Introduction
Neuroinflammation plays an important role in the
pathological process of neurological disease. Microglial activation
is considered to be the hallmark of neuroinflammation (1). Previous data derived from in
vitro or chronic neurodegenerative disease have shown that
activation of microglia contributes to neuroinflammation (2) and to exacerbation of neuronal injury
(3). By contrast, several recent
studies have indicated that activated microglia provided a
neuroprotective role and prevented neuronal loss after brain injury
(4). However, the role of
activated microglia in response to subacute cerebral ischemia (SCI)
remains unknown. Moreover, previous findings revealed that the type
of stimulus and the local microenvironment critically affect the
phenotypes (5,6) and role (7) of microglia.
Since the function of activated microglia is
associated with activated microenvironments, while the condition
induced by SCI is significantly different from environments induced
by in vitro stimulation (e.g. with LPS or OGP) or by chronic
neurodegenerative disease, microglial role after SCI is also
different from that in chronic neurodegenerative disease or in
vitro. Hence, further in vivo investigations are key for
exploring the effects of activated microglia after SCI.
Previous data have shown that
N-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide
(PJ34) can inhibit activation of microglia in vivo by
suppressing the activation of poly (ADP-ribose) polymerase-1
(PARP-1) which is a signaling pathway of the activation of
microglia (8). Therefore, we
selected PJ34 as the inhibitor of microglial activation.
In the present study, 156 male adult Sprague-Dawley
(SD) rats were first subjected to permanent cerebral middle artery
occlusion (pMCAO) and were then treated with PJ34 (an inhibitor of
microglial activation), or vehicle alone. Finally, the differences
between the PJ34-and vehicle-treated rats were observed with
respect to neurological deficits, infarct volume, neuronal survival
by NeuN staining and the expression of CD11b (the marker of
microglial activation) and the mRNA and protein expressions of
glial cell line-derived neurotrophic factor (GDNF) and tumor
necrosis factor-α (TNF-α) at 1, 3 and 7 days following treatment.
This study provides evidence that can be used to further
investigate the role of activated microglia.
Materials and methods
Animal model of cerebral ischemia
Animal experiments were conducted in accordance with
the guidelines published in the NIH Guide for the Care and Use of
Laboratory Animals (US Department of Health and Human Services,
Publication no. 85-23, 1985), and all efforts were made to minimize
animal suffering as well as the number of animals used. One hundred
and fifty six male Sprague-Dawley rats (250–300 g) were obtained
from the Experimental Animal Center of Zhengzhou University. They
were first subjected to pMCAO surgery and were then randomly
divided into two groups with 78 rats in each group: the PJ34 group
rats were treated with PJ34, and the vehicle group rats were
treated with vehicle. The animals were maintained at a controlled
temperature (20±2°C) and group-housed (12 h light/dark cycle) with
access to food and water ad libitum. Infarct volume (%) was
expressed as a percentage of the contralateral hemisphere.
The cerebral ischemic model was induced using a
pMCAO rat model (9) under a
microscope. Rats were anesthetized by inhalation of 5% isoflurane
and maintained with 2% isoflurane in a mixture of 70%
N2O and 30% O2. After the external carotid
artery, the internal carotid artery (ICA) and the pterygopalatine
artery of the ICA were exposed, a piece of monofilament nylon
suture (diameter, 220 μm), with its tip rounded by gentle heating
(diameter, 270±10 μm), was introduced via the lumen of the left
external carotid artery stump and left ICA to embed into the left
anterior cerebral artery so that the right middle cerebral artery
was occluded at its origin. After the operation, rats were
transferred to their cage in which the temperature was kept at 37°C
until the animals woke up completely.
Drug treatment
In the PJ34-treated group, rats were
intraperitoneally injected with PJ34 at the dose of 15 mg/kg
(Sigma, Mississippi; diluted in 1.0 ml sterile saline vehicle)
immediately after the operation and every 12 h thereafter (8). The vehicle-treated rats were
administered the same volume of saline after pMCAO.
Measurement of neurological deficits
The measurements of neurological deficits were
performed at 1, 3 and 7 days after pMCAO. All measurements were
completed in a dedicated behavioral study facility during this
interval to minimize environmental impact related to transfer
between home cage and the testing arenas. The measured results were
analyzed by individuals blinded to the experimental conditions. The
battery consisted of two tests to evaluate neurological deficits:
the cylinder test and the Zea Longa test. In the cylinder test,
rats were placed in a clear Plexiglas cylinder (95×180 mm height)
(10) and were observed for 3 min
and the number of wall touches with each forelimb was scored for
each rat. Normal rats touch the walls equally often with each
forelimb, but hemiparetic rats touch the walls less often with the
affected limb (10,11). The relative percentage of left
(unimpaired) forepaw contacts was calculated as follows:
(left-right)/(right+left+both) ×100%. In the Zea Longa test,
neurological deficits were performed using a neurological grade
score (0, no observable deficit; 1, torso flexion to the right; 2,
spontaneous circling to the right; 3, leaning/falling to the right;
and 4, no spontaneous movement) (12).
Measurement of infarct volume
After the evaluation of neurological function, rats
(n=6/group) were sacrificed with an overdose of sodium
pentobarbital (75 mg/kg) on Day 7 after treatment. Brains were
quickly removed and were cut into 5 coronal sections, 2 mm thick.
The sections were immersed in a 2% solution of
2,3,5-triphenyltetrazolium chloride (TTC; Sigma) in saline for 20
min at 37°C and then fixed with 4% paraformaldehyde. The infarct
area was measured in each slice using computerized planimetry
(PC-based image tools software). The infarct volume
(mm3) was calculated as 2 mm (thickness of each slice) ×
(sum of infarct area in all brain slices).
Immunohistochemistry and
immunofluorescence confocal microscopy
At 1, 3 and 7 days after pMCAO, rats (n=6/experiment
subgroup/time-point) were deeply anesthetized with 5% isoflurane
and perfused intracardially with saline and 5% buffered
formaldehyde. Brains were paraffin-embedded and sliced into coronal
sections (5 μm) starting at 3 mm posterior to the anterior pole.
Parallel sections were used to stain individually for the following
primary antibodies: goat polyclonal antibody to CD11b (1:200; Santa
Cruz Biotechnology, Inc., Santa Cruz, CA), mouse monoclonal
antibody to NeuN (1:1,000; Sigma). Following extensive rinsing
steps in 0.1 M PBS, the sections were reincubated in biotinylated
rabbit anti-goat or goat anti-mouse immunoglobulin (Boster
Biological Technology, Ltd., Wuhan, China) for 1 h at room
temperature and then the Vector ABC system followed. For negative
controls, the primary antibody was omitted. The positive cells of
the 3 visual fields in the peri-necrotic cortex were calculated at
×400 magnification (one visual field=0.196 mm2).
