Introduction
The incidence of obesity worldwide has increased
significantly during recent decades. Consequently, obesity and its
associated disorders constitute a serious threat to the current and
future health of populations (1).
It is now well established that obesity results in a state of
chronic low-grade inflammation thought to contribute to several
metabolic disorders, including insulin resistance, type II diabetes
and cardiovascular disease (2,3).
In individuals with these metabolic disorders, different
cardiovascular disorders often develop simultaneously, thus obese
patients present with an increased risk of suffering from insulin
resistance and type II diabetes, these being frequently associated
with cardiovascular disease including hypertension,
atherosclerosis, arrhythmias and heart failure, suggesting the
sharing of similar pathogenic mechanisms (4,5).
Nevertheless, the exact underlying mechanisms involved remains
elusive.
Transcription factor nuclear factor-κB (NF-κB) is a
potent transcriptional activator and plays an important role in a
variety of physiological and disease processes, including immune
and inflammatory responses, cardiovascular diseases, insulin
resistance and type II diabetes (6–8).
The NF-κB proteins are a family of ubiquitously expressed
transcription factors that, in mammals, comprise five members:
NF-κB1 (p50; precursor protein: p105), NF-κB2 (p52; precursor
protein: p100), p65 (RelA), c-Rel (Rel) and RelB, which share the
so-called Rel homology domain that mediates DNA binding,
dimerization and nuclear translocation (9,10).
NF-κB activation is triggered by the phosphorylation of the
regulatory protein IκB, and regulated through the canonical IκB
kinases [kinase IκB kinase (IKK)α, IKKβ] and non-canonical
IKK-related kinases (IKKɛ, TBK1). Findings of previous studies
suggested a role for IKKα and β in NF-κB activation that leads to
an increase in transcription of inflammation, and cell
growth/survival of genes (11–13). However, the kinase domain of IKKɛ
only exhibits 27 and 24% identity to IKKα and β, respectively
(14), suggesting that IKKɛ may
have a different role to IKKα and β.
The stress-activated protein kinase IKKɛ is a
central signal transducer that regulates immune response, cell
proliferation and transformation, and oncogenesis (15–20). Results of a previous study showed
that a high-fat diet (HFD) can increase NF-κB activation in mice,
which leads to a sustained elevation of IKKɛ levels in the liver,
adipocytes and adipose tissue macrophages. Deletion of the IKKɛ
gene rendered mice partially resistant to the HFD-dependent
development of obesity, insulin sensitivity, hepatic steatosis and
inflammation (15). Based on this
established connection between obesity-induced insulin resistance
and enhanced NF-κB activity in insulin target tissues, we
investigated whether IKKɛ has a similar role in HFD-induced
cardiovascular disorders.
To address this issue, murine models of
apolipoprotein E-deficient [ApoE(−/−)] mice and ApoE/IKKɛ
double-knockout mice [ApoE(−/−)/IKKɛ(−/−)] were established. One
group of ApoE(−/−)/IKKɛ(−/−) mice was switched from normal diet
(ND) to HFD for 12 weeks from 8 weeks of age, respectively, and
another control group [ApoE(−/−) mice] also maintained the same
diet. Therefore, IKKɛ modulates HFD-induced obesity, inflammation
by activation of NF-κB-dependent gene transcription and may be
useful as a therapeutic target in the treatment of
metabolism-associated cardiovascular diseases.
Materials and methods
Animal models
IKKɛ knockout
(B6.Cg-Ikbke<tm1Tman>/J) mice, purchased from
Jackson Laboratory (Bar Harbor, ME, USA), underwent rederivation to
achieve pathogen-free status in the Model Animal Research Center of
Nanjing University (Nanjing, China). ApoE knockout [ApoE(−/−)] mice
were obtained from the Model Animal Research Center of Nanjing
University at the age of 8 weeks. IKKɛ knockout mice were bred into
the ApoE knockout genetic background to obtain the
ApoE(−/−)/IKKɛ(−/−) group of mice, as reported previously (21). At the age of 8 weeks, the male
ApoE(−/−) and ApoE(−/−)/IKKɛ(−/−) mice were placed on a HFD
containing 60% calories of lipid (soybean oil, 5.5%; lard, 54.5%;
D12492; Research Diets, Inc., New Brunswick, NJ, USA) and fed for
12 weeks, whereas the remaining mice were kept as controls on a
standard ND containing 10% calories for 12 weeks. All the mice were
housed in specific pathogen-free box cages at a constant
temperature of 23±2°C and humidity of 60±10%. An inverted
light-dark cycle of 12:12 h was used and the animals had free
access to food and water. Both male and female mice were 12 weeks
old at the time of experiments. Animal experiments were performed
in compliance with the Institute of Laboratory Animal Research
Guide for the Care and Use of Laboratory Animals of the National
Institutes of Health and approved by the Institutional Animal Care
and Use Committee of Nanjing Medical University.
