was calculated by adding up the extent scores
on the four representative layers of each hemithorax.
Pulmonary function
The patients and normal subjects underwent pulmonary
function testing using a MasterScreen spirometer (Jaeger GmbH,
Würzburg, Germany). Pulmonary function testing, including forced
vital capacity/predicted value (FVC% Pred), forced expiratory
volume in one second/predicted value (FEV1% Pred),
FEV1/FVC and diffusing capacity of the lung for carbon
monoxide/predicted value (DLCO% Pred), was performed.
Blood samples were analyzed for arterial partial pressure of oxygen
(PaO2) values in room air using a Blood-gas analyzer
(Radiometer, Copenhagen, Denmark).
Immunological function and
biochemistry
Immunocytes in the peripheral blood of 19 patients
and normal subjects were detected using a flow cytometer (Beckman
Coulter, Inc., Miami, FL, USA). IgG, IgA, IgM, C3, C4, LDH and CK
in peripheral blood using a biochemical analyzer (Olympus Optical
Co, Ltd., Japan), and CYFR211 in peripheral blood using a full
automatic dual-head radioimmunoassay Gamma Counter (Shanghai Hesuo
Rihuan Photoelectric Instrument Co., Ltd., China) were detected in
all study participants.
Statistical analysis
Differences between the parameters of patients with
idiopathic PAP and normal subjects were tested using a paired
t-test. P<0.05 was considered statistically significant. All
tests were two-sided. Results were reported as the mean ± SD. The
correlation between variables was determined by Pearson’s
correlation analysis. Statistical analysis was performed using SPSS
version 15.
Results
Patient characteristics
The characteristics of the idiopathic PAP group were
compared with those of the normal subjects group (Table I). The mean dyspnea score was shown
as 1.17±1.04. There were various symptoms, including dyspnea
(n=14), dry cough (n=11), expectoration (n=10) and thoracalgia
(n=5) in 24 patients, but no symptoms occurred in other patients.
The results of pulmonary function and PaO2 are listed in
Table I. FVC% Pred,
FEV1% Pred, DLCO% Pred and PaO2
were higher in the idiopathic PAP patients than those of normal
subjects (all P<0.01). No differences were found in FEV1/FVC
between the two groups (P>0.05). The mean score of chest HRCT of
patients was 19.10±9.01. Significant correlations between the score
of chest HRCT and the dyspnea score (r=−0.857; P=0.000), FVC% Pred
(r=−0.657; P=0.001), FEV1% Pred (r=−0.565; P=0.004),
DLCO% Pred (r=−0.733; P=0.000) and PaO2
(r=−0.685; P=0.000) were observed in 21 patients. However, no
apparent correlations were observed between the score of chest HRCT
and FEV1/FVC (r=0.349; P=0.143).
Immunological and biochemical indices of
the patients and normal subjects, and Pearson’s correlation
coefficient
The results of immunocytes in peripheral blood are
listed in Table II. The peripheral
blood of 19 patients demonstrated a decrease in
CD4+/CD8+ (P<0.05), the percentage of
CD4+ T lymphocyte (P<0.01) and helper T lymphocyte
(P<0.01), and an increase in the percentage of suppressor T
lymphocyte (P<0.01) compared to that of normal subjects. No
correlations were found in the percentage of CD3+ T
lymphocyte, natural killer and CD8+ T lymphocyte between
the two groups (all P>0.05). No correlations were observed
between any of factor including CD4+ T lymphocyte,
CD4+/CD8+, helper T lymphocyte and suppressor
T lymphocyte, and the dyspnea score, score of chest HRCT, FVC%
Pred, FEV1% Pred, DLCO% Pred and
PaO2, respectively (all P>0.05).
| Table IIImmunological and biochemical index of
the patients and normal subjects. |
Table II
Immunological and biochemical index of
the patients and normal subjects.
