Biotransformation refers to a number of different
enzyme-catalyzed processes, in which the body converts endogenous
compounds, xenobiotics and toxic substances into harmless or easily
excreted metabolites. These compounds may become active
metabolites, inactive metabolic products, or metabolites with
higher or lower activity (1,2).
The reactions associated with biotransformation are
traditionally classified into two main groups. Phase I
(non-synthetic) reactions are responsible for oxidation, reduction,
hydrolysis and hydrogen removal reactions. These reactions
typically occur in the liver. The oxidation reactions include
cytochrome P450, nicotinamide adenine dinucleotide phosphate and
oxygen. Phase II (conjugation) reactions are biosynthetic and
require energy and certain cofactors. These reactions add a
relatively large polar group (typically sulfate, amino acids,
glutathione, methyl and glucuronic acid) to phase I reaction
products (3). An alternative
classification for biotransformation has been proposed according to
the nature of the reaction as functionalization reactions and
conjugation reactions. Whereas phase I reactions are associated
with unmasking a polar functional group, phase II reactions link an
endogenous polar group to a specific substrate (4).
In the majority of animal species, a set of enzymes
catalyzes these conjugation reactions. The uridine 5′-diphospho
(UDP)-glucuronosyltransferase (UGT) superfamily, which primarily
catalyzes conjugation reactions, is one of these enzyme families.
The UGT superfamily is located in the microsomal fraction of
various tissues, including the liver, kidney, skin, intestine and
brain and is quantitatively important in the liver. The catalytic
reaction of the UGT superfamily is the incorporation of a glycosyl
group (glucuronic acid, glucose, xylose or galactose) to a range of
acceptors. A number of frequently used drugs are conjugated with
glucuronic acid, which is synthesized from glucose in the soluble
fraction of the liver (5).
UDP-glucuronic acid (UDPGA) serves as the glucuronic
acid donor to various acceptors that consist of drugs and
metabolites. Although glucuronidation frequently inactivates
xenobiotics, there are exceptions, including morphine and
12-retinoic acid, which become pharmacologically active. The
conjugation with glucuronic acid is an important qualitative and
quantitative reaction due to the number of substrates that can be
modified and the wide availability of UDPGA (6,7).
The human UGT superfamily is divided into four major
families: UGT1, 2, 3 and 8, with the UGT1 and 2 families being most
important for glucuronidation reactions. The UGT2 family is
subdivided into two subfamilies (UGT2A and UGT2B). The function and
catalytic activity of the UGT3 remains unknown and the UGT8 gene
product is a UDP-galactose ceramide galactosyltransferase (8).
The role of the genetic variants of the UGT1 family,
its associated syndromes and altered drug metabolism have been well
documented. Changes in the nucleotide sequence may be located in
the promoter, regulatory, intronic or coding regions. Specific
genetic variants are associated with a number of pathologies are
described in Table I (9–31) and
those associated with drug metabolism are described in Table II (10,32–46).
The UGT1A subfamily is typically responsible for the
conjugation of bilirubin, phenols, anthraquinones, flavones,
estriol and estradiol (47). The
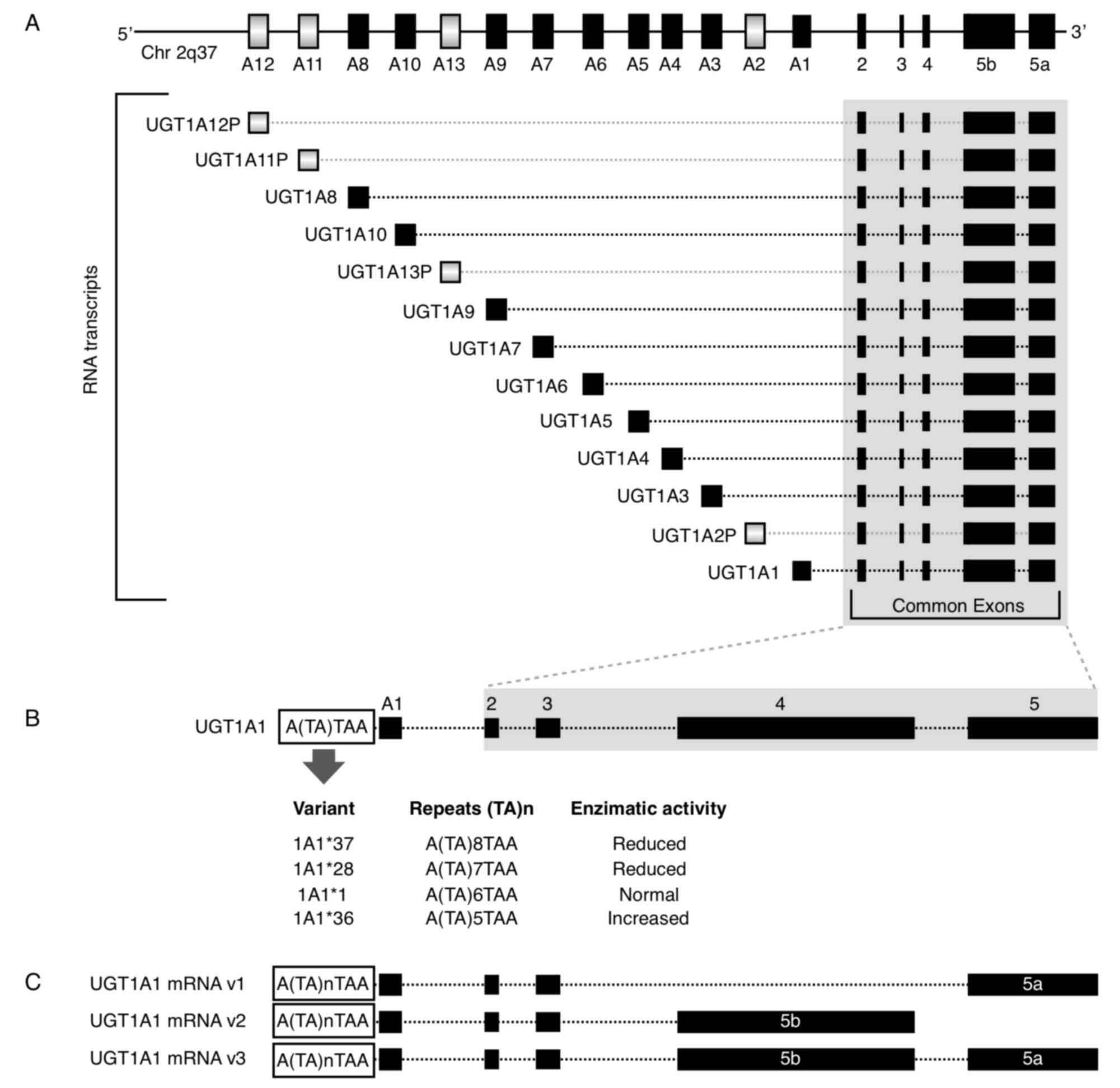
UGT1A locus is located at 2q37.1 and it contains four common exons
2–5 and 13 alternative exons (A1-A13). All combinations of one
alternative exon in addition to the four common exons have the
potential to generate 13 transcription units (Fig. 1A). However, the UGT1A locus
potentially encodes for only nine functional proteins: UGT1A1 and
UGT1A3-10 as 1A2P, 1A11P-1A13P belong to pseudogenes that do not
encode for proteins (9,48). For the mRNA 3′ region, there are two
alternative exons, termed 5a and 5b. Each of the nine potential
coded protein RNA transcripts with A1-A13 exons have the
possibility to include the 5a, 5b or 5a plus 5b variant exons,
resulting in three possible mRNAs and three putative transcripts
(49) (Fig. 1C). When only the 5a variant is
incorporated at the mRNA 3′ region; the mRNA is termed the V1
isoform and encodes for the catalytically active form. When the 5b
or 5a plus 5b variants are incorporated, they form inactive
isoforms termed v2 and v3, respectively (50). Thus, the 5b variant, alone or in
combination with 5a, results in an enzymatically inactive protein,
but it acts as a negative modulator of the 5a variant.
