Introduction
Taxanes are microtubule-stabilizing agents (MSAs)
that exhibit anticancer activity in several human solid tumors
(1). Among these taxanes,
docetaxel (Doc) has an increased affinity for tubulin (2) and has higher antitumor activity than
paclitaxel (3). Doc has been
approved for use in the clinical treatment of lung, breast and
prostate cancer (4). Although the
primary action mechanism of this drug is well understood, the
signaling pathways that confer resistance to these agents in
certain types of cancer remain poorly understood.
Several signal transduction pathways are involved in
apoptosis evoked by taxanes (5).
The upregulation of p53 and the cyclin-dependent kinase inhibitor
(p21/Waf1) (6), phosphorylation
of Bcl-2 (7) and activation of
MAP kinases (8) have been
implicated as mediators of Taxol-induced apoptosis. Mitotic
catastrophe resulting from micronucleation or aberrant mitosis also
appears to be a critical step during Doc-induced apoptosis
(9). Recently, we demonstrated
that binding of Smac/Diablo to survivin in the nucleus plays an
important role in Doc-induced apoptosis of prostate cancer cells
(10).
Although p53 is implicated in the modulation of
taxane-induced cancer cell death, it remains controversial whether
it plays a key role in MSA-mediated apoptosis. Several studies have
suggested that Taxol-induced apoptosis is independent of the p53
status (11–16). In contrast, increased sensitivity
to Taxol was observed in non-transformed cells in the absence of
functional p53 (17). Similar
results were observed in ovarian carcinoma cells (18). However, no further study has
evaluated the hypothesis that deficiency or mutation of p53 may
render cancer cells more susceptible to taxane-induced
apoptosis.
In this study, we report that chemical or genetic
knockout of p53 enhances susceptibility to Doc-induced apoptosis in
both prostate and colorectal cancer cells. Our data suggest that
p53 plays a pivotal role in suppressing Doc-induced apoptosis in
prostate and colorectal cancer cells and that the p53 status could
be used to stratify patients for Doc-containing treatment
regimens.
Materials and methods
Reagents
Docetaxel (Taxotere) was obtained from Aventis
Pharmaceuticals (Bridgewater, NJ, USA). Hoechst 33258, RNase A and
propidium iodide (PI) were from Sigma Chemical Co. (St. Louis, MO,
USA). 3,3′-Dihexyloxacarbocyanine iodide [DiOC6(3)] was from Invitrogen Molecular Probes
(Carlsbad, CA, USA).
Antibodies
The antibodies used included rabbit anti-p53 (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA); rabbit anti-cleaved
caspase-3, -7, -8 and -9 (Cell Signaling Technology, Inc., Beverly,
MA, USA); rabbit anti-tBID (p15) cleavage site-specific antibody
(BioSource International, Camarillo, CA, USA); mouse anti-PARP and
rabbit anti-phosphorylated p53 (Calbiochem, San Diego, CA, USA);
mouse anti-β-actin (Sigma); mouse anti-XIAP (BD Pharmingen, San
Diego, CA, USA). Horseradish peroxidase-conjugated anti-rabbit IgG
and anti-mouse IgG (Amersham Pharmacia Biotech, Piscataway, NJ,
USA) and FITC-conjugated anti-rabbit IgG (Vector Laboratories,
Burlingame, CA, USA) were also used.
Cell culture and drug treatment
LNCaP, DU145 and PC3 (human prostate carcinoma cell
lines; American Type Culture Collection, Rockville, MD, USA) were
maintained in Dulbecco’s modified Eagle’s medium/ Nutrient Mixture
F-12 HAM (Sigma) containing 10% heat-inactivated fetal bovine serum
(FBS) (Invitrogen) and 1.2 g/l sodium bicarbonate supplemented with
10 μg/ml penicillin-streptomycin (Invitrogen). HCT-116
p53+/+ and p53−/− cells were provided by Dr
Bert Vogelstein (Howard Hughes Medical Institute, Baltimore, MD,
USA) and maintained in RPMI-1640 (Invitrogen) containing 10%
heat-inactivated FBS and 2 g/l sodium bicarbonate supplemented with
10 μg/ml penicillin-streptomycin. Cells were incubated in a
humidified incubator at 37°C with 5% CO2. Doc (10 mg)
was dissolved in 7.8 ml of 1X PBS (pH 7.4), 7.8 ml of DMSO and 15.6
ml of absolute ethanol as 375 μM stock, and it was added to
cells at 50% confluency.
