Introduction
As a barrier between the vessel lumen and the
surrounding tissue, the vascular endothelium is the primary
participant in, and regulator of, vascular inflammatory reactions
(1). Vascular inflammation has been
shown to be involved in the progression of various cardiovascular
diseases, including atherosclerosis (2). Tumor necrosis factor (TNF)-α is a
pleiotropic, pro-inflammatory cytokine. It serves a critical role
in the disruption of vascular function and subsequent inflammatory
responses by triggering several intracellular signaling pathways;
this may ultimately stimulate the expression of adhesion molecules,
particularly vascular cell adhesion molecule-1 (VCAM-1) and
cytokines such as interleukin (IL)-1β, IL-6 and monocyte
chemotactic protein 1 (MCP-1) (3–5). These
molecules recruit monocytes to the endothelial cell surface,
resulting in inflammation and, ultimately, atherosclerosis
(6). NF-κB and mitogen-activated
protein kinases (MAPKs) serve pivotal roles in the progression of
vascular inflammation by mediating the expression of adhesion
molecules and chemokines in vascular endothelial cells (7,8). Both
are essential for the transcriptional regulation of factors induced
by TNF-α (9); therefore, compounds
that can suppress TNF-α-induced MAPK and NF-κB activation represent
promising candidates for the treatment and prevention of vascular
endothelial dysfunction and inflammation.
Salidroside, a principal active ingredient isolated
from the root of the Rhodiola rosea plant, has been reported
to exert various physiological and pharmacological effects. A
review summarized that salidroside was of benefit to patients with
diabetes mellitus, as it was able to regulate 5′-AMP-activated
protein kinase pathway-mediated glycolipid metabolism, oxidative
stress and inflammatory responses (10). A previous study also demonstrated
that salidroside retarded the proliferation of breast cancer cells
by inhibiting MAPK pathway activation (11). In terms of cardiac protection, one
report confirmed that salidroside alleviates lipopolysaccharide
(LPS)-induced cardiac injury by regulating the PI3K/AKT/mTOR
pathway (12). Notably, salidroside
has been shown to possess strong anti-inflammatory properties. A
previous study proposed that salidroside exerts anti-inflammatory
effects on macrophages by blocking MAPK and NF-κB activation, and
subsequently reducing the secretion of inflammatory cytokines
(13). However, the effect of
salidroside against TNF-α-stimulated vascular inflammation in CMECs
remains to be elucidated. The present study details an
investigation into the protective effects of salidroside on CMECs
by regulating the production of pro-inflammatory cytokines and
adhesion molecules. The results may provide novel ideas and
intervention targets for the recognition and treatment of
pathogenesis associated with vascular inflammation.
Materials and methods
Reagents
Salidroside was purchased from Yuanye Biotechnology
Co., Ltd. Recombinant rabbit TNF-α was purchased from Novoprotein.
High-glucose DMEM, RPMI 1640 medium, FBS and trypsin were purchased
from Thermo Fisher Scientific, Inc. The primary antibody against
factor VIII (cat. no. sc-14014) was purchased from Santa Cruz
Biotechnology, Inc., and anti-VCAM-1 (cat. no. ab134047) was
obtained from Abcam. Antibodies against p38 (cat. no. 8690),
phospho-p38 (cat. no. 4511), p44/42 (cat. no. 4695), phospho-p44/42
(cat. no. 4370), stress-activated protein kinase (SAPK)/JNK (cat.
no. 9252), phospho-SAPK/JNK (cat. no. 9255), inhibitor of NF-κB
(IκB)α (cat. no. 4814), phospho-IκBα (cat. no. 2589), NF-κB p65
(cat. no. 8242) and phospho-NF-κB p65 (cat. no. 3033) were obtained
from Cell Signaling Technology, Inc. The PrimeScript™ RT Reagent
kit and SYBR® Premix Ex Taq™ kit were purchased from
Takara Bio, Inc. The ELISA kits were purchased from MultiSciences
Biotech Co., Ltd. All other chemicals were of analytical grade.
Isolation and culture of CMECs
The isolation and primary culture of CMECs were
performed as previously described (14). All experiments related to animals
were conducted in accordance with the Guidelines to Laboratory
Animal Research of Fudan University and were approved by the
Institutional Animal Care and Use Committee of Fudan University. A
total of 50 2-week-old Sprague Dawley rats were purchased from
Shanghai Jiesijie Experimental Animal Co., Ltd., and were housed
under a 12:12 h light: Dark cycle with controlled temperature
(21–25°C), humidity (50±5%) and free access to food and water in
the Department of Laboratory Animal Science at Fudan University.