Variations in UGT1A1 have been studied and 136
allelic variants have been described. The variants were associated
with diminished or absent enzyme activity, resulting in clinical
implications. The ClinVar database contains a dataset with
clinically significant variants (51).
Mutations in the UGT1A1 exons or promoter region
produce structural or functional deficiencies in the enzyme, which
may result in deterioration of the conjugation. A commonly
described variant is the TA dinucleotide insertion in the TATA
element of the gene promoter (Fig.
1B). The 7 TA repeats instead of the 6 normal TA repeats
(UGT1A1*1) is designated the UGT1A1*28 allele. This variant is
associated with Gilbert's syndrome (GS), prenatal
hyperbilirubinemia and adverse events due to the metabolism of
certain drugs, including irinotecan, FOLFIRI, atazanavir,
tamoxifen, belinostat and acetaminophen (Tables I and II).
Besides the seven TA repeats variant in the UGT1A1
gene, there are additional 5 (UGT1A1*36) and 8 (UGT1A1*37) TA
repeats. It has been demonstrated that the greater the number of
repetitions, the lower the enzyme activity. Therefore, the 5, 7 and
8 repeat variants exhibit 130, 65 and 50% activity, respectively,
compared with the normal 6 repeat version (Fig. 1B) (52). The 211G>A variant (Arg71Gly,
UGT1A1*6 allele) in exon 1 has also been described and exhibits 30%
of the normal activity. This variant affects the metabolism of
7-ethyl-10-hydroxycamptothecin (SN-38), an active metabolite of
irinotecan, which is commonly employed in colon cancer treatment
and associated with GS and neonatal hyperbilirubinemia (36,53).
Genotyping patients with UGT1A1 variants is important and alerts
must be taken into account for screening in pharmacogenomics and
prior to certain drugs treatments, including irinotecan and
atazanavir (10).
The two main diseases associated with UGT1A1
variants are Crigler-Najjar syndrome (CNS) type I (-I) and type II
(-II) and GS. A previous review reported that these diseases were
associated with 77 point missense mutations, 14 point nonsense
mutations, 21 deletions, 10 insertions and 8 promoter/intronic
mutations (polymorphisms) in the UGT1A1 gene (54).
A range of diseases are associated with bilirubin
clearance, the majority of which are inherited (55); however, the elevation of serum
bilirubin is a common finding during the first week of life. This
phenomenon should be evaluated, as it may be a transitory condition
that spontaneously resolves or a serious illness. Neonatal
non-conjugated hyperbilirubinemia is a common condition in
pediatric medicine. Hemoglobin is metabolized to heme and globin
groups; heme becomes biliverdin, which in turn becomes bilirubin
(non-conjugated). Bilirubin is conjugated with glucuronic acid in
the liver, becoming conjugated bilirubin; the conjugated form
returns to the water-soluble bilirubin molecule that may be
excreted in bile (47). Failure in
bilirubin conjugation leads to an increased level of non-conjugated
bilirubin, which is less hydrosoluble and has the ability to cross
the blood-brain barrier. Although non-conjugated hyperbilirubinemia
usually is self-limiting and benign, occasionally the severe
non-conjugated hyperbilirubinemia leads to encephalopathy or
kernicterus. The causes of the non-conjugated hyperbilirubinemia
may be excessive production of billirubin during a hemolytic
process, inadequate clarification of bilirubin or a combination of
the two (56).
In newborns, the UGT1A1*28 allele is associated with
hyperbilirubinemia and jaundice. In Spain, a study of 136 newborns,
21 of them with jaundice, demonstrated that newborns with jaundice
had a tendency to have a higher prevalence of the UGT1A1*28, but
this result was not statistically significance (57). A Chinese study concluded that
different variants in the UGT1A1 gene, including UGT1A1*6,
UGT1A1*28 and minor allele T of rs887829, are associated with
bilirubin levels in the first days of life (11). Long-term studies are necessary to
identify any diseases associated with bilirubin or drug metabolism
that patients may develop in the future.
GS is a benign hereditary condition, typically
diagnosed in adolescence. This disease is characterized by
moderated non-conjugated or indirect hyperbilirubinemia, which is
defined as a bilirubin concentration between 1 and 6 mg/dl.
Bilirubin typically increases with fasting and in the presence of
normal liver enzyme levels (47). It
has been postulated that the homozygous UGT1A1*28 variant is
necessary, but not sufficient, for the clinical expression of GS.
In the general Caucasian population, ~15% are homozygous and 50%
are heterozygous for the UGT1A1*28 polymorphism; however, only
10.3% are clinically diagnosed as patients with GS (9,58). This
differential clinical manifestation may be associated with
environmental factors and individual genetic variants that exert
influence on global glucuronidation activity (41,59). GS
has also been associated with defects in the conjugation of certain
other compounds (60). Total
bilirubin levels in patients with GS are also influenced by the
−3279T>G variant (61).