For double immunofluorescence, the sections were
incubated with a mixture of goat polyclonal antibody to CD11b
(1:200) and mouse polyclonal antibody to GDNF (1:100), or with a
mixture of goat polyclonal antibody to CD11b (1:200) and mouse
monoclonal antibody to TNF-α (1:100) (all were from Santa Cruz
Biotechnology, Inc.) overnight at 4°C, followed by a mixture of
FITC-conjugated donkey anti-goat IgG and Cy3-conjugated donkey
anti-mouse IgG (1:100; Jackson Laboratory, Bar Harbor, ME, USA).
The sections were washed in TBS (0.2% Triton X-100 PBS) and mounted
with Vectashield mounting medium (Vector Laboratories, Burlingame,
CA, USA). Confocal images were captured by an Olympus FV300
confocal spectral microscope (Olympus, Tokyo, Japan) at ×200
magnification (one visual field=0.392 mm2). Image
analysis was performed with ImageJ 1.39u (National Institutes of
Health, Bethesda, MD, USA). The number of cells showing double
immunostaining was estimated by counting cells in 3 random fields
of the peri-necrotic cortex.
RT-PCR
After evaluation of neurological deficits, rats
(n=6/experiment group/time-point) were sacrificed with an overdose
of sodium pentobarbital (75 mg/kg). Total RNA was extracted from
the peri-necrotic cortex with the TRIzol kit (Fermentas Inc., Glen
Burnie, MD, USA). Primers were designed with primer premier 5.0
software (Premier Biosoft International, Palo Alto, CA, USA) for
CD11b (NP_036843/351bp), GDNF (NM_019139/206bp) and TNF-α
(NM_012675/255bp) and β-actin (NM_031144/341bp). RT-PCR experiments
followed standard protocols. Briefly, the cDNA strand first was
synthesized from the total RNA and then the mixture of the primers
and the cDNA products was amplified by PCR. Finally, the PCR
products were separated on 0.8% agarose gel. Analysis was carried
out using a gel imaging system (Bio-Rad Laboratories Inc.,
Hercules, CA, USA). Optical density (OD) of CD11b, GDNF and TNF-α
was measured by the electrophoresis gel imaging system and their
expression levels were calculated as percentage relative to β-actin
control (OD of these proteins/OD of β-actin).
Western blotting
The total sample adjacent necrotic cortex was
dissociated from the rats which were sacrificed with an overdose of
sodium pentobarbital (75 mg/kg) at 1, 3 and 7 days after treatment
(n=6/experiment group/time-point). Brain tissues were lysed in RIPA
lysis buffer, and the lysates were harvested by centrifugation
(12,000 rpm) at 4°C for 30 min. Next, the protein samples (20 μg)
were separated by electrophoresis in a 12% sodium dodecyl sulfate
polyacrylamide gel and were transferred onto a polyvinylidene
difluoride (PVDF) membrane. The membrane was placed in 5% non-fat
milk for 1 h to block the nonspecific binding sites and was then
incubated with the mouse polyclonal antibody anti-CD11b (1:1,000)
or GDNF (1:1,000), or TNF-α (1:1,000). Then, the membranes were
incubated with horseradish peroxidase-conjugated goat anti-mouse
antibody (1:5,000) (all were from Santa Cruz Biotechnology, Inc.)
at 37°C for 60 min. After four washes, the bands were detected with
the enhanced chemiluminescence reagent (Cell Signaling Technology,
Danvers, MA, USA). Band density was measured with ImageJ software
(National Institutes of Health) and was standardized to that of
GAPDH detected using mouse anti-rat GAPDH monoclonal antibody
(Hangzhou GoodHere, Hangzhou, China).
Statistical analysis
Observers blinded to experimental conditions
evaluated outcome measures. A repeated measurement ANOVA was
employed to analyze the behavioral measurement data. Student's
t-test was used when comparing 2 groups. Differences were
considered statistically significant when P-values were <0.05.
All data are expressed as the means ± SEM.
Results
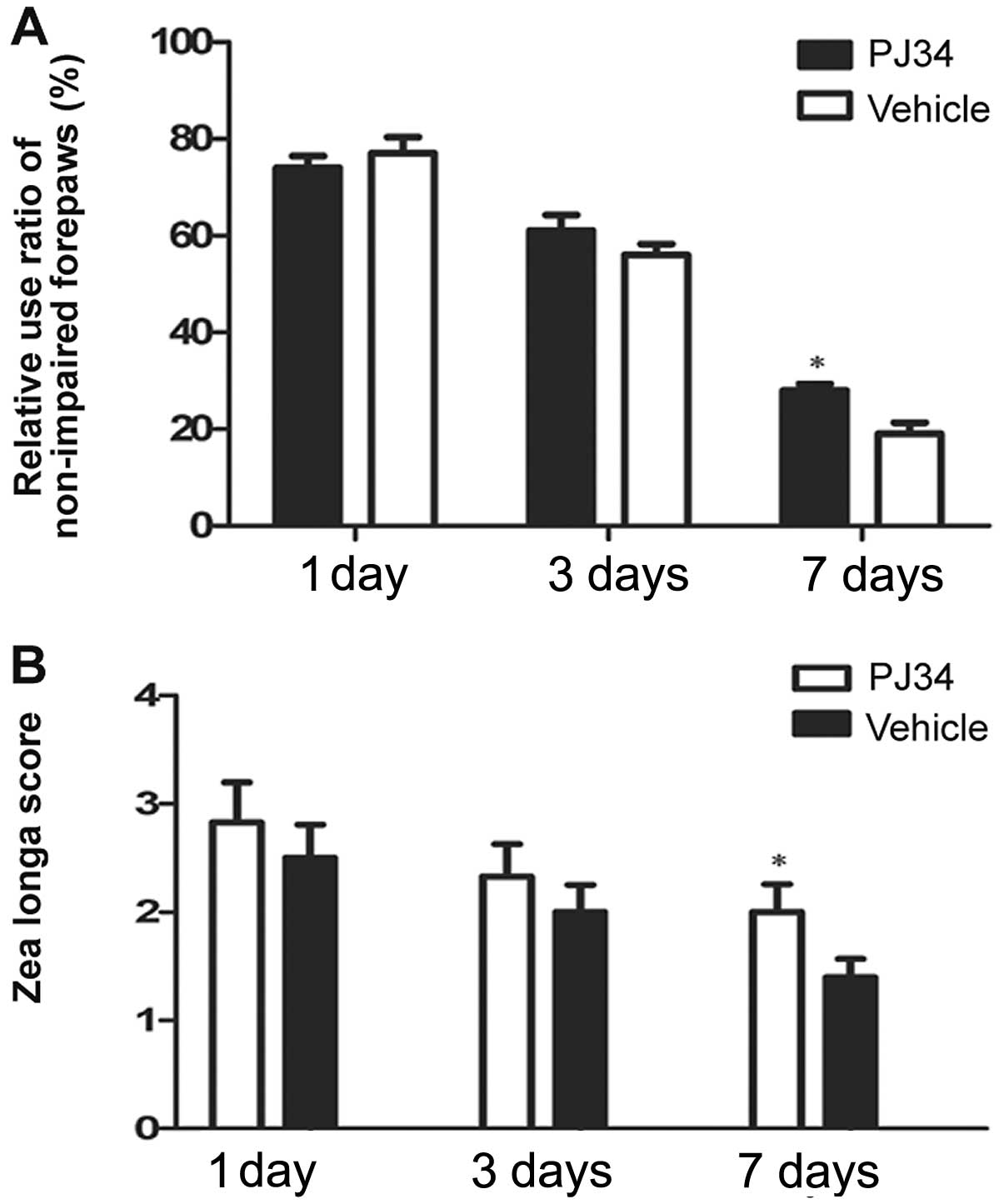
Measurement of neurological deficits
Neurological deficits were evaluated by the cylinder
and the Zea Longa test at 1, 3 and 7 days following pMCAO. In the
cylinder test, the PJ34-treated rats used the hemiplegic forepaws
less than the vehicle-treated rats at 3 and 7 days after treatment.