Animal health and behavior were monitored every day. The 2-week-old
male Sprague Dawley rats (30–40 g) were sacrificed by cervical
dislocation after anesthesia with 5% isoflurane, and the heart was
immediately excised and rinsed with PBS pre-cooled to 4°C. After
removal of the atrial tissues, great vessels, epicardium and
endocardium, the remaining ventricular tissues were cut into
1-mm3 pieces and plated in a 10-cm culture dish
pre-coated with 1 ml FBS. The tissues were then incubated at 37°C
(5% CO2) for 4 h, and for an additional 48 h in
high-glucose DMEM (10% FBS). After a further 48 h, when the cells
had reached a confluence of 80%, the tissue pieces were removed and
the cells were passaged using trypsin (0.25%). The second
generation of cells was used for subsequent experimentation. THP-1
monocytic cells (American Type Culture Collection) were cultured in
RPMI-1640 containing 10% FBS, and maintained in conditions
identical to those of the CMECs.
Cell viability assay
The Cell Counting Kit-8 (CCK-8; Beyotime Institute
of Biotechnology) was used to determine cell viability. Briefly,
CMECs (1×104 cells/well) were seeded into a 96-well
plate and incubated with 10, 50 or 100 µM salidroside for 24 h.
Following salidroside treatment, CCK-8 reagent was added to each
well and the plate was incubated for 2 h at 37°C. The optical
density was measured at 450 nm using a microplate reader (Synergy™
H4; BioTek Instruments, Inc.).
Monocyte adhesion assay
To determine the degree of monocyte THP-1 cell
adhesion to CMECs, CMECs (1×105 cells/well) were seeded
into 6-well plates, treated with salidroside (according to the
aforementioned protocol) and 10 ng/ml TNF-α for 12 h, and incubated
until 100% confluence is achieved. THP-1 cells were labeled with 5
µM 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbo-cyanine
perchlorate for 10 min and resuspended in high-glucose DMEM (1%
FBS); the cells were then added to the confluent CMECs and
incubated for 1 h. The cells were washed in PBS to remove
non-adherent THP-1 cells, and the adherent monocytes were
visualized using a fluorescence microscope at ×200 magnification
(Olympus Corporation).
Western blot analysis
Total protein was extracted from the CMECs using
RIPA buffer (Beyotime Institute of Biotechnology), and the protein
concentration was determined using the Bradford method. Equal
amounts (30 µg) of total protein were separated by SDS-PAGE using a
10% gel, and transferred onto PVDF membranes (EMD Millipore). After
being blocked with 5% non-fat dry milk for 1 h at room temperature,
the membranes were incubated with the following primary antibodies
at a 1:1,000 dilution, overnight at 4°C: VCAM-1, p38, phospho-p38,
p44/42, phospho-P44/42, SAPK/JNK, phospho-SAPK/JNK, IκBα,
phospho-IκBα, NF-κB p65 and phospho-NF-κB p65. A further incubation
with a horseradish peroxidase-conjugated secondary antibody (cat.
no. 31460; Invitrogen; Thermo Fisher Scientific, Inc.; dilution,
1:5,000) was then conducted at room temperature for 2 h. The bands
were visualized using the Gel Doc™ XR+System (Bio-Rad Laboratories,
Inc.), and the band intensity was quantified using ImageJ 1.6.0
software (National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the CMECs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and 1 µg/sample of total RNA was reverse transcribed using
the PrimeScript™ RT Reagent kit. The reverse transcribed conditions
were as follows: 37°C for 15 min, 85°C for 5 sec and 4°C for 5 min.
The resulting cDNA was then amplified using the SYBR®
Premix Ex Taq™ kit on a CFX Connect™ Real-Time System (Bio-Rad
Laboratories, Inc.). The thermal cycler conditions were as follows:
Initial denaturation at 95°C for 30 sec, followed by 40 cycles at
95°C for 5 sec and 60°C for 30 sec. The relative quantification of
the mRNA expression levels was normalized to that of the β-actin
endogenous control, and calculated using the PCR system software
via the 2−ΔΔCq relative quantification method (15). The primers used for PCR were as
follows: Rat VCAM-1 forward, 5′-GCTGCTGTTGGCTGTvGACTCTC-3′ and
reverse, 5′-GCTCAGCGTCAGTGTGGATGTAG-3′; rat IL-6 forward,
5′-AGACTTCCATCCAGTTGCCTTCTTG-3′ and reverse,
5′-CATGTGTAATTAAGCCTCCGACTTGTG-3′; rat IL-1β forward,
5′-AACTGTGAAATAGCAGCTTTCG-3′ and reverse,
5′-CTGTGAGATTTGAAGCTGGATG-3′; rat MCP-1 forward,
5′-GCAGGTCTCTGTCACGCTTCTG-3′ and reverse,
5′-GAATGAGTAGCAGCAGGTGAGTGG-3′; rat β-actin forward,
5′-TACAACCTTCTTGCAGCTCC-3′ and reverse,
5′-ATCTTCATGAGGTAGTCTGTC-3′.