UGT1A variants have been associated with an
increased risk of developing colorectal, breast, laryngeal,
orolaryngeal and proximal digestive tract cancer and hepatocellular
carcinoma (Table I). The role of
different types of UGT1A proteins in the metabolism of carcinogenic
compounds is indirectly associated with the risk of cancer
development. The UGT1A subfamily is responsible for the
glucuronidation of carcinogenic tobacco compounds, such as
benzo(α)pyrene (BaP). The BaP-trans-7R,8R-dihydrodiol [BPD(−)], the
precursor of the mutagenic compound
anti-(+)-BaP-7R,8S-dihydrodiol-9S,10R-epoxide, is primarily
metabolized by UGT1A1 and UGT1A9 gene products; and both are
expressed in the liver (10,68). Experiments conducted in normal liver
microsomes isolated from individuals with *1/*1, 1*/28 and *28/28
genotypes revealed that bilirubin glucuronidation activity and
BPD(−) glucuronide levels decreased, which suggests that the
decreased activity of UDP glucoronosyltransferases serves a role in
the detoxification of BaP and therefore, the risk of developing
cancer (68).
UGT1A1*28 and UGT1A7 polymorphisms have been
associated with the risk of colorectal cancer. The frequency of
genotypes containing the UGT1A1*28 allele in the homozygous or
heterozygous state was reported to be significantly higher in
patients with colorectal cancer compared with controls (12). UGT1A7 is expressed in
gastrointestinal and lung tissues. The UGT1A7 gene product is
associated with the metabolism of carcinogens found in diets,
including polycyclic or heterocyclic aromatic hydrocarbons and
heterocyclic amines. A previous study demonstrated that UGT1A7*2
(Lys129, Lys131 and Trp208) and *3 (Lys129, Lys131 and Arg208)
alleles are significantly associated with the risk of colorectal
cancer and this is affected by alcohol intake and cigarette smoking
(22).
Estrogen-sensitive cancers have been associated with
the production of hydroxylated estrogen metabolites, termed
catechol estrogens. Estradiol (E2) derived metabolites exhibit
different biological properties. In breast cancer (BC), E2 and its
oxidized and methoxylated metabolites are conjugated with
glucuronic acid via the UGT1A1, 3 and 8–10 and 2B7 enzymes and
these glucuronides are devoid of biologic activities. The genetic
variants of these UGT genes may influence estrogen metabolism and
the risk of BC (69,70). Also, a 150 kb deletion polymorphism
in UGT2B17, which is considered a null genotype, inhibited the
expression of the gene product and thus there was no enzyme
activity. It has been reported that this null genotype is
associated with BC; it has also been suggested that the UGT2B17
enzyme serves a role in cancer drug metabolism (71). Finally, a study reported that smoking
or alcohol consumption combined with the G allele of the UGT1A1*6
gene (rs4148323 A/G) increased the risk of laryngeal cancer
(27). Further examples of the
association between cancer and UGT variants are cited in Table I.
The aforementioned genetic variants in the UGT
families are associated with increased levels of plasma
non-conjugated bilirubin. McCarty (72) previously suggested that bilirubin
served a role as a potent antioxidant that scavenges superoxide,
peroxyl radicals, hydroxyl radicals, hypochlorous acid, singlet
oxygen and the reactive nitrogen species nitroxyl and
peroxynitrite. Initially considered as a toxic compound,
information has emerged regarding the protective role of moderately
high levels of bilirubin, as observed in patients with GS and
chronic diseases. Examples of the aforementioned protective role
include an altered lipid profile and a reduced pro-inflammatory
status (73), an inverse correlation
between serum bilirubin concentrations and the risk of certain
types of cancer (74) and a reduced
risk of ischemic heart disease and hypertension (72).
Irinotecan, an inhibitor of DNA topoisomerase I, is
used to treat patients with metastatic colorectal cancer, which is
a commonly diagnosed malignancy and one of the leading causes of
mortality associated with cancer worldwide (75). Irinotecan has also been employed in
ovarian (76), non-small lung cell
(77) and pancreatic and biliary
tract cancers (78). Irinotecan is
prescribed alone, or combined with: i) 5-fluorouracil and
Leucovorin (FOLFIRI), ii) FOLFIRI plus oxaliplatin, iii) Cetuximab,
a chimeric immunoglobulin G1 anti-epidermal growth factor receptor
monoclonal antibody (37,79), or iv) capecitabine (80). The combined effects of the genetic
variants in these drug-metabolizing enzymes need to be considered
to reduce undesirable effects and to increase the effectiveness of
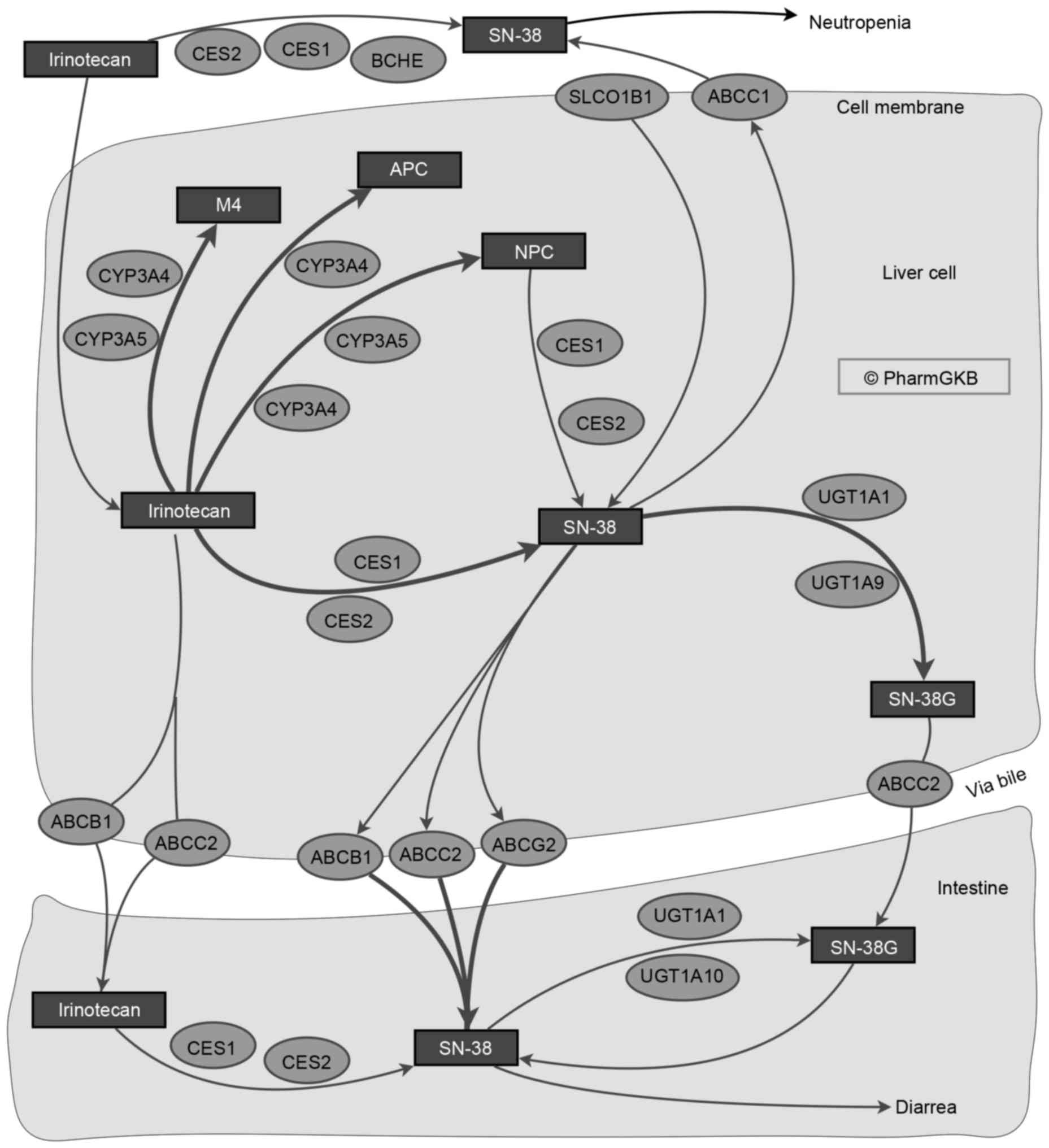
the drugs. Irinotecan is a prodrug that requires metabolism to the
active form, SN-38, which has 100-fold higher antitumor activity,
through carboxyesterases (10).