On Day 7 post surgery, there was a significant difference between
the PJ34- and vehicle-treated rats in the frequencies of using the
impaired forepaws (F=4.89, P=0.033) (Fig. 1A). In the Zea Longa test, the rats
treated with PJ34 showed higher grade score than the rats treated
with vehicle at 3 and 7 days post treatment. The difference between
the two groups was significant in the grade score on Day 7 (F=7.50,
P=0.021) (Fig. 1B).
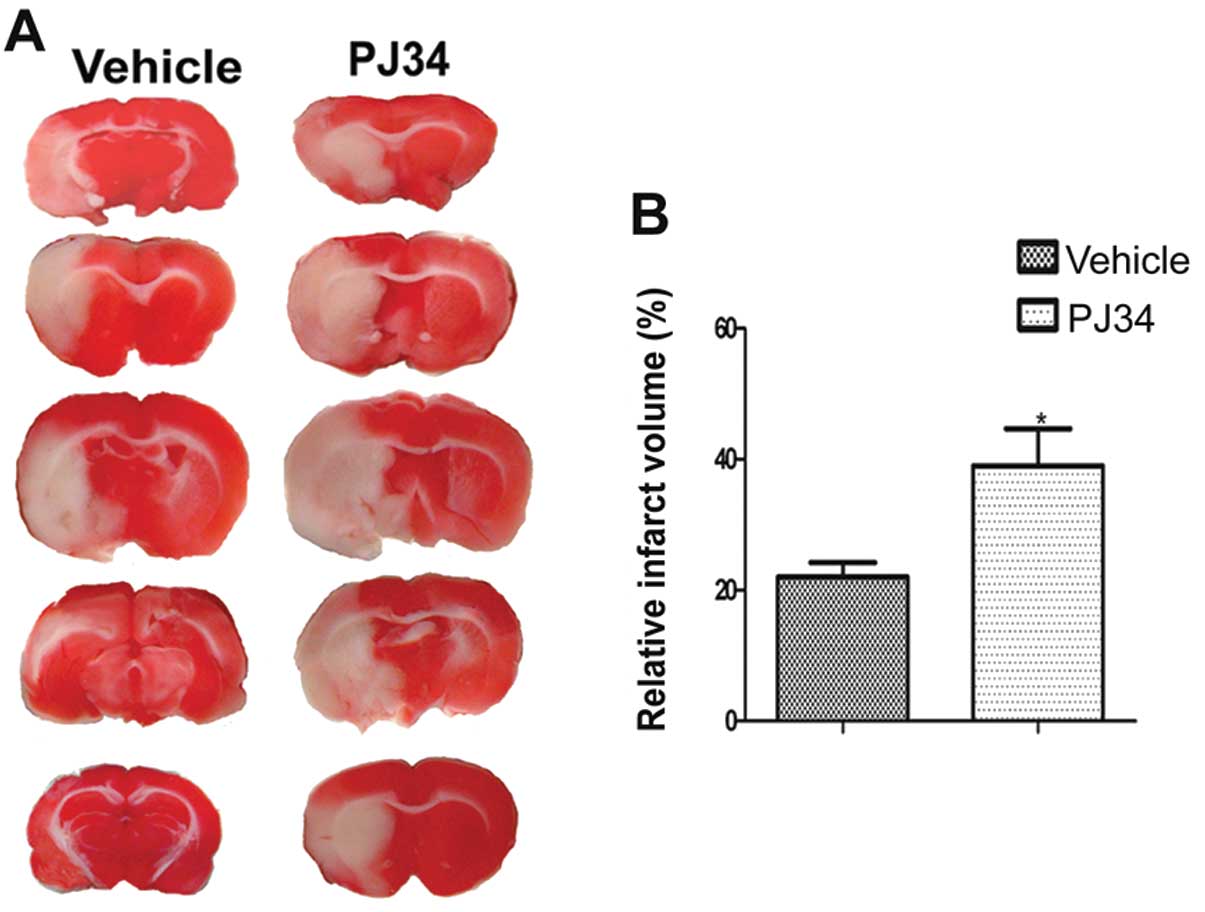
Lack of activated microglia increases
infarct volume
The infarct volumes of the PJ34-and vehicle-treated
rats (n=6/experiment group) were detected by TTC on Day 7 after the
operation. Compared with the vehicle-treated rats, the PJ34-treated
rats had a larger infarct volume (t=1.730, P=0.032)(Fig. 2).