Immunocytochemistry
CMECs were cultured on glass coverslips and exposed
to TNF-α (10 ng/ml) and salidroside (10, 50 and 100 µM), before
being washed with PBS and fixed in 4% paraformaldehyde for 10 min
at room temperature. The cells were incubated in 3% bovine serum
albumin (Beyotime Institute of Biotechnology)-PBS blocking solution
for 30 min at room temperature, with anti-factor VIII (1:250),
anti-VCAM-1 (1:700) and anti-p65 (1:800) antibodies, overnight at
4°C, and then with a secondary antibody labeled with Alexa Fluor
488 (dilution, 1:200; cat. no. A11034; Invitrogen; Thermo Fisher
Scientific, Inc.) for 2 h at room temperature. Cell nuclei were
stained with DAPI (Sigma-Aldrich; Merck KGaA) for 10 min at room
temperature, and all samples were observed under a fluorescence
microscope at ×400 magnification (Olympus Corporation).
ELISA
CMECs, seeded into 6-well plates at 1×105
cells/well, were pre-treated with 10, 50 or 100 µM salidroside for
12 h, followed by 10 ng/ml TNF-α for a further 12 h in a 37°C
thermostatic cell incubator. The cell supernatants were collected,
and IL-1β (cat. no. KGERC007-1), IL-6 (cat. no. KGERC003-1) and
MCP-1 (cat. no. KGERC113-1) secretion was quantified using the
corresponding ELISA kits (Nanjing KeyGen Biotech Co., Ltd.),
according to the manufacturer's instructions.
Statistical analysis
All data are expressed as the mean ± standard
deviation. Each experiment was repeated at least 3 times. The data
were analyzed using SPSS 19.0 software (SPSS, Inc.). The
differences between groups were determined using one-way ANOVA and
Tukey's test, and P<0.05 was considered to indicate a
statistically significant difference.
Results
Isolation and identification of
CMECs