SN-38 may subsequently be inactivated by UGT via glucuronidation
(Fig. 2) (81,82).
Severe toxicity has been reported in <36% of patients treated
with irinotecan and the UGT1A1*28 allele is associated with
toxicity in a dose-dependent manner. However, other UGTA1 variants
may be associated with this toxicity (83).
Patients homozygous for UGT1A1*28 or UGT1A1*6 allele
may receive irinotecan at an initial dose of 150 mg/m2,
but a reduction in the dose of subsequent cycles or a delay in the
treatment is required (84). The
aforementioned alleles have been associated with irinotecan-induced
neutropenia in patients with colon cancer (85). Routine genotyping prior to
chemotherapy has been used to prevent febrile neutropenia in
patients with metastatic colorectal cancer at a reasonable cost
(85). Furthermore, the
administration of granulocyte-colony stimulating factor to patients
with homozygous UGT1A1*28 may prevent the development of
neutropenia (86). Finally, the
UGT1A7 gene product, which is expressed in extrahepatic tissues,
including the esophagus, stomach and lung, has been demonstrated to
be associated with the metabolism of irinotecan to its non-toxic
metabolite, SN-38 (87).
PEG-V is a pegylated recombinant analog of human
growth hormone (hGH), with covalently bonded polyethylene glycol
polymer chains that reduce immunogenicity and the rate of clearance
from the body, prolonging half-life (88). PEG-V is a modified version of hGH
designed to bind to and inhibit the hGH receptor. In patients with
acromegalia, PEG-V alone or in combination with a somatostatin
analog has an efficacy of >90% for the control insulin-like
growth factor (89,90). Liver injury has been reported with
PEG-V in patients with Gilberts´Syndrome or the UGT1A1*28 genotype
(91–94).
The UGT1A1*28 allele is associated with atazanavir
metabolism. The UGT1A1*28 allele, a low CD4 cell count and the
presence of the G2677T/A variant of the multi-drug resistance gene
(MDR)1, were independent risk factors for severe hyperbilirubinemia
in Korean patients infected with human immunodeficiency virus,
while the normal (MDR)1 and UGT1A1 alleles did not exhibit this
condition (95). The UGT1A1*28
allele, a low CD4 cell count and the presence of the variant MDR1
G2677T/A in a 30 months follow-up study suggested that
hyperbilirubinemia associated with atazanavir was common, but
transient in Korean population that exhibit a low frequency of the
UGT1A1*28 allele (96).
Studies continue to provide information regarding
the association between the family of UGT enzymes, which are
associated with the metabolism of drugs, xenobiotics and endogenous
compounds and the effects of DNA variants on enzyme activity. In
the current review, the importance of the UGT complex, which is
associated with drug and xenobiotic metabolism and diseases
associated with anomalies in the conjugation of bilirubin, was
described. Also, the frequencies of genetic variants in population
studies suggest that clinical significance depends on ethnicity.
The diversity in the frequency of certain genetic variants by
ethnicity may lead to a greater understanding of the role of
patients' genetic backgrounds, the development of therapies
according to pharmacogenomics profiles of patients and improved
adverse event prediction due to drug metabolism. The
pharmacogenomics profiles of UGT1A1 may improve the quality of life
of patients, prevent adverse effects and reduce the cost of
treating patients with associated diseases. The design of
nanoparticles for the treatment of diseases, such as cancer,
according to the pharmacogenomics profile and ethnicity of a
patient is a notable opportunity for advancement in treatment
options (97). In certain cases, the
Food and Drug Administration has made changes to the labels of
specific prescription drugs, such as irinotecan, warning that there
may be a need for genotyping variants of UGT1A1 enzymes prior to
the administration of the chemotherapeutic agent. In the future,
the recommendation for individual genotyping prior to drug
administration may often be prescribed.
Not applicable.
No funding was received.
Not applicable.
CNSD and ROL were responsible for the conception of
the work, acquisition, analysis and interpretation of data for the
review, drafting the work, revising it critically for important
intellectual content, and gave final approval of the version to be
published. MASS was responsible for the analysis and interpretation
of data for the review, drafting the work and gave final approval
of the version to be published. HLGB was responsible for the
analysis and interpretation of data for the review, drafting the
work, figure editing, permissions, revising it critically for
important intellectual content, and gave final approval of the
version to be published.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Obach RS: Pharmacologically active drug
metabolites: Impact on drug discovery and pharmacotherapy.
Pharmacol Rev. 65:578–640. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Laizure SC, Herring V, Hu Z, Witbrodt K
and Parker RB: The role of human carboxylesterases in drug
metabolism: Have we overlooked their importance? Pharmacotherapy.