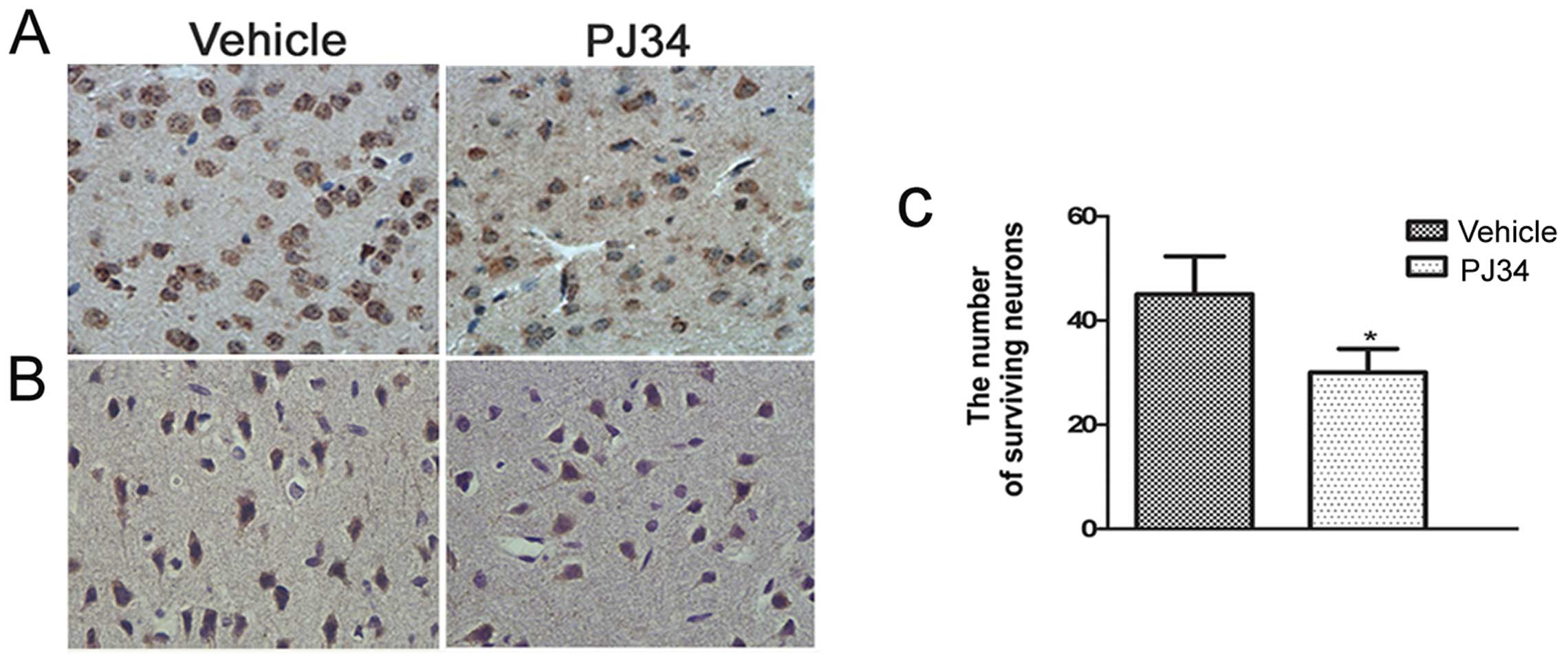
Greater neuronal loss in the absence of
activated microglia
Immunohistochemistry was used to detect the CD11b-
and NeuN-positive cells in the peri-necrotic cortex of pMCAO rats
(n=6/experiment subgroup/time-point) at 1, 3 and 7 days following
treatment. The number of CD11b- and NeuN-positive cells in the
brain of the PJ34-treated rats was similar to the vehicle-treated
rats at 1 day after treatment. Compared with the vehicle-treated
rats, the number of CD11b- and NeuN-positive cells of the
PJ34-treated rats was reduced on Day 3 and was reduced further on
Day 7. The difference between the PJ34- and vehicle-treated rats
was significant in the number of the CD11b-positive cells (11.4±1.1
and 21.6±1.3/field, respectively; t=2.471, P<0.01) (Fig. 3A) and the number of the
NeuN-positive cells (t=2.283, P=0.029) (Fig. 3B and C) on Day 7.
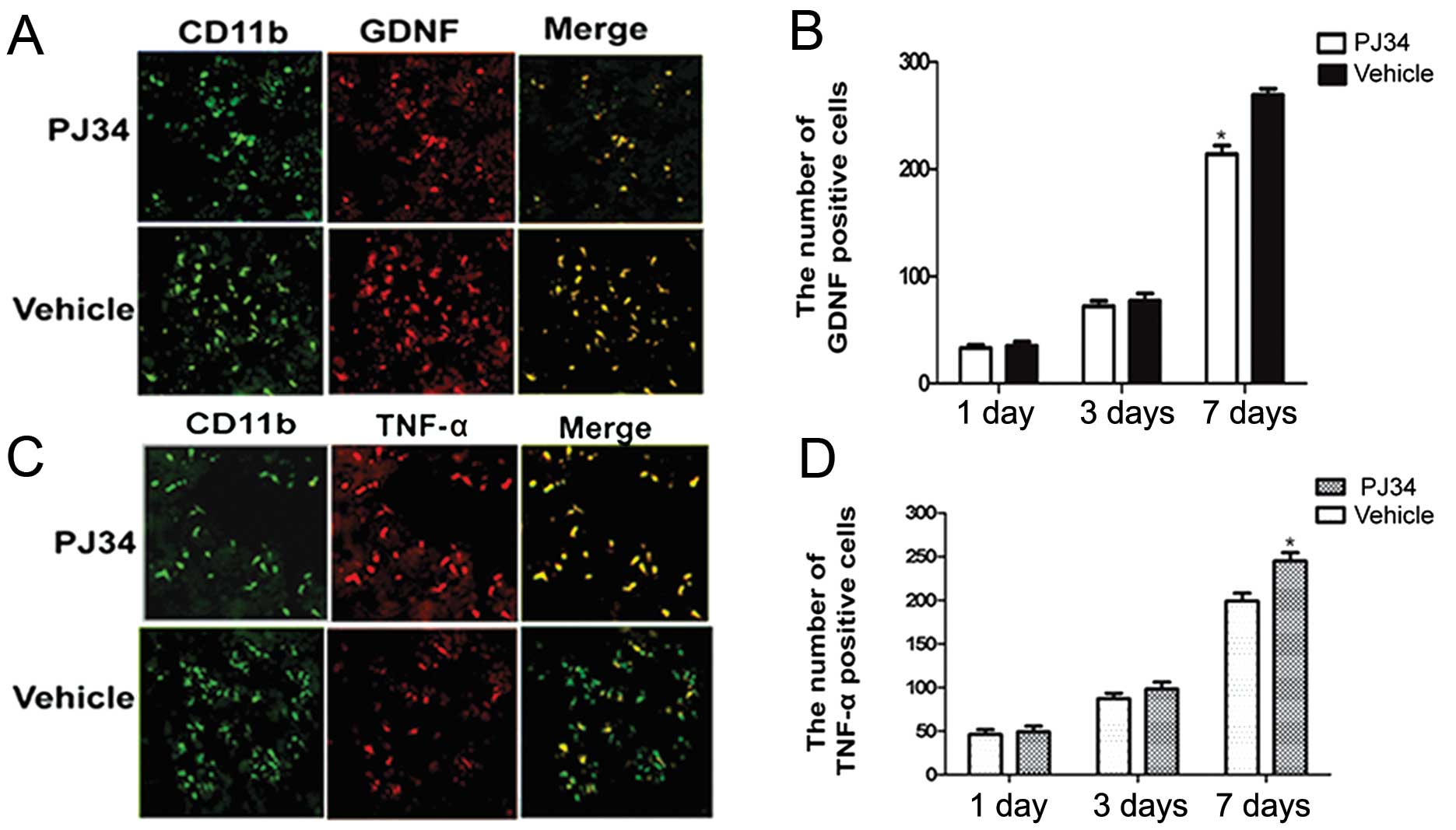
Expression of GDNF and TNF-α in activated
microglia after SCI
The GDNF- and TNF-α-expressions in activated
microglia in the peri-necrotic cortex of rats treated with PJ34 or
vehicle (n=6/experiment subgroup/time-point) were examined by
double immunofluorescence at 1, 3 and 7 days after treatment. There
was no difference between the PJ34- and vehicle-treated rats with
respect to the expression of GDNF and TNF-α at 1 day after the
treatment. Compared with the vehicle-treated rats, the PJ34-treated
rats showed a reduction in GDNF expression in activated microglia
at 3 days after treatment and a further reduction at 7 days
(t=2.105, P=0.043) (Fig. 4A and
B).