After incubation in vitro for 24–48 h,
primary rat CMECs of the ventricular tissue were spindle-like or
polygonal in shape. After 72–96 h incubation, when the CMECs had
reached 95–100% confluence, a ‘cobblestone’ appearance was
observed. Most of the CMECs were positively stained with the
microvascular endothelial cell specific anti-factor VIII antibody
(Fig. 1).
Salidroside inhibits TNF-α-induced
binding of monocytes to CMECs
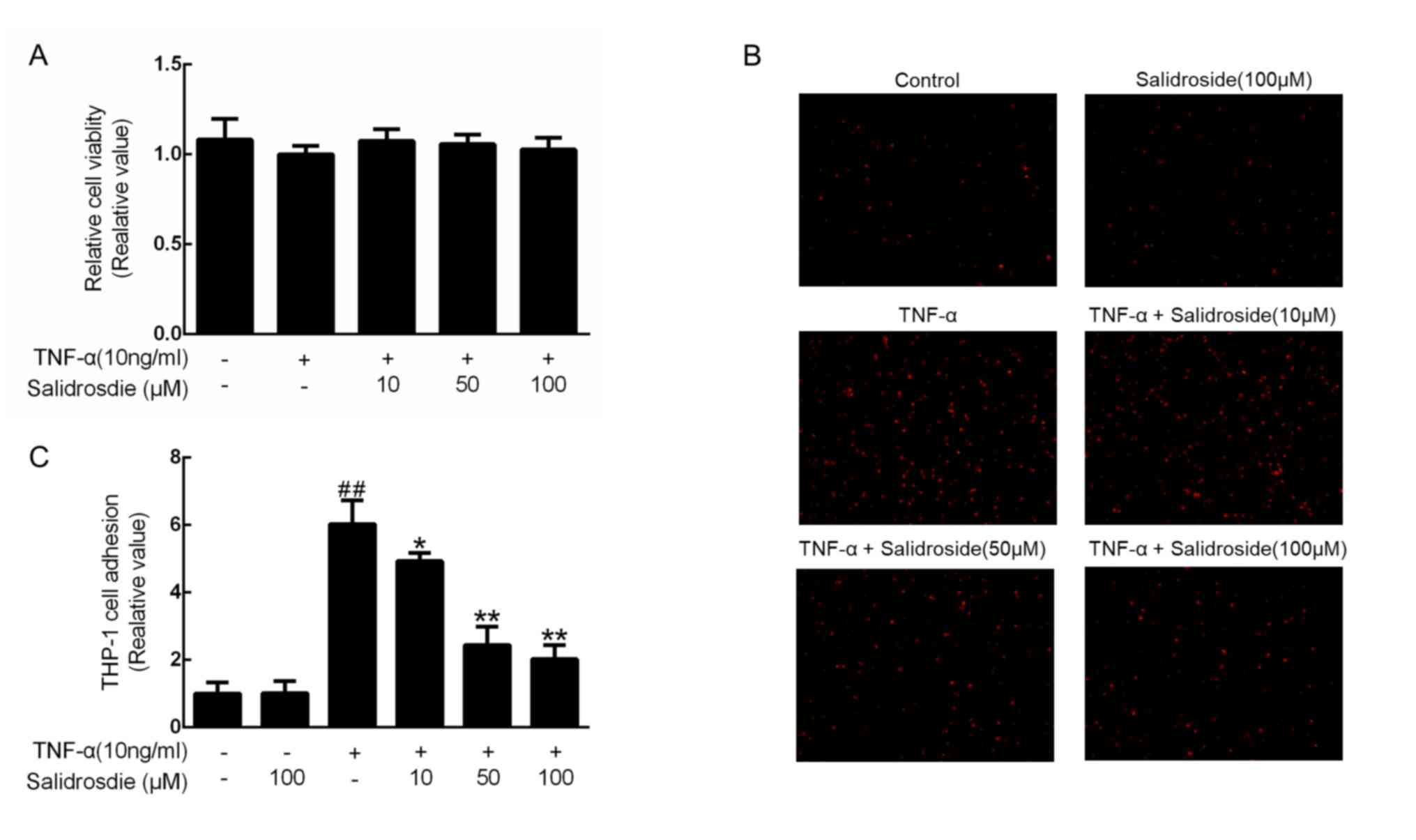
To ensure that a non-cytotoxic concentration of
salidroside was used, a CCK-8 assay was performed at three
predetermined concentrations of salidroside (10, 50 and 100 µM) in
TNF-α-stimulated CMECs. Since cell viability was not affected by up
to 100 µM salidroside (Fig. 2A),
CMECs were treated with 10, 50 and 100 µM in the subsequent
experiments. Considering that monocyte-to-endothelial cell adhesion
is an essential step in the development of atherosclerosis
(6), the inhibitory effects of
salidroside on the adhesion of THP-1 monocytes to TNF-α-stimulated
CMECs were further investigated. As indicated, 100 µM salidroside
did not alter the number of THP-1 cells adhering to the CMECs, and
10 ng/ml TNF-α significantly increased these numbers; however,
salidroside pretreatment prior to TNF-α administration decreased
the degree of monocyte adhesion to TNF-α-treated CMECs in a
concentration-dependent manner (Fig. 2B
and C).
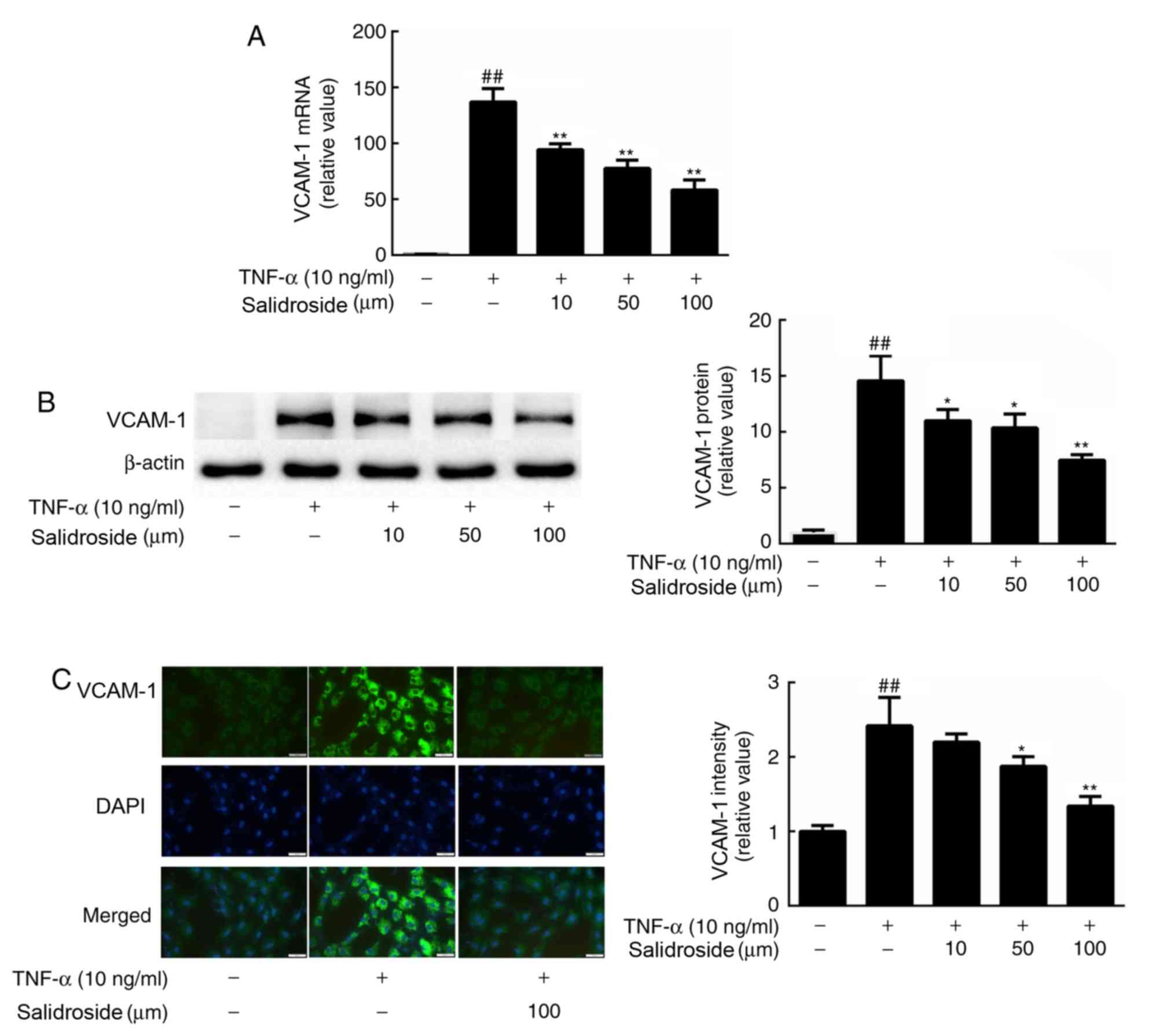
Salidroside suppresses TNF-α-induced
VCAM-1 protein expression
To determine whether VCAM-1 was involved in the
salidroside-associated inhibition of TNF-α-induced monocyte-to-CMEC
binding, the VCAM-1 mRNA expression level was quantified, and
protein abundance and immunofluorescence intensity were determined.