33:210–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gustavsson L: Pharmacogenomics in drug
developmentgenomics and proteomics for clinical discovery and
development. Springer; New York, NY: pp. 225–241. 2014
|
|
4
|
Rowland A, Miners JO and Mackenzie PI: The
UDP-glucuronosyltransferases: Their role in drug metabolism and
detoxification. Int J Biochem Cell Biol. 45:1121–1132. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaivosaari S, Finel M and Koskinen M:
N-glucuronidation of drugs and other xenobiotics by human and
animal UDP-glucuronosyltransferases. Xenobiotica. 41:652–669. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ishii Y, Nurrochmad A and Yamada H:
Modulation of UDP-glucuronosyltransferase activity by endogenous
compounds. Drug Metab Pharmacokinet. 25:134–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang TKH: Drug-metabolizing
enzymesHandbook of drug-nutrient interactions. Boullata IJ and
Armenti TV: Humana Press; Totowa, NJ: pp. 85–117. 2010
|
|
8
|
Mackenzie PI, Bock KW, Burchell B,
Guillemette C, Ikushiro S, Iyanagi T, Miners JO, Owens IS and
Nebert DW: Nomenclature update for the mammalian UDP
glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics.
15:677–685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bosma PJ, Chowdhury JR, Bakker C, Gantla
S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Elferink
Oude RP, et al: The genetic basis of the reduced expression of
bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N
Engl J Med. 333:1171–1175. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barbarino JM, Haidar CE, Klein TE and
Altman RB: PharmGKB summary: Very important pharmacogene
information for UGT1A1. Pharmacogenet Genomics. 24:177–183. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Y, Wang SN, Li H, Zha W, Peng Q, Li
S, Chen Y and Jin L: Quantitative trait analysis of polymorphisms
in two bilirubin metabolism enzymes to physiologic bilirubin levels
in Chinese newborns. J Pediatr. 165:1154–1160.e1. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bajro MH, Josifovski T, Panovski M,
Jankulovski N, Nestorovska AK, Matevska N, Petrusevska N and
Dimovski AJ: Promoter length polymorphism in UGT1A1 and the risk of
sporadic colorectal cancer. Cancer Genet. 205:163–167. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Monaghan G, Ryan M, Seddon R, Hume R and
Burchell B: Genetic variation in bilirubin
UPD-glucuronosyltransferase gene promoter and Gilbert's syndrome.
Lancet. 347:578–581. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shatalova EG, Loginov VI, Braga EA,
Kazubskaia TP, Sudomoina MA, Blanchard RL and Favorova OO:
Association of polymorphisms in SULT1A1 and UGT1A1 Genes with
breast cancer risk and phenotypes in Russian women. Mol Biol
(Mosk). 40:263–270. 2006.(In Russian). View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen YH, Hung SC and Tarng DC: Serum
bilirubin links UGT1A1*28 polymorphism and predicts long-term
cardiovascular events and mortality in chronic hemodialysis
patients. Clin J Am Soc Nephrol. 6:567–574. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Petersen JP, Ebbesen F, Hollegaard MV,
Andersson S, Hougaard DM, Thorlacius-Ussing O and Henriksen TB:
UGT1A1*28 genotypes and respiratory disease in very preterm
infants: A cohort study. Neonatology. 109:124–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
do Sameiro-Faria M, Kohlova M, Ribeiro S,
Rocha-Pereira P, Teixeira L, Nascimento H, Reis F, Miranda V,
Bronze-da-Rocha E, Quintanilha A, et al: Potential cardiovascular
risk protection of bilirubin in end-stage renal disease patients
under hemodialysis. Biomed Res Int. 2014:1752862014.PubMed/NCBI
|
|
18
|
Torrecilla Lodoso B, Atance Palomo E,
Grande Camarena C, Díaz Fernández MC, Llanillo Hierro L, De la Vega
Bueno A, Remacha Frauca E, Bartolo Muñoz G and Vega Jara P:
Crigler-Najjar syndrome: Diagnosis and treatment. An Pediatr
(Barc). 65:73–78. 2006.(In Spanish). View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ciotti M, Chen F, Rubaltelli FF and Owens
IS: Coding defect and a TATA box mutation at the bilirubin
UDP-glucuronosyltransferase gene cause Crigler-Najjar type I
disease. Biochim Biophys Acta. 1407:40–50. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Petit FM, Hébert M, Gajdos V, Capel L,
M'Rad R and Labrune P: Large deletion in UGT1A1 gene encompassing
the promoter and the exon 1 responsible for Crigler-Najjar type I
syndrome. Haematologica. 93:1590–1591. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ko JS, Chang JY, Moon JS, Yang HR and Seo
JK: Molecular analysis of the UGT1A1 gene in Korean patients with
Crigler-Najjar syndrome type II. Pediatr Gastroenterol Hepatol
Nutr. 17:37–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen K, Jin M, Zhu Y, Jiang Q, Yu W, Ma X
and Yao K: Genetic polymorphisms of the uridine diphosphate
glucuronosyltransferase 1A7 and colorectal cancer risk in relation
to cigarette smoking and alcohol drinking in a Chinese population.
J Gastroenterol Hepatol. 21:1036–1041. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vogel A, Kneip S, Barut A, Ehmer U, Tukey
RH, Manns MP and Strassburg CP: Genetic link of hepatocellular
carcinoma with polymorphisms of the UDP-glucuronosyltransferase
UGT1A7 gene. Gastroenterology. 121:1136–1144. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng Z, Park JY, Guillemette C, Schantz
SP and Lazarus P: Tobacco carcinogen-detoxifying enzyme UGT1A7 and
its association with orolaryngeal cancer risk. J Natl Cancer Inst.
93:1411–1418. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Strassburg CP, Vogel A, Kneip S, Tukey RH
and Manns MP: Polymorphisms of the human
UDP-glucuronosyltransferase (UGT) 1A7 gene in colorectal cancer.
Gut. 50:851–856. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vogel A, Ockenga J, Ehmer U, Barut A,
Kramer FJ, Tukey RH, Manns MP and Strassburg CP: Polymorphisms of
the carcinogen detoxifying UDP-glucuronosyltransferase UGT1A7 in
proximal digestive tract cancer. Z Gastroenterol. 40:497–502. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huangfu H, Pan H, Wang B, Wen S, Han R and
Li L: Association between UGT1A1 polymorphism and risk of laryngeal
squamous cell carcinoma. Int J Environ Res Public Health.
13:E1122016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maruo Y, Morioka Y, Fujito H, Nakahara S,
Yanagi T, Matsui K, Mori A, Sato H, Tukey RH and Takeuchi Y:
Bilirubin uridine diphosphate-glucuronosyltransferase variation is
a genetic basis of breast milk jaundice. J Pediatr. 165:36–41.e1.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang H, Wang Q, Zheng L, Zheng XB, Lin M,
Zhan XF and Yang LY: Clinical significance of UGT1A1 genetic
analysis in chinese neonates with severe hyperbilirubinemia.