Although the total number of CD11b-positive cells in
the brains of PJ34-treated rats was reduced at 3 and 7 days after
treatment, TNF-α expression in the remaining CD11b-positive cells
was increased at 3 days after treatment and further increased at 7
days relative to the TNF-α expression in CD11b-positive cells in
the brains of vehicle-treated rats (t=3.741, P<0.05) (Fig. 4C and D).
Inhibition of microglial activation
influences the expression of the GDNF and TNF-α mRNA
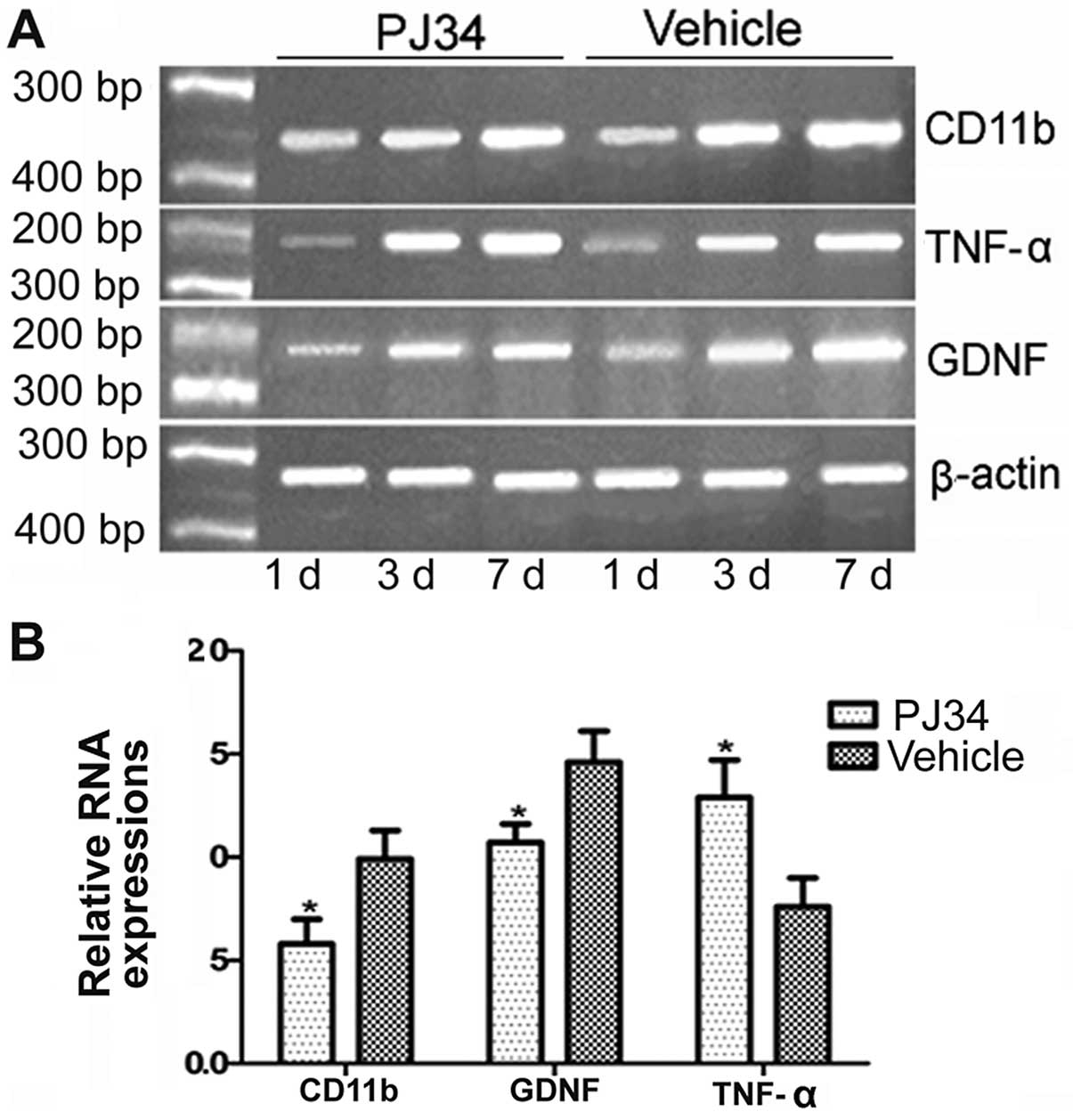
The expression of the CD11b, GDNF and TNF-α mRNAs
(n=6/group/time-point) was detected at 1, 3 and 7 days after
treatment. There was no difference between of the PJ34- and
vehicle-treated rats in the levels of CD11b, GDNF and TNF-α mRNAs
on Day 1. The mRNA level of CD11b and GDNF was decreased on Day 3
after treatment and further decreased on Day 7 in the brains of the
PJ34-treated rats compared with the vehicle-treated rats. However,
the mRNA expression of TNF-α was increased on Day 3 after pMCAO and
further increased on Day 7 in the brains of the PJ34-treated rats
compared with the vehicle-treated rats (P<0.05) (Fig. 5).