According to the observed results, treatment of CMECs with 10 ng/ml
TNF-α strongly induced VCAM-1 gene expression (Fig. 3A). By contrast, pretreatment with
salidroside inhibited the induction of VCAM-1 by TNF-α. In
accordance with these results, western blot analysis and
immunocytochemistry revealed that salidroside treatment also
decreased the protein expression levels of VCAM-1 in CMECs,
compared with a single dose of TNF-α (Fig. 3B and C).
Salidroside reduces the production of
pro-inflammatory cytokines in CMECs
In order to identify the effect of salidroside on
the production of TNF-α-induced pro-inflammatory cytokines, the
expression levels of IL-1β, IL-6 and MCP-1 in CMECs were measured.
After a 12-h incubation with 10, 50 or 100 µM salidroside, CMECs
were treated with 10 ng/ml TNF-α for a further 12 h. RT-qPCR
analysis and ELISA revealed that the TNF-α-induced mRNA expression
levels and secretion of IL-1β, IL-6 and MCP-1 were reduced by
salidroside treatment in CMECs (Fig.
4).
Salidroside regulates MAPK and NF-κB
signaling in CMECs
In order to verify the possible downstream changes
stimulated by salidroside, CMECs were pretreated with salidroside
for 12 h and then incubated with TNF-α for an additional 30 min.
The salidroside concentration was regulated, and MAPK and NF-κB
activation were investigated. The levels of p38, Jnk and Erk
phosphorylation were markedly upregulated following TNF-α
treatment. However, salidroside effectively suppressed the
TNF-α-induced activation of p38, Jnk and Erk, as indicated by a
reduction in the phosphorylation of these proteins (Fig. 5A). Also, the effect of salidroside on
NF-κB nuclear accumulation was detected by p65 immunostaining.