Pediatr Neonatol. 57:310–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Azlin I, Wong FL, Ezham M, Hafiza A and
Ainoon O: Prevalence of uridine glucuronosyl transferase 1A1
(UGT1A1) mutations in Malay neonates with severe jaundice. Malays J
Pathol. 33:95–100. 2011.PubMed/NCBI
|
|
31
|
Yu Z, Zhu K, Wang L, Liu Y and Sun J:
Association of neonatal hyperbilirubinemia with UGT1A1 gene
polymorphisms: A meta-analysis. Med Sci Monit. 21:3104–3114. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ando Y, Saka H, Asai G, Sugiura S,
Shimokata K and Kamataki T: UGT1A1 genotypes and glucuronidation of
SN-38, the active metabolite of irinotecan. Ann Oncol. 9:845–847.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iyer L, King CD, Whitington PF, Green MD,
Roy SK, Tephly TR, Coffman BL and Ratain MJ: Genetic predisposition
to the metabolism of irinotecan (CPT-11). Role of uridine
diphosphate glucuronosyltransferase isoform 1A1 in the
glucuronidation of its active metabolite (SN-38) in human liver
microsomes. J Clin Invest. 101:847–854. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Innocenti F, Undevia SD, Iyer L, Chen PX,
Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM,
et al: Genetic variants in the UDP-glucuronosyltransferase 1A1 gene
predict the risk of severe neutropenia of irinotecan. J Clin Oncol.
22:1382–1388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lankisch TO, Schulz C, Zwingers T,
Erichsen TJ, Manns MP, Heinemann V and Strassburg CP: Gilbert's
syndrome and irinotecan toxicity: Combination with
UDP-glucuronosyltransferase 1A7 variants increases risk. Cancer
Epidemiol Biomarkers Prev. 17:695–701. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jinno H, Tanaka-Kagawa T, Hanioka N, Saeki
M, Ishida S, Nishimura T, Ando M, Saito Y, Ozawa S and Sawada J:
Glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38), an
active metabolite of irinotecan (CPT-11), by human UGT1A1 variants,
G71R, P229Q, and Y486D. Drug Metab Dispos. 31:108–113. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wen F and Li Q: Treatment dilemmas of
cetuximab combined with chemotherapy for metastatic colorectal
cancer. World J Gastroenterol. 22:5332–5341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu C, Tang X, Qu Y, Keyoumu S, Zhou N and
Tang Y: UGT1A1 gene polymorphism is associated with toxicity and
clinical efficacy of irinotecan-based chemotherapy in patients with
advanced colorectal cancer. Cancer Chemother Pharmacol. 78:119–130.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pacheco PR, Brilhante MJ, Ballart C,
Sigalat F, Polena H, Cabral R, Branco CC and Mota-Vieira L: UGT1A1,
UGT1A6 and UGT1A7 genetic analysis: Repercussion for irinotecan
pharmacogenetics in the Sao Miguel Island population (Azores,
Portugal). Mol Diagn Ther. 13:261–268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rotger M, Taffe P, Bleiber G, Gunthard HF,
Furrer H, Vernazza P, Drechsler H, Bernasconi E, Rickenbach M and
Telenti A: Swiss HIV Cohort Study: Gilbert syndrome and the
development of antiretroviral therapy-associated
hyperbilirubinemia. J Infect Dis. 192:1381–1386. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lankisch TO, Moebius U, Wehmeier M,
Behrens G, Manns MP, Schmidt RE and Strassburg CP: Gilbert's
disease and atazanavir: From phenotype to
UDP-glucuronosyltransferase haplotype. Hepatology. 44:1324–1332.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Romero-Lorca A, Novillo A, Gaibar M,
Bandrés F and Fernández-Santander A: Impacts of the glucuronidase
genotypes UGT1A4, UGT2B7, UGT2B15 and UGT2B17 on tamoxifen
metabolism in breast cancer patients. PLoS One. 10:e01322692015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sutiman N, Lim JS, Muerdter TE, Singh O,
Cheung YB, Ng RCH, Yap YS, Wong NS, Ang PCS, Dent R, et al:
Pharmacogenetics of UGT1A4, UGT2B7 and UGT2B15 and their influence
on tamoxifen disposition in asian breast cancer patients. Clin
Pharmacokinet. 55:1239–1250. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Goey AK and Figg WD: UGT genotyping in
belinostat dosing. Pharmacol Res. 105:22–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Goey AK, Sissung TM, Peer CJ, Trepel JB,
Lee MJ, Tomita Y, Ehrlich S, Bryla C, Balasubramaniam S, Piekarz R,
et al: Effects of UGT1A1 genotype on the pharmacokinetics,
pharmacodynamics, and toxicities of belinostat administered by
48-hour continuous infusion in patients with cancer. J Clin
Pharmacol. 56:461–473. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Court MH, Freytsis M, Wang X, Peter I,
Guillemette C, Hazarika S, Duan SX, Greenblatt DJ and Lee WM: Acute
Liver Failure Study Group: The UDP-glucuronosyltransferase (UGT) 1A
polymorphism c.2042C>G (rs8330) is associated with increased
human liver acetaminophen glucuronidation, increased UGT1A exon
5a/5b splice variant mRNA ratio, and decreased risk of
unintentional acetaminophen-induced acute liver failure. J
Pharmacol Exp Ther. 345:297–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kaplan M, Hammerman C and Maisels MJ:
Bilirubin genetics for the nongeneticist: Hereditary defects of
neonatal bilirubin conjugation. Pediatrics. 111:886–893. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jancova P, Anzenbacher P and
Anzenbacherova E: Phase II drug metabolizing enzymes. Biomed Pap
Med Fac Univ Palacky Olomouc Czech Repub. 154:103–116. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tourancheau A, Margaillan G, Rouleau M,
Gilbert I, Villeneuve L, Lévesque E, Droit A and Guillemette C:
Unravelling the transcriptomic landscape of the major phase II
UDP-glucuronosyltransferase drug metabolizing pathway using
targeted RNA sequencing. Pharmacogenomics J. 16:60–70. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Girard H, Lévesque E, Bellemare J,
Journault K, Caillier B and Guillemette C: Genetic diversity at the
UGT1 locus is amplified by a novel 3′ alternative splicing
mechanism leading to nine additional UGT1A proteins that act as
regulators of glucuronidation activity. Pharmacogenet Genomics.
17:1077–1089. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Landrum MJ, Lee JM, Riley GR, Jang W,
Rubinstein WS, Church DM and Maglott DR: ClinVar: Public archive of
relationships among sequence variation and human phenotype. Nucleic
Acids Res. 42:(Database Issue). D980–D985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Beutler E, Gelbart T and Demina A: Racial
variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter:
A balanced polymorphism for regulation of bilirubin metabolism?