Inhibition of microglia activation
influences the expression of the GDNF and TNF-α proteins
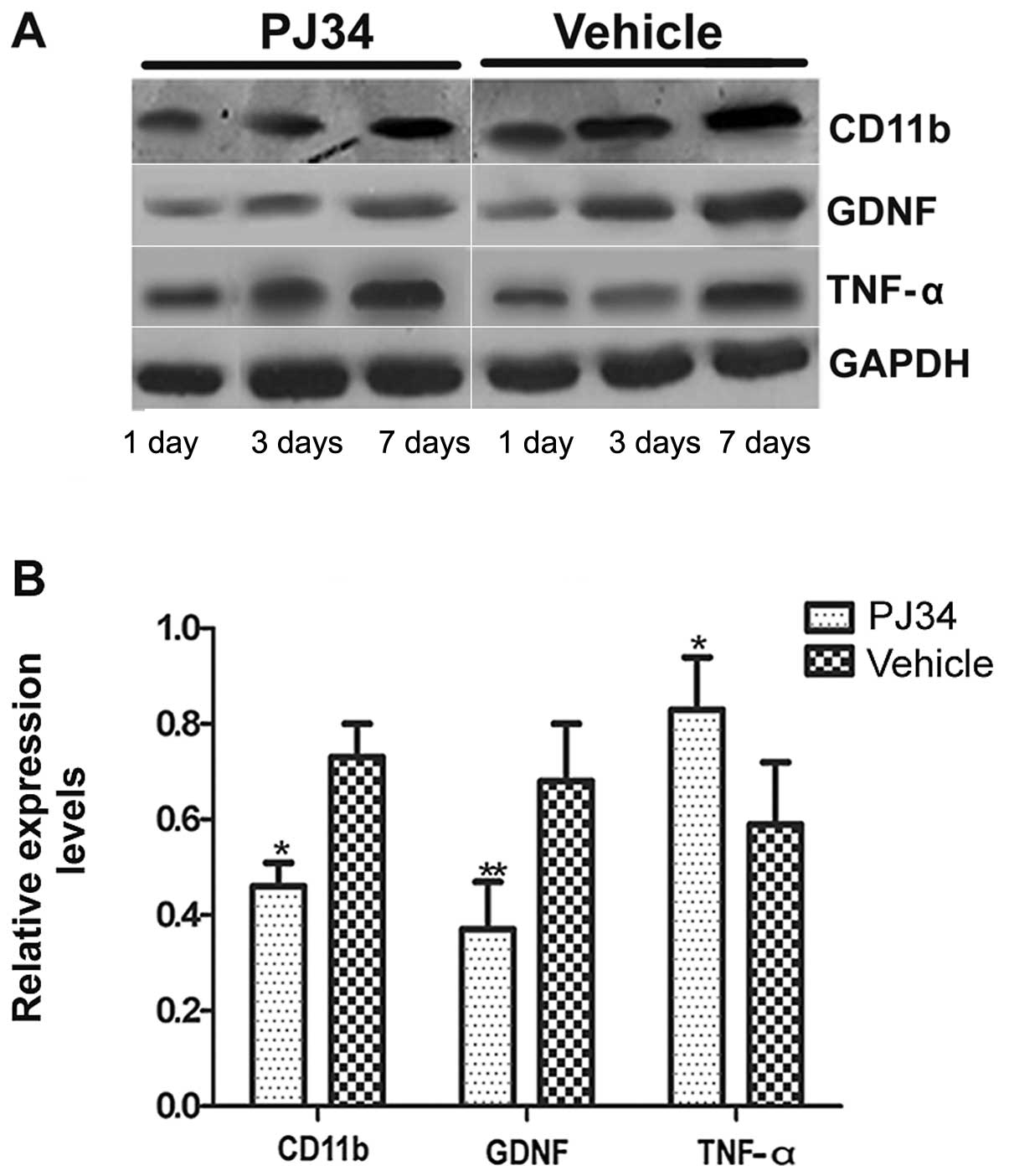
Western blotting (n=6/group/time-point) was used to
determine the expression level of CD11b, GDNF and TNF-α proteins
around lesion cortex of rats treated with vehicle or PJ34 at 1, 3
and 7 days following pMCAO. There was no difference between the
PJ34- and vehicle-treated rats in the expression levels of CD11b,
GDNF and TNF-α at 1 day after treatment. However, on Days 3 and 7
after the operation, the rats treated with PJ34 showed that
accompanied by a decrease in the CD11b expression, GDNF expression
was decreased but TNF-α expression was increased (Fig. 6A). There was a significant
difference between the PJ34- and vehicle-treated groups with
respect to the expression of CD11b, GDNF and TNF-α on Day 7
following treatment (P<0.05) (Fig.
6B).
Discussion
The present study showed for the first time that
activated microglia provide a neuroprotective role through
balancing the GDNF and the TNF-α expressions following SCI.
Compared with the vehicle-treated rats, the PJ34-treated rats had
more severe neuronal deficits and a larger infarct volume and
exhibited fewer activated microglia, greater neuronal loss,
decreased GDNF expression and increased TNF-α expression at 3 and 7
days following ischemic stroke.
Currently, most findings related to the role of
activated microglia in central nervous system disease (CND) are
derived from in vitro analysis or a chronic
neurodegenerative disease model (13). Previous data suggested that the
different microenvironments inducing microglial activation lead to
the different roles played by microglia in CND (14–16) while the condition after SCI is
significantly different from the microenvironments in chronic
neurodegenerative disease or the in vitro model (17). Therefore, in vivo
investigations contribute more to knowledge of the effects of
microglia after SCI. We hypothesized that the complete depletion of
activated microglia is impossible in vivo. Thus, it is
preferable to select an inhibitor to suppress the microglial
activation as a control. Previous studies have shown that
microglial activation correlates with PARP-1 which is an important
co-activating factor and plays a crucial role in the nuclear factor
(NF)-κB signaling pathway (18–20). Therefore, the inhibition of PARP-1
activity or NF-κB-dependent gene transcription by the inhibitor of
PARP-1 blocks the signaling paths related to microglial activation
(18,21,22) and hence microglial activation is
suppressed (23,24). In addition, it has been well
documented that PJ34, a PARP inhibitor, can effectively inhibit
microglial activation (8,25,26).