Wild-type p53 transfection
For transfection, PC3 cells were seeded in 6-well
plates and grown until they attained 50% confluency, and then 1.5
μg of p53 plasmid was introduced into the cells using
Transfectin (Bio-Rad, Richmond, CA, USA) according to the
manufacturer’s instructions. After 24 h, the cells were treated
with 5 nM Doc and then incubated at 37°C for an additional 24
h.
Knockdown of p53 with small interfering
RNA (siRNA)
For transfection, cells were seeded in 6-well plates
and grown until they attained 50% confluency, after which 6
μl of 10 μM stock siRNA (SignalSilence®
p53 siRNA; Cell Signaling Technology, Inc.) was introduced into the
cells using RNAiFect (Qiagen, Valencia, CA, USA) according to the
manufacturer’s instructions.
Sub-G1 analysis
Cells were harvested for cell cycle analysis, fixed
with 95% ethanol (with 0.5% Tween-20) for 24 h, incubated with 0.05
mg/ml PI and 1 μg/ml RNase A at 37°C for 30 min, and
analyzed by flow cytometry using an Epics XL flow cytometer with
analysis software (EXPO32™; Beckman Coulter, Miami, FL, USA). The
cells in the sub-G1 population were considered
apoptotic.
Annexin V cell death assay
Cells were stained with Annexin V-FITC and PI using
the Annexin V-FITC Apoptosis Detection kit (BD Biosciences,
Franklin Lakes, NJ, USA) according to the manufacturer’s protocol.
Briefly, 1×105 cells were pelleted and resuspended in
100 μl of binding buffer. Subsequently, 5 μl of
Annexin V-FITC and PI (0.05 mg/ml) were added to the cells, and
cells were incubated for 15 min at room temperature (RT). After
incubation, 400 μl of the binding buffer was added to the
stained cells, and the cells were analyzed by flow cytometry.
Measurement of mitochondrial membrane
potential
Cells (5×105) were incubated with 100 nM
DiOC6 at 37°C for 30 min and then washed and resuspended
with PBS; thereafter, the fluorescence was measured by flow
cytometry.
Western blot analysis
For western blot analysis, whole cell lysates were
prepared by incubating cell pellets in lysis buffer [30 mM NaCl,
0.5% Triton X-100, 50 mM Tris-HCl (pH 7.4), 1 mM
Na3VO4, 25 mM NaF, 10 mM
Na4P2O7] for 30 min on ice. After
insoluble fractions were removed by centrifugation at 14,000 rpm at
4°C for 40 min, the supernatants were collected, and the protein
concentration was determined using a BCA protein assay kit (Pierce
Biotechnology, Inc., Woburn, MA, USA). Cell lysates (50 μg)
were subjected to SDS-PAGE and transferred onto a nitrocellulose
membrane (Amersham Pharmacia Biotech). Membranes were incubated for
1 h at RT with a primary antibody in Tris-buffered saline
containing 0.05% Tween-20 (TBS-T; pH 7.4) in the presence of 5%
nonfat dry milk. After the membranes were washed in TBS-T,
secondary antibody reactions were performed with an appropriate
source of antibody conjugated with horseradish peroxidase. The
signals were detected with an Enhanced-Chemiluminescence Detection
kit (Amersham Pharmacia Biotech) in the LAS-3000 detector (Fuji,
Japan). Immunoblotting for β-actin was performed in every
experiment as an internal control.
Immunocytochemistry
Collected cells were attached on slide glass by
cytospin centrifugation using a Cellspin™ (Hanil, Korea). Cells
were fixed with 4% paraformaldehyde at RT for 30 min, washed with
PBS for 10 min, and incubated with 0.2% Triton X-100 for 10 min.
Then, cells were washed, incubated with the appropriate primary
antibody in 1% bovine serum albumin at RT for 2 h. For the
secondary antibody reaction, cells were incubated with the
appropriate fluorescein (FITC)-conjugated secondary antibody at RT
for 1 h. For counterstaining of the nucleus (when required), cells
were incubated with PI (50 μg/ml) at RT for 15 min. Finally,
cells were washed, mounted, and observed using a confocal
microscope (LSM510; Carl Zeiss, Germany).