Proc Natl Acad Sci USA. 95:8170–8174. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sai K and Saito Y: Ethnic differences in
the metabolism, toxicology and efficacy of three anticancer drugs.
Expert Opin Drug Metab Toxicol. 7:967–988. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Canu G, Minucci A, Zuppi C and Capoluongo
E: Gilbert and Crigler Najjar syndromes: An update of the
UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database.
Blood Cells Mol Dis. 50:273–280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Memon N, Weinberger BI, Hegyi T and
Aleksunes LM: Inherited disorders of bilirubin clearance. Pediatr
Res. 79:378–386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Maisels MJ: Managing the jaundiced
newborn: A persistent challenge. CMAJ. 187:335–343. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Seco ML, del Río E, Barceló MJ, Remacha A,
Ginovart G, Moliner E and Baiget M: Interest in the study of
genetic variants of the promoter region of the UGT1A1 gene in
neonatal jaundice. An Esp Pediatr. 56:139–143. 2002.(In Spanish).
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ramos-Leví AM, Bernabeu I, Sampedro-Núñez
M and Marazuela M: Genetic predictors of response to different
medical therapies in acromegaly. Prog Mol Biol Transl Sci.
138:85–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Köhle C, Möhrle B, Münzel PA, Schwab M,
Wernet D, Badary OA and Bock KW: Frequent co-occurrence of the TATA
box mutation associated with Gilbert's syndrome (UGT1A1*28) with
other polymorphisms of the UDP-glucuronosyltransferase-1 locus
(UGT1A6*2 and UGT1A7*3) in Caucasians and Egyptians. Biochem
Pharmacol. 65:1521–1527. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bosma PJ: Inherited disorders of bilirubin
metabolism. J Hepatol. 38:107–117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rodrigues C, Vieira E, Santos R, de
Carvalho J, Santos-Silva A, Costa E and Bronze-da-Rocha E: Impact
of UGT1A1 gene variants on total bilirubin levels in Gilbert
syndrome patients and in healthy subjects. Blood Cells Mol Dis.
48:166–172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ciotti M, Werlin SL and Owens IS: Delayed
response to phenobarbital treatment of a Crigler-Najjar type II
patient with partially inactivating missense mutations in the
bilirubin UDP-glucuronosyltransferase gene. J Pediatr Gastroenterol
Nutr. 28:210–213. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Maruo Y, Nakahara S, Yanagi T, Nomura A,
Mimura Y, Matsui K, Sato H and Takeuchi Y: Genotype of UGT1A1 and
phenotype correlation between Crigler-Najjar syndrome type II and
Gilbert syndrome. J Gastroenterol Hepatol. 31:403–408. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bosma PJ, Chowdhury NR, Goldhoorn BG,
Hofker MH, Elferink Oude RP, Jansen PL and Chowdhury JR: Sequence
of exons and the flanking regions of human
bilirubin-UDP-glucuronosyltransferase gene complex and
identification of a genetic mutation in a patient with
Crigler-Najjar syndrome, type I. Hepatology. 15:941–947. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bosma PJ, Goldhoorn B, Elferink Oude RP,
Sinaasappel M, Oostra BA and Jansen PL: A mutation in bilirubin
uridine 5′-diphosphate-glucuronosyltransferase isoform 1 causing
Crigler-Najjar syndrome type II. Gastroenterology. 105:216–220.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Petit F, Gajdos V, Capel L, Parisot F,
Myara A, Francoual J and Labrune P: Crigler-Najjar type II syndrome
may result from several types and combinations of mutations in the
UGT1A1 gene. Clin Genet. 69:525–527. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Servedio V, d'Apolito M, Maiorano N,
Minuti B, Torricelli F, Ronchi F, Zancan L, Perrotta S, Vajro P,
Boschetto L and Iolascon A: Spectrum of UGT1A1 mutations in
Crigler-Najjar (CN) syndrome patients: Identification of twelve
novel alleles and genotype-phenotype correlation. Hum Mutat.
25:3252005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fang JL and Lazarus P: Correlation between
the UDP-glucuronosyltransferase (UGT1A1) TATAA box polymorphism and
carcinogen detoxification phenotype: significantly decreased
glucuronidating activity against benzo(a)pyrene-7,8-dihydrodiol(−)
in liver microsomes from subjects with the UGT1A1*28 variant.
Cancer Epidemiol Biomarkers Prev. 13:102–109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Thibaudeau J, Lépine J, Tojcic J, Duguay
Y, Pelletier G, Plante M, Brisson J, Têtu B, Jacob S, Perusse L, et
al: Characterization of common UGT1A8, UGT1A9, and UGT2B7 variants
with different capacities to inactivate mutagenic 4-hydroxylated
metabolites of estradiol and estrone. Cancer Res. 66:125–133. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Guillemette C, Bélanger A and Lépine J:
Metabolic inactivation of estrogens in breast tissue by
UDP-glucuronosyltransferase enzymes: An overview. Breast Cancer
Res. 6:246–254. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
71
|
Eskandari-Nasab E, Hashemi M, Rezaei H,
Fazaeli A, Mashhadi MA, Moghaddam SS, Arbabi F, Jahantigh M and
Taheri M: Evaluation of UDP-glucuronosyltransferase 2B17 (UGT2B17)
and dihydrofolate reductase (DHFR) genes deletion and the
expression level of NGX6 mRNA in breast cancer. Mol Biol Rep.
39:10531–10539. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
McCarty MF: ‘Iatrogenic Gilbert
syndrome’-a strategy for reducing vascular and cancer risk by
increasing plasma unconjugated bilirubin. Med Hypotheses.
69:974–994. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wallner M, Marculescu R, Doberer D, Wolzt
M, Wagner O, Vitek L, Bulmer AC and Wagner KH: Protection from
age-related increase in lipid biomarkers and inflammation
contributes to cardiovascular protection in Gilbert's syndrome.
Clin Sci (Lond). 125:257–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zucker SD, Horn PS and Sherman KE: Serum
bilirubin levels in the U.S. population: Gender effect and inverse
correlation with colorectal cancer. Hepatology. 40:827–835. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Glimelius B, Garmo H, Berglund A,
Fredriksson LA, Berglund M, Kohnke H, Byström P, Sørbye H and
Wadelius M: Prediction of irinotecan and 5-fluorouracil toxicity
and response in patients with advanced colorectal cancer.
Pharmacogenomics J. 11:61–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ushijima K, Kamura T, Tamura K, Kuzuya K,
Sugiyama T, Noda K and Ochiai K: Docetaxel/irinotecan combination
chemotherapy in platinum/taxane-refractory and -resistant ovarian
cancer: JGOG/WJGOG intergroup study. Int J Clin Oncol. 18:126–131.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Osawa K: Gene polymorphisms and
chemotherapy in non-small cell lung cancer. Zhongguo Fei Ai Za Zhi.