Plasma parameters
After being fed with ND and HFD for 12 weeks,
respectively, mice were fasted for 12 h prior to being anesthetized
through administration of intraperitoneal injection of
pentobarbital (50 mg/kg body weight), and the adequacy of
anesthesia was evaluated by monitoring hind limb reflexes. Blood
samples were obtained from the retro-orbital plexus. Total
cholesterol (TC), triglycerides (TG), low-density lipoprotein (LDL)
and high-density lipoprotein (HDL) in the serum were determined
using colorimetric enzymatic assays that were adjusted to the
96-well format (Sigma, St. Louis, MO, USA).
Tissue collection and histological
analysis
After euthanasia, murine heart tissues were
collected, fixed in 4% formalin for 48 h and then embedded in
paraffin. Serial aortic sections (4 μm) were then prepared and
stained with hematoxylin and eosin (H&E) for histopathology.
Formalin-fixed, paraffin-embedded sections were subjected to
antigen retrieval by boiling in 0.01 M sodium citrate buffer (pH
6.0) in a microwave oven after dewaxing and rehydration. The
sections were treated with 3% hydrogen peroxide for 15 min to block
endogenous peroxidase activity and incubated in buffered normal
horse serum to prevent non-specific binding of antibodies. The
sections were then incubated separately for 14 h with antibodies
against tumor necrosis factor-α (TNF-α) (1:800), macrophage-colony
stimulating factor (M-CSF) (1:200), CD4 T cells (CD4) (1:200) (all
from Abcam, Cambridge, MA, USA), IKKɛ (1:500; Novus Biologicals,
Littleton, CO, USA) followed by incubation with horseradish
peroxidase (HRP)-conjugated goat anti-rabbit IgG (Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd., Beijing, China) for 1 h at
37°C in a humidified box. The signal of each antibody was developed
using the substrate diaminobenzidine (DAB; Beijing Zhongshan Golden
Bridge Biotechnology Co.). Sections were counterstained with
hematoxylin and photomicrographs were obtained using an Olympus
BX-URA2 camera.
Quantitative reverse transcription PCR
(qRT-PCR)
For qPCR, total RNA was extracted from the frozen
mouse tissue using TRIzol (Invitrogen, Carlsbad, CA, USA) and
reverse-transcribed into cDNA using oligo(dT) primers with a
transcriptor first-strand cDNA synthesis kit. PCR amplifications
were quantified using the SYBR-Green PCR Master mix (Applied
Biosystems, Foster City, CA, USA) and normalized to GAPDH gene
expression. The primers for quantitative PCR are shown in Table I.
 | Table IThe primers for vector construct. |
Table I
The primers for vector construct.