Statistics
Data are expressed as the mean ± SD of 3 or 4
separate experiments and analyzed by the Student’s t-test. Mean
values were considered statistically significant at P<0.05.
Results
Doc does not effectively induce apoptosis
in p53 wild-type prostate cancer cells
To monitor the role of p53 in Doc-induced cancer
cell death, three types of prostate cancer cells [LNCaP (p53
wild-type), DU145 (p53 mutant) and PC3 (p53 null)] with a different
p53 status were primarily employed in this study. According to our
previous study (10), 5 nM Doc
(for 48 h) was considered as the optimal condition for the
induction of apoptosis in these prostate cancer cell lines. Flow
cytometric sub-G1 analysis showed that significant cell
death occurred in DU145 and PC3 cells, but not in LNCaP cells
(Fig. 1A). The type of cell death
induced by Doc was mostly apoptotic, as evaluated by Annexin V
binding assay (Fig. 1B).
Moreover, Doc-induced apoptosis was accompanied by depolarization
of mitochondrial membrane potential (MMP; ΔΨm) (Fig. 1C). These results indicate that Doc
effectively induces apoptosis in prostate cancer cells that harbor
defective p53.
Doc-induced apoptosis in prostate cancer
cells is accompanied by activation of multiple caspases
Changes in total p53 levels and phosphorylated p53
(at serine 15) after exposure to Doc were detected only in LNCaP
cells (Fig. 2). While significant
activation of caspase-9, -8, -7 and -3 was not observed in LNCaP
cells, all caspases were cleaved in both DU145 and PC3 cells
(Fig. 2). Appearance of tBID
supports the evidence that the caspase-8 pathway was also
associated with Doc-induced apoptosis, in conjunction with the
caspase-9 pathway (Fig. 2).
Cleavage of PARP and decreased levels of XIAP protein indicate that
Doc-induced apoptosis in prostate cancer cells was caspase-3
dependent (Fig. 2).
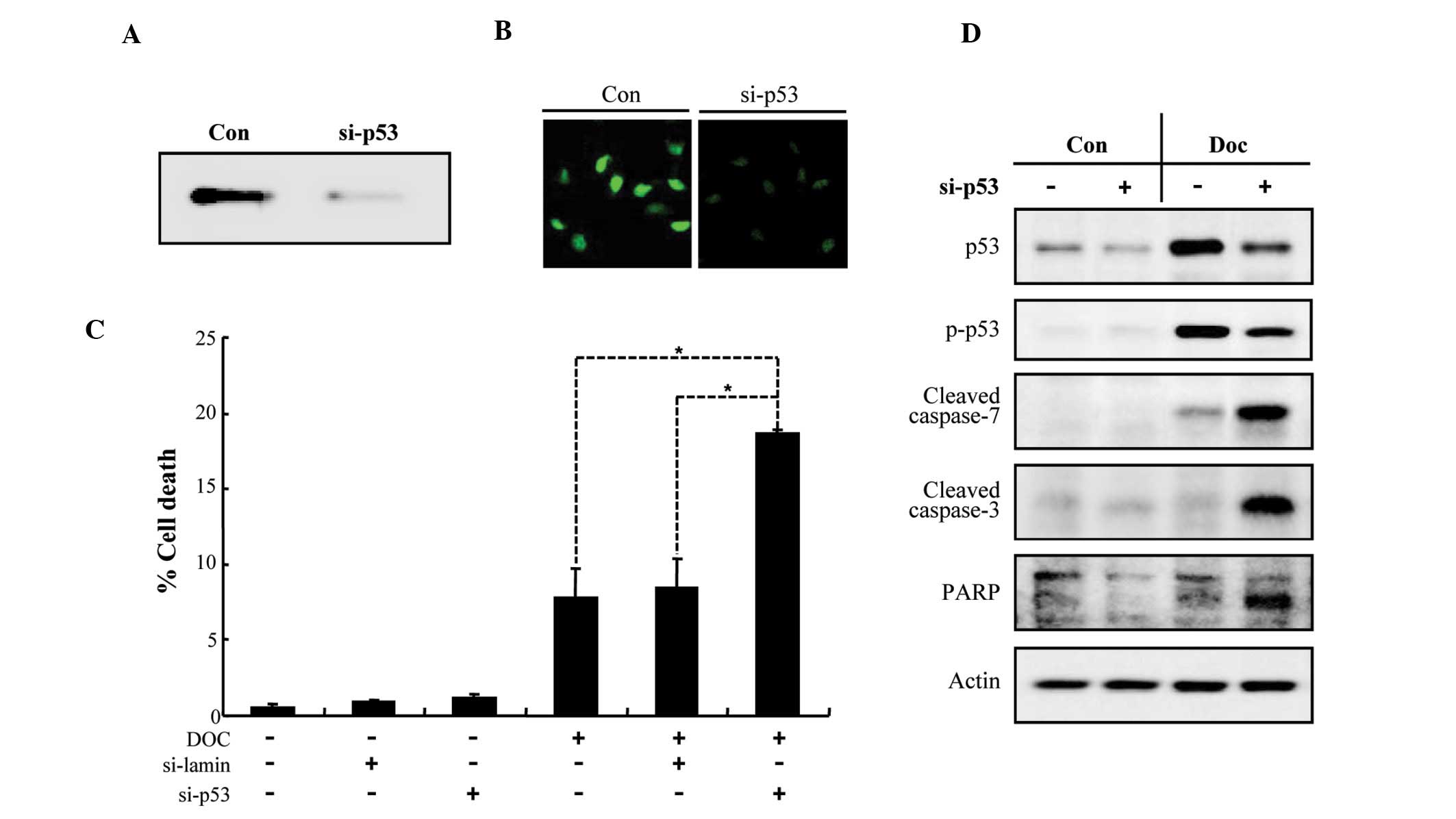
Knockdown of p53 sensitizes LNCaP cells
to Doc-induced apoptosis
In order to examine whether the ablation of p53 in