| Idiopathic PAP
patients
| Normal subjects
| |
|---|
| Immunological and
biochemical index | n | Mean ± SD | n | Mean ± SD | P-value |
|---|
| Immunocytes | | | | | |
| CD3+
(%) | 19 | 70.5±6.0 | 30 | 68.3±8.9 | 0.338 |
| NK (%) | 19 | 16.2±17.6 | 30 | 8.4±5.5 | 0.074 |
| CD4±
(%) | 19 | 34.6±10.6 | 30 | 44.0±6.8 | 0.002 |
| CD8±
(%) | 19 | 28.1±10.0 | 30 | 24.9±7.5 | 0.210 |
|
CD4±/CD8± | 19 | 1.5±0.8 | 30 | 2.0±0.8 | 0.033 |
| Th (%) | 19 | 0.5±0.16 | 30 | 0.6±0.09 | 0.001 |
| Ts (%) | 19 | 0.4±0.1 | 30 | 0.3±0.08 | 0.009 |
| Immunoglobulin | | | | | |
| IgG (g/l) | 29 | 10.7±3.8 | 30 | 10.9±2.1 | 0.781 |
| IgA (g/l) | 29 | 2.7±1.8 | 30 | 2.4±1.8 | 0.571 |
| IgM (g/l) | 29 | 1.4±0.8 | 30 | 1.1±0.6 | 0.104 |
| Complement | | | | | |
| C3 (g/l) | 29 | 1.5±1.7 | 30 | 1.2±0.3 | 0.330 |
| C4 (g/l) | 29 | 0.4±0.1 | 30 | 0.4±0.2 | 0.491 |
| Biochemical
index | | | | | |
| CYFRA211
(ng/ml) | 29 | 7.0±4.5 | 30 | 1.0±0.6 | 0.000 |
| LDH (IU/l) | 29 | 239.9±75.1 | 30 | 155.3±31.1 | 0.000 |
| CK (IU/l) | 29 | 84.3±69.0 | 30 | 66.4±22.4 | 0.193 |
Pearson’s correlation coefficients (r) between the
two variables are listed in Table
III. The results of immunoglobulin and complement in peripheral
blood are listed in Table II. No
differences were observed in the concentration of IgG, IgA, IgM, C3
and C4 between the two groups (all P>0.05). The serum level of
LDH, CYFRA211 and CK are listed in Table II. An increase was found in the LDH
(t=7.191; P<0.01) and CYFR211 (t=5.622; P<0.01) in the
peripheral blood of the patients compared to that of normal
subjects, but there were no differences in the concentration of CK
between the two groups (P>0.05). The serum level of LDH was
negatively correlated with FVC% Pred (r=−0.400; P=0.032),
DLCO% Pred (r=−0.604; P=0.001), PaO2 (r=−0.395;
P=0.034), positively associated with the dyspnea score (r=0.645;
P=0.000) in 29 patients with idiopathic PAP, and positively
correlated with the score of chest HRCT in 21 patients (r=0.494;
P=0.023). There was no correlation between LDH and any of the
FEV1% Pred (r=−0.345; P=0.067) or FEV1/FVC (r=−0.140;
P=0.468), respectively. The serum level of CYFR211 was negatively
correlated with DLCO% Pred (r=−0.401; P=0.031), and
positively associated with the dyspnea score (r=0.403; P=0.030).
However, no correlation was found between FVC% Pred (r=−0.256;
P=0.180), FEV1% Pred (r=−0.224; P=0.243), FEV1/FVC
(r=0.309; P=0.103) and PaO2 (r=−0.171; P=0.374) in any
of the patients with idiopathic PAP and the score of chest HRCT
(r=0.341; P=0.131) of 21 patients.
| Table IIIPearson’s correlation coefficient (r)
between the two variables. |
Table III
Pearson’s correlation coefficient (r)
between the two variables.
| Characteristic | CD4+
(%) |
CD4+/CD8+ | Th (%) | Ts (%) |
|---|
| Dyspnea score | 0.180 | 0.294 | 0.232 | 0.402 |
| FVC% Pred | 0.167 | 0.099 | 0.317 | 0.165 |
| FEV1%
Pred | 0.113 | 0.007 | 0.230 | 0.233 |
| DLCO%
Pred | 0.264 | 0.270 | 0.286 | 0.434 |
| PaO2
(mmHg) | 0.080 | 0.025 | 0.062 | 0.098 |
| Score of chest | | | | |
| HRCT | 0.002 | 0.050 | 0.167 | 0.043 |
Discussion
By summarizing previous studies for over 50 years
Carey and Trapnell (2) showed that
idiopathic PAP was regarded as being strongly associated with high
levels of the neutralizing GM-CSF autoantibody (9,10).