To validate the inhibitive effect of PJ34 on
microglial activation, we used immunohistochemical staining, RT-PCR
and western blotting to examine the CD11b immunoreactivity and
expression levels in the peri-necrotic cortex of the PJ34- and
vehicle-treated rats post ischemia. Indeed, we observed that the
CD11b positive cells were markedly reduced and the CD11b expression
level was markedly decreased in the brains of rats treated with
PJ34 compared with that of rats treated with vehicle 7 days after
treatment. The results suggest that PJ34 can effectively inhibit
microglial activation following brain injury.
We next considered whether activated microglia play
a neuroprotective role after ischemic stroke. To address this, we
compared the changes in motor function, the infarct volume and
neuronal survival under the conditions of microglial activation or
inhibition at 1, 3 and 7 days following treatment. The present
results demonstrated that compared with the vehicle-treated rats,
PJ34-treated rats had more severe neurological deficits, a larger
infarct volume and exhibited fewer CD11b positive cells and greater
neuronal loss on Days 3 and 7 after treatment. The lack of
activated microglia in the penumbra regions leads to greater neuron
loss and the enhancement of infarct volume. Therefore, there were
more severe neurological deficits. These results indicate that
activated microglia rescue the neurons in the penumbra area after
SCI.
To explore the neuroprotective mechanisms of
microglia, we detected GDNF and TNF-α immunoreactivity and their
expression levels at 1, 3 and 7 days after surgery. GDNF and TNF-α
are often investigated as they are two of the primary cytokines
related to brain injury. Previous data have revealed that GDNF
provides a potent neuroprotective role in animal models of
Parkinson's disease (27),
cerebral ischemia (28) and motor
neuron degeneration (29). In
vitro studies have shown that the phosphorylated extracellular
signal-regulated kinase (ERK) is present in activated microglia
(30). Previous investigations
revealed that GDNF expression is associated with the
phosphorylation of ERK (31). In
the present study, we found that, along with the reduction in CD11b
positive cells or the decrease in CD11b expression level, the
number of GDNF positive cells and the expression levels of GDNF
mRNA and protein were decreased at 3 and 7 days after treatment.
These results indicate that the inhibition of microglial activation
attenuates GDNF production. With regard to the mechanisms
underlying the role of microglial modulation in the GDNF secretion,
we speculated that reacted microglia induce the phosphorylation of
ERK and further promote the GDNF secretion. However, the inhibition
of microglial activation attenuates the phosphorylation level of
ERK and the GDNF secretion is reduced further.
TNF-α is often considered a neurotoxic
pro-inflammatory factor. Previous in vitro studies have
demonstrated that upon stimulation with LPS, BV2 cells become
overactivated and largely express the TNF-α protein (32). Contrary to these findings, our
results indicated that in the absence of the activation of
microglia, the number of TNF-α positive cells and the expression
levels of TNF-α mRNA and protein were markedly increased. With
regard to the regulating role of microglia in the TNF-α secretion,
recent studies have revealed that the activation of P2X receptor
(purinergic receptor) in activated microglia induced by ATP leads
to the reduction of TNF-α release (33) while astrocytes can release ATP
after brain injury (34). We
inferred that activated microglia protect astrocytes (35) and promote ATP release from
astrocytes after SCI. Subsequently, the increase of extracellular
ATP activates P2X receptor of microglia and TNF-α secretion from
microglia is decreased. However, when the activation of microglia
is inhibited after SCI, the number of dead astrocytes is increased.
Thus, ATP release is also reduced accompanied by the inhibition of
P2X receptor activation. Therefore, TNF-α expression in microglia
is also increased. Moreover, recent data have indicated that the
presence of CD4+ T cells at sites of motoneuron injury
in mSOD1 mice shifts the balance of microglial response from
decreased GNDF expression and increased TNF-α expression to
increased GDNF expression and decreased TNF-α expression after
inherited amyotrophic lateral sclerosis (ALS) (36). However, further studies are
required to ascertain whether the same role of CD4+ T
cells is present in the brain of SCI.
We do not completely exclude the possibility that
PJ34 affects the present results. However, several previous studies
demonstrated that PJ34 provides neuroprotection after cerebral
insult (8). Moreover, the results
from double immunofluorescence and western blotting showed that the
alterations of microglial activation are accompanied by the changes
in GDNF and TNF-α expression levels. The time consistency between
microglial activation and the secretions of GDNF and TNF-α further
verifies that the present results are mainly attributed to the role
of activated microglia rather than to PJ34.
In summary, the present study investigated the
effects of activated microglia after SCI for the first time. Our
results demonstrate that activated microglia exert a
neuroprotective role through balancing the expressions of GDNF and
TNF-α after SCI. These data suggest that microglia may be a
potential target and vehicle for cerebral ischemic stroke
therapy.
Acknowledgements
The authors thank Huaping Guo for his assistance
with the language and the writing of this study, and Yuanhong He
and Hengfang Liu for their technical assistance. This study was
supported by the Science and Research Foundation of National
Institutes of Health (WKJ2008-2-031).
Abbreviations:
|
SCI
|
subacute cerebral ischemia
|
|
CND
|
central nervous disease
|
|
GDNF
|
glial cell line-derived neurotrophic
factor
|
|
TNF-α
|
tumor necrosis factor-α
|
|
pMCAO
|
permanent middle cerebral artery
occlusion
|
References
|
1
|
Lv M, Liu Y, Zhang J, et al: Roles of
inflammation response in microglia cell through Toll-like receptors
2/interleukin-23/interleukin-17 pathway in cerebral
ischemia/reperfusion injury. Neuroscience. 176:162–172. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Block ML and Hong JS: Microglia and
inflammation-mediated neurodegeneration: multiple triggers with a
common mechanism. Prog Neurobiol. 76:77–98. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu B, Du L and Hong JS: Naloxone protects
rat dopaminergic neurons against inflammatory damage through
inhibition of microglia activation and superoxide generation. J
Pharmacol Exp Ther. 293:607–617. 2000.PubMed/NCBI
|
|
4
|
Napoli I and Neumann H: Protective effects
of microglia in multiple sclerosis. Exp Neurol. 225:24–28. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Petersen MA and Dailey ME: Diverse
microglial motility behaviors during clearance of dead cells in
hippocampal slices. Glia. 46:195–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lalancette-Hebert M, Gowing G, Simard A,
Weng YC and Kriz J: Selective ablation of proliferating microglial
cells exacerbates ischemic injury in the brain. J Neurosci.