LNCaP cells increases their susceptibility to Doc-induced
apoptosis, we used siRNA-mediated knockdown of p53. As shown in
Fig. 3A and B, the expression of
endogenous p53 was dramatically reduced by siRNA transfection. p53
knockdown synergistically increased LNCaP cell death induced by Doc
(Fig. 3C), which was associated
with a caspase-dependent pathway (Fig. 3D). These results indicate that p53
plays a crucial role in the suppression of Doc-dependent apoptosis
in prostate cancer cells.
Ectopic expression of p53 attenuates
Doc-induced apoptosis in PC3 cells
We next tested whether transient transfection of p53
into PC3 cells suppresses Doc-induced apoptosis. An increase in
total and phospho-p53 was detected in PC3 cells following
transfection (Fig. 4A and C) and
was associated with a significant repression of Doc-induced
apoptosis (Fig. 4B). The
inhibitory role of p53 in Doc-induced apoptosis was confirmed by
the inhibition of the cleavage of caspase-3 and -7, and PARP that
are provoked by Doc (Fig. 4C).
These results imply that p53 interferes with Doc-induced apoptosis
in prostate cancer cells.
p53-deficient colorectal cancer cells are
more susceptible to Doc-induced apoptosis than those with wild-type
p53
To determine whether p53 plays an inhibitory role in
Doc-induced apoptosis in cells other than prostate cancer cells, an
isogenic pair of colorectal carcinoma (HCT-116) cell lines
differing only in their p53 status was investigated. Doc induced
cell death in a higher number HCT-116 p53−/− cells when
compared to the number in the HCT-116 p53+/+ cells
(Fig. 5A). Apoptosis occurred in
a caspase-dependent manner, as revealed by cleavage of caspase-3
and -7, and PARP (Fig. 5B).
Furthermore, the functional knockdown of p53 in HCT-116
p53+/+ cells by either p53 siRNA or pifithrin-α (PFT-α),
a p53 inhibitor, sensitized cells to Doc-induced apoptosis, which
was concomitant with caspase-3 and -7, and PARP cleavage (Fig. 5C). Taken together, these results
indicate that p53 also inhibits colorectal cancer cell apoptosis
induced by Doc.