Additionally, secondary PAP was associated with various diverse
clinical disorders and various toxic inhalation syndromes. Findings
of Costabel and Nakata (11)
indicated that inhalation of dusts may be the initiating cause
which resulted in the elevation of the neutralizing GM-CSF
autoantibody. Thus, the cause of idiopathic PAP and the reason for
the elevated levels of the GM-CSF autoantibody remain to be
investigated.
Dyspnea was observed as the most dominant symptom of
patients with idiopathic PAP in Korean and Japanese populations
(9,12) and 17 patients in this study (58.6%).
Results of pulmonary function in this study were similar to those
of previous studies (12), i.e., a
restrictive defect, with a reduction in the diffusing capacity.
Arterial blood gas analysis was found to exhibit hypoxemia in
previous studies as well as this one (10,12).
Chest HRCT objectively showed the degree, extent and severity of
lung opacity and was a key external index used to evaluate the
severity of PAP. Correlations between pulmonary function testing
and HRCT parameters were found by Chen et al(13), with significance in average lung
density and FVC, total lung mass and FEV1, the ratio of air-filling
lung volume to total lung volume and peak expiratory flow,
DLCO and the ratio of DLCO to alveolar
volume. Correlation analyses that were performed between
quantitative CT parameters and DLCO and PaO2
by Guan et al(14) indicated
that DLCO correlated well with the total lung weight,
airspace volume, mean lung density, and mean lung inflation, and
that PaO2 also correlated well with the total lung weight, mean
lung density, mean lung inflation and airspace volume/total lung
volume ratio. In this study, significant correlations were noted
between the score of chest HRCT, dyspnea score, and FVC% Pred,
FEV1% Pred, DLCO% and PaO2,
respectively. Thus, the severity of idiopathic PAP was reflected by
the dyspnea score, score of chest HRCT, pulmonary function and
PaO2 to a great extent.
CD4+ T lymphocyte is able to promote the
multiplication and differentiation of B lymphocytes, T lymphocytes
and other immunocytes, and coordinate the interaction between
variants of immunocytes. The helper T lymphocyte is the regulatory
T cell. The suppressor T lymphocyte is one of the CD8+ T
lymphocytes which released inhibitory factors and acted on the
antigen-specific helper T and/or B lymphocyte and led to
immunological function suppression. The peri pheral blood of the
patients showed a decrease in CD4+/CD8+, the
percentage of CD4+ T lymphocyte and helper T lymphocyte,
and an increase in the percentage of suppressor T lymphocyte. These
results indicated that cellular immunity was inhibited. The
correlation analysis between the significant immunocytes and
dyspnea score, score of chest HRCT, lung function index and
PaO2, indicated that these immunocytes were not
associated with the severity of PAP. However, a predominance of
CD4+ cells in some idiopathic PAP cases was reported
previously (15). Thus, more
studies should be conducted to determine the role of these cells in
idiopathic PAP. Results obtained from the analysis of the
immunoglobulin and complement indicated that humoral immunity had
no correlation with PAP.
The serum level of LDH was increased in idiopathic
PAP patients in previous studies (16,17). A
significant correlation between LDH and alveolar artery oxygen
gradient was detected by Xu et al(16). In this study, it was also found that
LDH was elevated and was correlated with FVC% Pred,
DLCO% Pred, PaO2, dyspnea score and the score
of chest HRCT, suggesting a correlation with the severity of
idiopathic PAP. A case of PAP was previously reported in which the
value of cytokeratin 19 fragment in the serum was initially
elevated and decreased to the normal range after the lung lavage
(18). CYFRA211 was the soluble
fragment of cytokeratin 19. In this study, the result showed that
CYFRA211 increased in PAP, and had a significant cotrelation with
DLCO% Pred and the dyspnea score. Those studies
indicated that CYFRA211 may regarded as one of the indices to
monitor the severity of idiopathic PAP.
In summary, the results of immunity (including
cellular and humoral immunity) and the chemistry index in
peripheral blood of patients with idiopathic PAP showed that
hypo-function of cellular immunity existed. By contrast, humoral
immunity was not involved with idiopathic PAP, LDH and CYFRA211,
which may be regarded as important indices to monitor the severity
of idiopathic PAP.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation (30971323) and partly
by the Project of Academic Leaders in Excellent Disciplines of
Shanghai in China (08XD1403400).