Salidroside was demonstrated to strongly suppress the degradation
and phosphorylation of IκBα, and the phosphorylation of NF-κB p65,
in a concentration-dependent manner (Fig. 5B and C).
 | Figure 5.Effect of salidroside on
TNF-α-induced MAPK and NF-κB activation in CMECs. (A) Western blot
analysis of the phosphorylation of p38, Jnk, Erk and total p38, Jnk
and Erk in CMECs treated with 10 ng/ml TNF-α for 15 min, after
treatment with the indicated concentrations of salidroside for 12
h. (B) Western blot analysis of the phosphorylation of NF-κB p65,
IκBα and total NF-κB p65 and IκBα in CMECs treated with 10 ng/ml
TNF-α for 15 min, after treatment with the indicated concentrations
of salidroside for 12 h. (C) CMECs were pre-incubated with 100 µM
salidroside for 12 h and TNF-α (10 ng/ml) for 30 min. NF-κB p65
(green) was detected by immunofluorescence using an anti-NF-κB p65
antibody, and the nuclei were stained with DAPI (magnification,
×400). The values are shown as the mean ± standard deviation from
three independent experiments. ##P<0.01 vs.
respective control group; *P<0.05 and **P<0.01 vs. respective
TNF-α-only group. TNF-α, tumor necrosis factor α; CMECs, cardiac
microvascular endothelial cells; IκBα, inhibitor of NF-κB; p,
phosphorylated; t, total. |
Discussion
Endothelial dysfunction is the cause of a number of
cardiovascular diseases (16).
Oxidative and endoplasmic reticulum stress, unfolded endothelial
cell protein responses and various other factors may lead to
endothelial dysfunction (17), and
vascular inflammation serves a key role in the pathogenesis of
endothelial dysfunction and associated cardiovascular diseases
(18,19). Attenuating endothelial inflammatory
responses alleviates endothelial dysfunction and reduces the
incidence of cardiovascular disease. Previous studies have
demonstrated that activation of the endothelium at sites of
inflammation results in the expression of a series of adhesion
molecules, including VCAM-1, and cytokines such as IL-1β, IL-6 and
MCP-1 (20,21). The present study demonstrated that
salidroside significantly reduced TNF-α-induced monocyte adherence
to CMECs by downregulating the expression of VCAM-1, as well as
IL-1β, IL-6 and MCP-1. Furthermore, it was indicated that these
pharmacological properties of salidroside were associated with its
inhibitory effects on MAPK and NF-κB activation.
VCAM-1 is an immunoglobulin-like adhesion molecule
expressed on activated endothelial cells. It is able to recruit
leukocytes and promote their infiltration into injured arteries;
this initiates atherosclerotic plaque development via its
interaction with α4β1 integrin, which is constitutively expressed
on lymphocytes, monocytes and eosinophils (6). Under normal physiological conditions,
VCAM-1 is not usually expressed, but is rapidly induced by stimuli
such as cytokine secretion and TLR activation (22). TNF-α is a pleiotropic
pro-inflammatory cytokines (5) and
was used to induce vascular inflammation in the present study. The
results demonstrated that salidroside attenuated the TNF-α-induced
expression of VCAM-1 in a concentration-dependent manner at both
the mRNA and protein levels. Additionally, the secretion of
pro-inflammatory cytokines (IL-1β, IL-6 and MCP-1) by CMECs was
decreased in a concentration-dependent manner following salidroside
treatment. Furthermore, salidroside significantly inhibited
THP-1-to-CMEC adhesion. Previous studies have demonstrated that a
reduction in the overexpression of VCAM-1 and other inflammatory
cytokines serves a preventative role in the development of
inflammatory diseases, resulting in improved patient prognosis
(23–26). Thus, the present study revealed that
salidroside modulates vascular inflammation by suppressing
inflammatory responses in TNF-α-activated CMECs.
The MAPK and NF-κB pathways are indispensable for
the activation of TNF-α-stimulated endothelial cells (27). Specifically, NF-κB is a major
transcription factor which participates in the inflammatory
regulation of endothelial cells by responding to pro-inflammatory
stimuli (28). During endothelial
inflammation, NF-κB regulates the expression of VCAM-1 and numerous
inflammatory cytokines (29–31). In the cytoplasm, NF-κB exists in its
inactive form associated with the inhibitory protein IκB via its
IκBα subunit. Once stimulated by cytokines, IκBα becomes
phosphorylated and degraded, which creates the optimal conditions
for NF-κB translocation into the nucleus, triggering gene
transcription (32). Also, the MAPK
signaling pathway is known to crucially regulate endothelial
inflammation, and serves an important role in the induction of
pro-inflammatory mediators (p38, Jnk and Erk) in endothelial cells
after stimulation with TNF-α. The MAPKs primarily constitute a
highly-conserved serine/threonine protein kinase family, which is
important for signal transduction from the cell surface to the
nucleus. MAPKs regulate cell growth, differentiation, environmental
stress adaptation, inflammatory responses, and other important
physiological and pathological processes within the cell (33). It has been shown that MAPKs also
regulate the expression of VCAM-1 and inflammatory cytokines
(8,34,35). A
previous study indicated that salidroside reduces cell mobility by
regulating NF-κB and MAPK signaling in LPS-treated-microglial cells
(36). The present study revealed
that salidroside significantly suppressed the TNF-α-induced
phosphorylation and degradation of IκBα, and the subsequent nuclear
translocation of NF-κB in CMECs. Simultaneously, salidroside was
observed to inhibit TNF-α-induced p38, Erk and Jnk1/2
phosphorylation in CMECs. These data suggest that salidroside may
suppress TNF-α-induced endothelial cell inflammation by inhibiting
NF-κB and MAPK activation.