Discussion
The p53 gene is frequently mutated in human cancers
(19), and p53 regulates cell
cycle arrest, apoptosis, and DNA repair in a variety of cells
(20). The role of p53 in the
ultimate sensitivity to various anticancer drugs remains
controversial. While several studies have demonstrated that cells
lacking functional p53 are resistant to anticancer agents (21–23), others have reported contrasting
findings (24–26). In this context, we examined
whether the p53 status modulates the sensitivity to apoptosis in
prostate and colorectal cancer cells after exposure to Doc.
In the present study, we demonstrated that p53
strongly inhibits Doc-induced apoptosis in prostate and colorectal
cancer cells. Caspases are central mediators of this process. Doc
treatment induced a graded apoptotic response in prostate cancer
cell apoptosis; specifically, apoptosis was lowest in cells with
wild-type p53 and highest in functionally null p53 PC3 cells. While
transient overexpression of p53 in PC3 cells blocked Doc-induced
apoptosis, knockdown of p53 in LNCaP cells enhanced apoptosis after
Doc exposure. This finding was strongly supported by the results
obtained with an isogenic pair of colorectal cancer cell lines,
HCT-116 p53−/− and p53+/+ cells. Taken
together, these results clearly indicate that p53 exhibits an
inhibitory role in the induction of apoptosis by Doc.
A co-relationship between defects in p53 and
increased sensitivity to Taxol has not been reported in ovarian
(12), colorectal (27), renal (16) and gastric (28) cancer cells. In contrast, loss of
normal p53 function did confer sensitization to Taxol by increasing
G2/M arrest and apoptosis (17), and similar results were found in
IGROV-1 ovarian carcinoma cells (18). This inconsistency may be
attributed to the different origin or cellular constituents of each
cancer cell type. Survivin is repressed by p53 (20) and is also implicated in the
assembly process of microtubule polymerization during Doc-induced
apoptosis in p53-mutated prostate cancer cells (10). Therefore, it is tempting to
speculate that p53-mediated repression of survivin may block the
MSA-induced cell death machinery in cancer cells. Further studies
of the potential role of p53 in regulating microtubule dynamics are
required in order to understand the relationship between p53 and
taxane resistance in various cancer cells.
The findings of this study suggest that p53 is a
potent inhibitor of Doc-induced apoptosis in both prostate and
colorectal cancer cells. Thus, the genetic and functional status of
p53 may be an important factor (biomarker) in guiding therapeutic
strategies for prostate and colorectal cancer patients. A
combinational treatment of Doc and p53-specific antagonists (i.e.,
PFT-α) could be considered for the development of a potential novel
chemotherapy for taxane-resistant cancers in the near future.
Acknowledgements
This study was supported by the Dong-A
University Research Fund.
References
|
1
|
Rowinsky EK: The development and clinical
utility of the taxane class of antimicrotubule chemotherapy agents.
Annu Rev Med. 48:353–374. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Diaz JF and Andreu JM: Assembly of
purified GDP-tubulin into microtubules induced by taxol and
taxotere: reversibility, ligand stoichiometry, and competition.
Biochemistry. 32:2747–2755. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lavelle F, Bissery MC, Combeau C, Riou JF,
Vrignaud P and André S: Preclinical evaluation of docetaxel
(Taxotere). Semin Oncol. 22:3–16. 1995.
|
|
4
|
Crown J and O’Leary M: The taxanes: an
update. Lancet. 355:1176–1178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang TH, Popp DM, Wang HS, et al:
Microtubule dysfunction induced by paclitaxel initiates apoptosis
through both c-Jun N-terminal kinase (JNK)-dependent and
-independent pathways in ovarian cancer cells. J Biol Chem.
274:8208–8216. 1999. View Article : Google Scholar
|
|
6
|
Blagosklonny MV, Schulte TW, Nguyen P,
Mimnaugh EG, Trepel J and Neckers L: Taxol induction of
p21WAF1 and p53 requires c-raf-1. Cancer Res.