12:837–840. 2009.PubMed/NCBI
|
|
78
|
Yang C, Liu Y, Xi WQ, Zhou CF, Jiang JL,
Ma T, Ye ZB, Zhang J and Zhu ZG: Relationship between UGT1A1*6/*28
polymorphisms and severe toxicities in Chinese patients with
pancreatic or biliary tract cancer treated with
irinotecan-containing regimens. Drug Des Devel Ther. 9:3677–3683.
2015.PubMed/NCBI
|
|
79
|
Phelip JM, Mineur L, De la Fouchardière C,
Chatelut E, Quesada JL, Roblin X, Pezet D, Mendoza C, Buc E and
Rivoire M: High resectability rate of initially unresectable
colorectal liver metastases after UGT1A1-adapted high-dose
irinotecan combined with LV5FU2 and cetuximab: A multicenter phase
II study (ERBIFORT). Ann Surg Oncol. 23:2161–2166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chiorean EG, Sanghani S, Schiel MA, Yu M,
Burns M, Tong Y, Hinkle DT, Coleman N, Robb B, LeBlanc J, et al:
Phase II and gene expression analysis trial of neoadjuvant
capecitabine plus irinotecan followed by capecitabine-based
chemoradiotherapy for locally advanced rectal cancer: Hoosier
oncology group GI03-53. Cancer Chemother Pharmacol. 70:25–32. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Whirl-Carrillo M, McDonagh EM, Hebert JM,
Gong L, Sangkuhl K, Thorn CF, Altman RB and Klein TE:
Pharmacogenomics knowledge for personalized medicine. Clin
Pharmacol Ther. 92:414–417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lowenberg D, Thorn CF, Whirl-Carrillo M,
Ramirez J, Gong L, Marsh S, Schuetz EG, Dolan ME, Innocenti F,
McLeod HL and Ratain MJ: Irinotecan Pathway, Pharmacokinetics:
Pharmacogenomics Knowledge Base (PharmGKB) and Stanford University.
https://www.pharmgkb.org/pathway/PA2001December
19–2016
|
|
83
|
Marsh S and Hoskins JM: Irinotecan
pharmacogenomics. Pharmacogenomics. 11:1003–1010. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Satoh T, Ura T, Yamada Y, Yamazaki K,
Tsujinaka T, Munakata M, Nishina T, Okamura S, Esaki T, Sasak Y, et
al: Genotype-directed, dose-finding study of irinotecan in cancer
patients with UGT1A1*28 and/or UGT1A1*6 polymorphisms. Cancer Sci.
102:1868–1873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Atasilp C, Chansriwong P, Sirachainan E,
Reungwetwattana T, Chamnanphon M, Puangpetch A, Wongwaisayawan S
and Sukasem C: Correlation of UGT1A1(*)28 and (*)6 polymorphisms
with irinotecan-induced neutropenia in Thai colorectal cancer
patients. Drug Metab Pharmacokinet. 31:90–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Pichereau S, Le Louarn A, Lecomte T,
Blasco H, Le Guellec C and Bourgoin H: Cost-Effectiveness of
UGT1A1*28 genotyping in preventing severe neutropenia following
FOLFIRI therapy in colorectal cancer. J Pharm Pharm Sci.
13:615–625. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Paulík A, Grim J and Filip S: Predictors
of irinotecan toxicity and efficacy in treatment of metastatic
colorectal cancer. Acta Medica (Hradec Kralove). 55:153–159. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Banerjee SS, Aher N, Patil R and Khandare
J: Poly(ethylene glycol)-prodrug conjugates: Concept, design, and
applications. J Drug Deliv. 2012:1039732012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Giustina A, Ambrosio MR, Peccoz Beck P,
Bogazzi F, Cannavo' S, De Marinis L, De Menis E, Grottoli S and
Pivonello R: Use of Pegvisomant in acromegaly. An Italian society
of endocrinology guideline. J Endocrinol Invest. 37:1017–1030.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Buchfelder M, Schlaffer S, Droste M, Mann
K, Saller B, Brübach K, Stalla GK and Strasburger CJ:
GermanPegvisomant Observational Study: The German ACROSTUDY: Past
and present. Eur J Endocrinol. 161 Suppl 1:S3–S10. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mallea-Gil MS, Bernabeu I, Spiraquis A,
Avangina A, Loidi L and Ballarino C: Pegvisomant-induced
cholestatic hepatitis in an acromegalic patient with UGT1A1 () 28
mutation. Case Rep Endocrinol. 2016:20871022016.PubMed/NCBI
|
|
92
|
Bernabeu I, Marazuela M, Lucas T, Loidi L,
Alvarez-Escolá C, Luque-Ramírez M, Fernandez-Rodriguez E, Paniagua
AE, Quinteiro C and Casanueva FF: Pegvisomant-induced liver injury
is related to the UGT1A1*28 polymorphism of Gilbert's syndrome. J
Clin Endocrinol Metab. 95:2147–2154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Filopanti M, Barbieri AM, Mantovani G,
Corbetta S, Gasco V, Ragonese M, Martini C, Bogazzi F, Colao A,
Ferone D, et al: Role of UGT1A1 and ADH gene polymorphisms in
pegvisomant-induced liver toxicity in acromegalic patients. Eur J
Endocrinol. 170:247–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bernabeu I, Cameselle-Teijeiro J,
Casanueva FF and Marazuela M: Pegvisomant-induced cholestatic
hepatitis with jaundice in a patient with Gilbert's syndrome. Eur J
Endocrinol. 160:869–872. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Park WB, Choe PG, Song KH, Jeon JH, Park
SW, Kim HB, Kim NJ, Oh MD and Choe KW: Genetic factors influencing
severe atazanavir-associated hyperbilirubinemia in a population
with low UDP-glucuronosyltransferase 1A1*28 allele frequency. Clin
Infect Dis. 51:101–106. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
96
|
Choe PG, Park WB, Song JS, Kim NH, Song
KH, Park SW, Kim HB, Kim NJ and Oh MD: Incidence of
atazanavir-associated hyperbilirubinemia in Korean HIV patients: 30
months follow-up results in a population with low
UDP-glucuronosyltransferase1A1*28 allele frequency. J Korean Med
Sci. 25:1427–1430. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sanchez-Dominguez CN, Gallardo-Blanco HL,
Rodriguez-Rodriguez AA, Vela-Gonzalez AV and Sanchez-Dominguez M:
Nanoparticles vs. cancer: A multifuncional tool. Curr Top Med Chem.
14:664–675. 2014. View Article : Google Scholar : PubMed/NCBI
|