In conclusion, the present study revealed the
suppressive effects of salidroside on TNF-α-induced CMEC
activation, which effectively inhibited TNF-α-induced monocyte/CMEC
interactions and the release of proinflammatory mediators,
including VCAM-1, IL-1β IL-6 and MCP-1. Such suppressive effects
are likely to have resulted from MAPK and NF-κB restriction. These
results provide novel insights into the therapeutic potential of
salidroside in preventing vascular inflammatory diseases, including
atherosclerosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81573710 and
81573711).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HS and XG contributed to the conception of the
study. RSL, XZ and RCL contributed significantly to analysis and
manuscript preparation. RSL and ZD also performed data analyses and
wrote the manuscript, and FY and YC helped perform the analysis
with constructive discussions. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
committee of Fudan University, Shanghai, China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pober JS and Sessa WC: Evolving functions
of endothelial cells in inflammation. Nat Rev Immunol. 7:803–815.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu S, Xu H, Peng J, Wang C, Jin Y, Liu K,
Sun H and Qin J: Potent anti-inflammatory effect of dioscin
mediated by suppression of TNF-α-induced VCAM-1, ICAM-1and EL
expression via the NF-κB pathway. Biochimie. 110:62–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang W, Huang M, Ouyang H, Peng J and
Liang J: Oridonin inhibits vascular inflammation by blocking NF-κB
and MAPK activation. Eur J Pharmacol. 826:133–139. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang H, Park Y, Wu J, Chen Xp, Lee S,
Yang J, Dellsperger KC and Zhang C: Role of TNF-alpha in vascular
dysfunction. Clin Sci (Lond). 116:219–230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cook-Mills JM, Marchese ME and
Abdala-Valencia H: Vascular cell adhesion molecule-1 expression and
signaling during disease: Regulation by reactive oxygen species and
antioxidants. Antioxid Redox Signal. 15:1607–1638. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Collins T, Read MA, Neish AS, Whitley MZ,
Thanos D and Maniatis T: Transcriptional regulation of endothelial
cell adhesion molecules: NF-kappa B and cytokine-inducible
enhancers. FASEB J. 9:899–909. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan LL and Dai M: Paeonol from Paeonia
suffruticosa prevents TNF-alpha-induced monocytic cell adhesion to
rat aortic endothelial cells by suppression of VCAM-1 expression.
Phytomedicine. 16:1027–1032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee CW, Lin WN, Lin CC, Luo SF, Wang JS,
Pouyssegur J and Yang CM: Transcriptional regulation of VCAM-1
expression by tumor necrosis factor-alpha in human tracheal smooth
muscle cells: Involvement of MAPKs, NF-kappaB, p300, and histone
acetylation. J Cell Physiol. 207:174–186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng T, Bian F, Chen L, Wang Q and Jin S:
beneficial effects of rhodiola and salidroside in diabetes:
Potential role of AMP-activated protein kinase. Mol Diagn Ther.
23:489–498. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao G, Shi A, Fan Z and Du Y: Salidroside
inhibits the growth of human breast cancer in vitro and
in vivo. Oncol Rep. 33:2553–2560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen L, Liu P, Feng X and Ma C:
Salidroside suppressing LPS-induced myocardial injury by inhibiting
ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J Cell Mol
Med. 21:3178–3189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guan S, Feng H, Song B, Guo W, Xiong Y,
Huang G, Zhong W, Huo M, Chen N, Lu J and Deng X: Salidroside
attenuates LPS-induced pro-inflammatory cytokine responses and
improves survival in murine endotoxemia. Int Immunopharmacol.