55:4623–4626. 1995.PubMed/NCBI
|
|
7
|
Haldar S, Chintapalli J and Croce CM:
Taxol induces bcl-2 phosphorylation and death of prostate cancer
cells. Cancer Res. 56:1253–1255. 1996.PubMed/NCBI
|
|
8
|
Bacus SS, Gudkov AV, Lowe M, et al:
Taxol-induced apoptosis depends on MAP kinase pathways (ERK and
p38) and is independent of p53. Oncogene. 20:147–155. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morse DL, Gray H, Payne CM and Gillies RJ:
Docetaxel induces cell death through mitotic catastrophe in human
breast cancer cells. Mol Cancer Ther. 4:1495–1504. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim JY, Chung JY, Lee SG, et al: Nuclear
interaction of Smac/DIABLO with Survivin at G2/M arrest prompts
docetaxel-induced apoptosis in DU145 prostate cancer cells. Biochem
Biophys Res Commun. 350:949–954. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Woods CM, Zhu J, McQueney PA, Bollag D and
Lazarides E: Taxol-induced mitotic block triggers rapid onset of a
p53-independent apoptotic pathway. Mol Med. 1:506–526.
1995.PubMed/NCBI
|
|
12
|
Debernardis D, Siré EG, De Feudis P, et
al: p53 status does not affect sensitivity of human ovarian cancer
cell lines to paclitaxel. Cancer Res. 57:870–874. 1997.PubMed/NCBI
|
|
13
|
Takahashi M, Kigawa J, Minagawa Y, et al:
Sensitivity to paclitaxel is not related to p53-dependent apoptosis
in ovarian cancer cells. Eur J Cancer. 36:1863–1868. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Osaki S, Nakanishi Y, Takayama K, Pei XH,
Ueno H and Hara N: Alteration of drug chemosensitivity caused by
the adenovirus-mediated transfer of the wild-type p53 gene in human
lung cancer cells. Cancer Gene Ther. 7:300–307. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sezgin C, Karabulut B, Uslu R, et al:
Potential predictive factors for response to weekly paclitaxel
treatment in patients with metastatic breast cancer. J Chemother.
17:96–103. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reinecke P, Kalinski T, Mahotka C, et al:
Paclitaxel/Taxol sensitivity in human renal cell carcinoma is not
determined by the p53 status. Cancer Lett. 222:165–171. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wahl AF, Donaldson KL, Fairchild C, et al:
Loss of normal p53 function confers sensitization to Taxol by
increasing G2/M arrest and apoptosis. Nat Med. 2:72–79. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cassinelli G, Supino R, Perego P, et al: A
role for loss of p53 function in sensitivity of ovarian carcinoma
cells to taxanes. Int J Cancer. 92:738–747. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vogelstein B: Cancer. A deadly
inheritance. Nature. 348:681–682. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bates S and Vousden KH: p53 in signaling
checkpoint arrest or apoptosis. Curr Opin Genet Dev. 6:12–18. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Clarke AR, Purdie CA, Harrison DJ, et al:
Thymocyte apoptosis induced by p53-dependent and independent
pathways. Nature. 362:849–852. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee JM and Bernstein A: p53 mutations
increase resistance to ionizing radiation. Proc Natl Acad Sci USA.
90:5742–5746. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lowe SW, Schmitt EM, Smith SW, Osborne BA
and Jacks T: p53 is required for radiation-induced apoptosis in
mouse thymocytes. Nature. 362:847–849. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brown R, Clugston C, Burns P, et al:
Increased accumulation of p53 protein in cisplatin-resistant
ovarian cell lines. Int J Cancer. 55:678–684. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fan S, Smith ML, Rivet DJ II, et al:
Disruption of p53 function sensitizes breast cancer MCF-7 cells to
cisplatin and pentoxifylline. Cancer Res. 55:1649–1654.
1995.PubMed/NCBI
|
|
26
|
Hawkins DS, Demers GW and Galloway DA:
Inactivation of p53 enhances sensitivity to multiple
chemotherapeutic agents. Cancer Res. 56:892–898. 1996.PubMed/NCBI
|
|
27
|
van Bree C, Savonije JH, Franken NA,
Haveman J and Bakker PJ: The effect of p53-function on the
sensitivity to paclitaxel with or without hyperthermia in human
colorectal carcinoma cells. Int J Oncol. 16:739–744.
2000.PubMed/NCBI
|
|
28
|
Matsuhashi N, Saio M, Matsuo A, Sugiyama Y
and Saji S: Apoptosis induced by 5-fluorouracil, cisplatin and
paclitaxel are associated with p53 gene status in gastric cancer
cell lines. Int J Oncol. 26:1563–1567. 2005.PubMed/NCBI
|