| Name | Forward | Reverse |
|---|
| IKKα |
GTCAGGACCGTGTTCTCAAGG |
CAGGCACCGTTCACACATAC |
| IKKβ |
AGCGAACGGTCATCCACGTCTT |
ACTGGTGATCTCTATGCTGTCA |
| IKKɛ |
ACTCCACTCACGGCAAATTC |
GCTTCTTTGATGTTACTGAGGGC |
| TBK1 |
CAAGATCCATGTCCAACGTG |
AGATGGCAATCGTGTTGTGGGC |
| GAPDH |
TTCTGGAAGTCCATACGCATTG |
TCTCCATGGTGGTGAAGACA |
Western blot analysis
Samples of total protein (50 μg) were extracted from
the murine heart of each group and separated by SDS-PAGE. Proteins
were then transferred to a polyvinylidene fluoride (PVDF) membrane
(Millipore Corp., Billerica, MA, USA), washed with a dilution of
1:1,000 in TBS with Tween (TBST; Promega Corp., Madison, WI, USA)
twice (10 min per wash) and blocked using 5% non-fat milk powder
for 1 h. The membranes were then probed with the following primary
antibodies in TBS with Tween plus 5% milk overnight at 4°C:
anti-IKKɛ (1:200; Novus Biologicals), anti-T-p50 (1:200),
anti-T-p65 (1:200), anti-IκB-α (1:200), anti-Pi-50 (1:300),
anti-Pi-65 (1:300), anti-IL-1β (1:500), anti-TGF-β (1:500), and
anti-GAPDH (1:5,000) (all from Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA). The following day, the PVDF membranes were
washed with TBST four times (10 min per wash) and incubated with
appropriately diluted peroxidase-conjugated secondary antibodies
and goat anti-rabbit IgG (anti-mouse and anti-rabbit
immunoglobulins, respectively; Santa Cruz Biotechnology, Inc.) at
room temperature for 1 h. The membranes were then washed with TBST
four times as previously described. Specific proteins were detected
using an ECL reagent and captured on Hyperfilm (both from GE
Healthcare, Piscataway, NJ, USA). The results were then analyzed
through the Quantity One software for semiquantification of the
mean gray value of each blot. Subsequently, the SPSS statistical
software was used to perform one-way analysis of variance (ANOVA)
to detect the differences among groups of mice. All presented
results are representative of at least three independent
experiments.
Immunofluorescence staining
Tissues collected for morphological analysis of the
heart were prepared as 4 μm thick serial optimum cutting
temperature (OCT) compound-embedded cryosections. The sections were
fixed in 4% paraformaldehyde for 30 min, washed in PBS, and
incubated in buffered normal goat serum to preclude non-specific
binding of antibodies at room temperature for 1 h. The sections
were then incubated separately overnight with antibodies against
Pi-p50, Pi-p65 (1:100; Santa Cruz Biotechnology, Inc.),
respectively, followed by incubation with Alexa Fluor 592 goat
anti-rabbit IgG (1:200; Invitrogen) at 37°C for 1 h in a humidified
box. Subsequently, the sections were washed in PBS and
counterstained with Hoechst DNA dye (10 mg/ml; Sigma) to irradiate
nuclei. Photomicrographs were captured at random using an Olympus
BX-URA2 camera in 5 sections per mouse sample.
Immunoprecipitation analysis
For immunoprecipitation analysis, tissue samples
were homogenized in ice-cold PBS and lysed for 2 h at 4°C in RIPA
buffer (Roche Diagnostics Norge AS, Oslo, Norway). Lysates were
cleared by centrifugation at 4°C for 10 min at 15,000 × g. Protein
A/G magnetic beads (Millipore) were washed and resuspended
individually according to the manufacturer’s instructions and
subsequently incubated overnight at 4°C with 10 μg anti-IKKɛ
(1:500) or anti-T-p50 (1:500), anti-T-p65 (1:500) (all from Santa
Cruz Biotechnology) antibody. Antibody-bound beads were washed
three times and incubated with cell lysate samples (500 μg) for 2 h
at 4°C in a volume of 1 ml. After washing, immunoprecipitated
proteins were eluted by heating at 100°C for 5 min in Laemmli
sample buffer. Samples of equal protein content were separated by
SDS-PAGE, transferred to PVDF and subsequently processed according
to the procedure described for western blot analysis.
Statistical analysis
The data are presented as the means ± SEM.
Differences among groups were determined by a two-way ANOVA
followed by SPSS 17.0 (SPSS, Inc., Chicago, IL, USA) software.
Comparisons between two groups were performed using an unpaired
Student’s t-test. The significance level was set at P<0.05.
Results
IKKɛ expression induced by HFD is
upregulated in ApoE(−/−) murine hearts
To investigate the potential response of the
inducible IKK family members to dietary stresses, we first analyzed
the mRNA levels of IKKs after ND or HFD for 12 weeks by qRT-PCR.
IKKɛ mRNA levels in the murine hearts were significantly
upregulated at 12 weeks after HFD, compared with the expression of
IKKα, β and TBK1 (Fig. 1A).