11:2194–2199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Han X, Fu M, Wang J, Song Y, Liu
Y, Zhang J, Zhou J and Ge J: Qiliqiangxin attenuates
hypoxia-induced injury in primary rat cardiac microvascular
endothelial cells via promoting HIF-1α-dependent glycolysis. J Cell
Mol Med. 22:2791–2803. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Widlansky ME, Gokce N, Keaney JF Jr and
Vita JA: The clinical implications of endothelial dysfunction. J Am
Coll Cardiol. 42:1149–1160. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amodio G, Moltedo O, Faraonio R and
Remondelli P: Targeting the Endoplasmic Reticulum Unfolded Protein
Response to Counteract the Oxidative Stress-Induced endothelial
dysfunction. Oxid Med Cell Longev. 2018:49462892018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gareus R, Kotsaki E, Xanthoulea S, van der
Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de
Winther MP and Pasparakis M: Endothelial cell-specific NF-kappaB
inhibition protects mice from atherosclerosis. Cell Metab.
8:372–383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Libby P: Inflammation in atherosclerosis.
Arterioscler Thromb Vasc Biol. 32:2045–2051. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Robinson A: Dimethyl fumarate (Tecfidera)
for multiple sclerosis. Nurse Pract. 39:10–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tian Y, Jain S, Kelemen SE and Autieri MV:
AIF-1 expression regulates endothelial cell activation, signal
transduction, and vasculogenesis. Am J Physiol Cell Physiol.
296:C256–C266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cybulsky MI, Iiyama K, Li H, Zhu S, Chen
M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW and Milstone
DS: A major role for VCAM-1, but not ICAM-1, in early
atherosclerosis. J Clin Invest. 107:1255–1262. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dansky HM, Barlow CB, Lominska C, Sikes
JL, Kao C, Weinsaft J, Cybulsky MI and Smith JD: Adhesion of
monocytes to arterial endothelium and initiation of atherosclerosis
are critically dependent on vascular cell adhesion molecule-1 gene
dosage. Arterioscler Thromb Vasc Biol. 21:1662–1667. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kirii H, Niwa T, Yamada Y, Wada H, Saito
K, Iwakura Y, Asano M, Moriwaki H and Seishima M: Lack of
interleukin-1beta decreases the severity of atherosclerosis in
ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 23:656–660.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Atreya R and Neurath MF: Involvement of
IL-6 in the pathogenesis of inflammatory bowel disease and colon
cancer. Clin Rev Allergy Immunol. 28:187–196. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gu L, Okada Y, Clinton SK, Gerard C,
Sukhova GK, Libby P and Rollins BJ: Absence of monocyte
chemoattractant protein-1 reduces atherosclerosis in low density
lipoprotein receptor-deficient mice. Mol Cell. 2:275–281. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pober JS: Endothelial activation:
Intracellular signaling pathways. Arthritis Res. 4 (Suppl
3):S109–S116. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Csiszar A, Wang M, Lakatta EG and Ungvari
Z: Inflammation and endothelial dysfunction during aging: role of
NF-kappaB. J Appl Physiol (1985). 105:1333–1341. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Z, Connell MC and MacEwan DJ:
TNFR1-induced NF-kappaB, but not ERK, p38MAPK or JNK activation,
mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial
cells. Cell Signal. 19:1238–1248. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wei G, Zhang X, Su Z and Li X: Glatiramer
acetate (GA) prevents TNF-α-induced monocyte adhesion to primary
endothelial cells through interfering with the NF-κB pathway.
Biochem Biophys Res Commun. 457:101–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li X, Tang Y, Ma B, Wang Z, Jiang J, Hou
S, Wang S, Zhang J, Deng M, Duan Z, et al: The peptide lycosin-I
attenuates TNF-α-induced inflammation in human umbilical vein
endothelial cells via IκB/NF-κB signaling pathway. Inflamm Res.
67:455–466. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Collins T and Cybulsky MI: NF-kappaB:
Pivotal mediator or innocent bystander in atherogenesis? J Clin
Invest. 107:255–264. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen YH, Lin SJ, Ku HH, Shiao MS, Lin FY,
Chen JW and Chen YL: Salvianolic acid B attenuates VCAM-1 and
ICAM-1 expression in TNF-alpha-treated human aortic endothelial
cells. J Cell Biochem. 82:512–521. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim KH, Lee EN, Park JK, Lee JR, Kim JH,
Choi HJ, Kim BS, Lee HW, Lee KS and Yoon S: Curcumin attenuates
TNF-α-induced expression of intercellular adhesion molecule-1,
vascular cell adhesion molecule-1 and proinflammatory cytokines in
human endometriotic stromal cells. Phytother Res. 26:1037–1047.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu H, Li Z, Zhu X, Lin R and Chen L:
Salidroside reduces cell mobility via NF-κ B and MAPK signaling in
LPS-induced BV2 microglial cells. Evid Based Complement Alternat
Med. 2014:3838212014. View Article : Google Scholar : PubMed/NCBI
|