27:2596–2605. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Figueiredo C, Pais TF, Gomes JR and
Chatterjee S: Neuron-microglia crosstalk up-regulates neuronal
FGF-2 expression which mediates neuroprotection against
excitotoxicity via JNK1/2. J Neurochem. 107:73–85. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hamby AM, Suh SW, Kauppinen TM and Swanson
RA: Use of a poly(ADP-ribose) polymerase inhibitor to suppress
inflammation and neuronal death after cerebral
ischemia-reperfusion. Stroke. 38:632–636. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xi GM, Wang HQ, He GH, Huang CF and Wei
GY: Evaluation of murine models of permanent focal cerebral
ischemia. Chin Med J (Engl). 117:389–394. 2004.PubMed/NCBI
|
|
10
|
van der Kooij MA, Ohl F, Arndt SS,
Kavelaars A, van Bel F and Heijnen CJ: Mild neonatal
hypoxia-ischemia induces long-term motor- and cognitive impairments
in mice. Brain Behav Immun. 24:850–856. 2010.PubMed/NCBI
|
|
11
|
Brooks SP and Dunnett SB: Tests to assess
motor phenotype in mice: a user's guide. Nat Rev Neurosci.
10:519–529. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bederson JB, Pitts LH, Tsuji M, Nishimura
MC, Davis RL and Bartkowski H: Rat middle cerebral artery
occlusion: evaluation of the model and development of a neurologic
examination. Stroke. 17:472–476. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
d'Avila JC, Lam TI, Bingham D, et al:
Microglial activation induced by brain trauma is suppressed by
post-injury treatment with a PARP inhibitor. J Neuroinflammation.
9:312012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hanisch UK and Kettenmann H: Microglia:
active sensor and versatile effector cells in the normal and
pathologic brain. Nat Neurosci. 10:1387–1394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frank-Cannon TC, Alto LT, McAlpine FE and
Tansey MG: Does neuroinflammation fan the flame in
neurodegenerative diseases? Mol Neurodegener. 4:472009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Polazzi E and Monti B: Microglia and
neuroprotection: from in vitro studies to therapeutic applications.
Prog Neurobiol. 92:293–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaushal V and Schlichter LC: Mechanisms of
microglia-mediated neurotoxicity in a new model of the stroke
penumbra. J Neurosci. 28:2221–2230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hassa PO and Hottiger MO: The functional
role of poly(ADP-ribose)polymerase 1 as novel coactivator of
NF-kappaB in inflammatory disorders. Cell Mol Life Sci.
59:1534–1553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hassa PO, Buerki C, Lombardi C, Imhof R
and Hottiger MO: Transcriptional coactivation of nuclear
factor-kappaB-dependent gene expression by p300 is regulated by
poly(ADP)-ribose polymerase-1. J Biol Chem. 278:45145–45153. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kauppinen TM and Swanson RA:
Poly(ADP-ribose) polymerase-1 promotes microglial activation,
proliferation, and matrix metalloproteinase-9-mediated neuron
death. J Immunol. 174:2288–2296. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kauppinen TM, Higashi Y, Suh SW, Escartin
C, Nagasawa K and Swanson RA: Zinc triggers microglial activation.
J Neurosci. 28:5827–5835. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ullrich O, Diestel A, Eyupoglu IY and
Nitsch R: Regulation of microglial expression of integrins by
poly(ADP-ribose) polymerase-1. Nat Cell Biol. 3:1035–1042. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yrjanheikki J, Keinanen R, Pellikka M,
Hokfelt T and Koistinaho J: Tetracyclines inhibit microglial
activation and are neuroprotective in global brain ischemia. Proc
Natl Acad Sci USA. 95:15769–15774. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fan R, Xu F, Previti ML, et al:
Minocycline reduces microglial activation and improves behavioral
deficits in a transgenic model of cerebral microvascular amyloid. J
Neurosci. 27:3057–3063. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mabley JG, Jagtap P, Perretti M, et al:
Anti-inflammatory effects of a novel, potent inhibitor of poly
(ADP-ribose) polymerase. Inflamm Res. 50:561–569. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mabley JG, Horvath EM, Murthy KG, et al:
Gender differences in the endotoxin-induced inflammatory and
vascular responses: potential role of poly(ADP-ribose) polymerase
activation. J Pharmacol Exp Ther. 315:812–820. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gash DM, Zhang Z, Ovadia A, et al:
Functional recovery in parkinsonian monkeys treated with GDNF.
Nature. 380:252–255. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li L, Wu W, Lin LF, Lei M, Oppenheim RW
and Houenou LJ: Rescue of adult mouse motoneurons from
injury-induced cell death by glial cell line-derived neurotrophic
factor. Proc Natl Acad Sci USA. 92:9771–9775. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Lin SZ, Chiou AL, Williams LR and
Hoffer BJ: Glial cell line-derived neurotrophic factor protects
against ischemia-induced injury in the cerebral cortex. J Neurosci.
17:4341–4348. 1997.PubMed/NCBI
|
|
30
|
Boscia F, Esposito CL, Di Crisci A, de
Franciscis V, Annunziato L and Cerchia L: GDNF selectively induces
microglial activation and neuronal survival in CA1/CA3 hippocampal
regions exposed to NMDA insult through Ret/ERK signalling. PLoS
One. 4:e64862009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hung SY, Liou HC and Fu WM: The mechanism
of heme oxygenase-1 action involved in the enhancement of
neurotrophic factor expression. Neuropharmacology. 58:321–329.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarit BS, Lajos G, Abraham D, Ron A and
Sigal FB: Inhibitory role of kinins on microglial nitric oxide and
tumor necrosis factor-alpha production. Peptides. 35:172–181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boucsein C, Zacharias R, Farber K,
Pavlovic S, Hanisch UK and Kettenmann H: Purinergic receptors on
microglial cells: functional expression in acute brain slices and
modulation of microglial activation in vitro. Eur J Neurosci.
17:2267–2276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nodin C, Zhu C, Blomgren K, Nilsson M and
Blomstrand F: Decreased oxidative stress during glycolytic
inhibition enables maintenance of ATP production and astrocytic
survival. Neurochem Int. 61:291–301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee GA, Lin CH, Jiang HH, Chao HJ, Wu CL
and Hsueh CM: Microglia-derived glial cell line-derived
neurotrophic factor could protect Sprague-Dawley rat astrocyte from
in vitro ischemia-induced damage. Neurosci Lett. 356:111–114. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Beers DR, Henkel JS, Zhao W, Wang J and
Appel SH: CD4+ T cells support glial neuroprotection,
slow disease progression, and modify glial morphology in an animal
model of inherited ALS. Proc Natl Acad Sci USA. 105:15558–15563.
2008.PubMed/NCBI
|