References
|
1
|
Rosen SH, Castleman B and Liebow AA:
Pulmonary alveolar proteinosis. N Engl J Med. 258:1123–1142. 1958.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carey B and Trapnell BC: The molecular
basis of pulmonary alveolar proteinosis. Clin Immunol. 135:223–235.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sakagami T, Beck D, Uchida K, et al:
Patient-derived granulocyte/ macrophage colony-stimulating factor
autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman
primates. Am J Respir Crit Care Med. 182:49–61. 2010. View Article : Google Scholar
|
|
4
|
Kitamura T, Tanaka N, Watanabe J, et al:
Idiopathic pulmonary alveolar proteinosis as an autoimmune disease
with neutralizing antibody against granulocyte/macrophage
colony-stimulating factor. J Exp Med. 190:875–880. 1999. View Article : Google Scholar
|
|
5
|
Uchida K, Nakata K, Trapnell BC, et al:
High-affinity auto-antibodies specifically eliminate
granulocyte-macrophage colony-stimulating factor activity in the
lungs of patients with idiopathic pulmonary alveolar proteinosis.
Blood. 103:1089–1098. 2004. View Article : Google Scholar
|
|
6
|
Kavuru MS, Sullivan EJ, Piccin R,
Thomassen MJ and Stoller JK: Exogenous granulocyte-macrophage
colony-stimulating factor administration for pulmonary alveolar
proteinosis. Am J Respir Crit Care Med. 161:1143–1148. 2000.
View Article : Google Scholar
|
|
7
|
Borie R, Danel C, Debray MP, et al:
Pulmonary alveolar proteinosis. Eur Respir Rev. 20:98–107. 2011.
View Article : Google Scholar
|
|
8
|
Lee KN, Levin DL, Webb WR, Chen D, Storto
ML and Golden JA: Pulmonary alveolar proteinosis: high-resolution
CT, chest radiographic, and functional correlations. Chest.
111:989–995. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Inoue Y, Trapnell BC, Tazawa R, et al:
Characteristics of a large cohort of patients with autoimmune
pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med.
177:752–762. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin FC, Chang GD, Chern MS, Chen YC and
Chang SC: Clinical significance of anti-GM-CSF antibodies in
idiopathic pulmonary alveolar proteinosis. Thorax. 61:528–534.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Costabel U and Nakata K: Pulmonary
alveolar proteinosis associated with dust inhalation: not secondary
but autoimmune? Am J Respir Crit Care Med. 181:427–428. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Byun MK, Kim DS, Kim YW, et al: Clinical
features and outcomes of idiopathic pulmonary alveolar proteinosis
in Korean population. J Korean Med Sci. 25:393–398. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen QL, Shen J, Gao Y, Guan YB, An JY and
Zheng JP: Evaluation of correlation between pulmonary function
testing and high resolution computed tomography in pulmonary
alveolar proteinosis. Zhonghua Jie He He Hu Xi Za Zhi. 31:505–508.
2008.(In Chinese).
|
|
14
|
Guan Y, Zeng Q, Yang H, et al: Pulmonary
alveolar proteinosis: Quantitative CT and pulmonary functional
correlations. Eur J Radiol. May 26–2011.(Epub ahead of print).
|
|
15
|
Schoch OD, Schanz U, Koller M, et al: BAL
findings in a patient with pulmonary alveolar proteinosis
successfully treated with GM-CSF. Thorax. 57:277–280. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu KF, Chen Y, Guo ZJ and Zhu YJ:
Autoantibody against granulocyte-macrophage colony-stimulating
factor and other serum markers in pulmonary alveolar proteinosis.
Zhonghua Jie He He Hu Xi Za Zhi. 27:824–828. 2004.(In Chinese).
|
|
17
|
Inoue Y, Nakata K, Arai T, et al:
Epidemiological and clinical features of idiopathic pulmonary
alveolar proteinosis in Japan. Respirology. 11:S55–S60. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Minakata Y, Kida Y, Nakanishi H, Nishimoto
T and Yukawa S: Change in cytokeratin 19 fragment level according
to the severity of pulmonary alveolar proteinosis. Intern Med.
40:1024–1027. 2001. View Article : Google Scholar : PubMed/NCBI
|