Consistent with these data, the protein expression levels of IKKɛ
in hearts from ApoE(−/−) mice after 12-week HFD also showed the
same trends by western blot analysis assays (Fig. 1B and C), suggesting that IKKɛ
plays a different role to IKKα, β and TBK1 in hearts. We then
examined expression in murine hearts by immunohistochemical
analysis. As expected, IKKɛ expression was frequently elevated in
the ApoE(−/−) group HFD-fed mice as compared to the ND controls.
Notably, we observed distinct IKKɛ staining, distributed diffusely
in the nucleus or cytoplasm, or both (Fig. 1D). However, no staining was
observed in the ApoE(−/−)/IKKɛ(−/−) group at the same time,
confirming that IKKɛ expression was absent in the hearts. Thus, the
altered pattern of IKKɛ expression in the hearts, suggested the
possibility that IKKɛ functions as a modulator of HFD-induced
metabolic and cardiovascular disorders.
ApoE(−/−)/IKKɛ(−/−) group mice gain less
weight although similar plasma lipid levels are produced by HFD
compared to ApoE(−/−)group mice
To examine the potential role of IKKɛ in obesity and
lipid metabolism, male ApoE(−/−) mice and ApoE(−/−)/IKKɛ(−/−) mice
were fed a HFD or a regular ND for 12 weeks starting at the age of
8 weeks, while monitoring body weight and food intake. Body weights
were determined every third week during the course of the study. As
shown in Fig. 2A (Table II), ApoE(−/−) mice gained almost
20 g following HFD, whereas the ApoE(−/−)/IKKɛ(−/−) group mice
gained less weight than the ApoE(−/−) group mice (final weight
34.1±0.45 g vs. 39.7±0.52 g; P<0.05). There was no obvious
alteration on food intake, either at the beginning or end of the
study (Fig. 2B, Table II), indicating that changes in
weight gain could not be attributed to greater food ingestion.
 | Figure 2Apolipoprotein E-deficient
[ApoE(−/−)]/IκB kinase (IKK) ɛ (−/−) murine group blunts weight
gain, however, similar plasma lipid levels were produced by
high-fat diet (HFD) compared to ApoE(−/−) murine group. (A) Body
weight measured for mice fed with normal diet (ND) or HFD is shown
at time-points of 0, 3, 6, 9 and 12 weeks. (B) Food intake per body
weight was measured each morning by weighing of ND or HFD, and the
mice of each group were measured at three different time-points
from week 0, 6–12 to obtain the average food intake, n=8–11 per
group. (C) Serum total cholesterol (TC), triglycerides (TG),
low-density lipoprotein (LDL) and high-density lipoprotein (HDL)
levels analysis after 12 weeks fed with ND or HFD as indicated,
n=8–11 per group. *P<0.05 vs. AKO ND;
§P<0.05 vs. DKO ND; #P<0.05 vs. AKO
HFD. |
| Table IIMetabolic parameters of different
groups of mice exposed to a ND or HFD diet. |
Table II
Metabolic parameters of different
groups of mice exposed to a ND or HFD diet.
| AKO | DKO |
|---|
|
|
|
|---|
| Parameter | ND | HFD | ND | HFD |
|---|
| Number | 9 | 11 | 8 | 10 |
| BW at week 0 (g) | 21.0±0.32 | 20.9±0.35 | 21.1±0.26 | 20.8±0.24 |
| BW at week 12
(g) | 28.4±0.29 | 39.7±0.52a | 28.9±0.47 | 34.1±0.45b,c |
| Food intake per BW
(mg/g) | 148.5±3.0 | 122.8±3.1 | 142.6±3.8 | 115.3±3.9 |
| TC (mmol/l) | 10.1±0.75 | 22.3±0.64a | 10.2±0.31 | 21.4±0.71b |
| TG (mmol/l) | 1.23±0.14 | 3.34±0.25a | 1.22±0.11 | 3.31±0.16b |
| LDL (mmol/l) | 0.96±0.09 | 6.22±0.31a | 1.17±0.06 | 6.50±0.20b |
| HDL (mmol/l) | 1.84±0.07 | 0.77±0.05a | 2.06±0.10 | 0.64±0.04b |
Although no significant serum lipid alterations were
observed between the ApoE(−/−) and ApoE(−/−)/IKKɛ(−/−) murine
groups following ND, HFD produced marked increase in the serum TC
and TG levels of the ApoE(−/−) and ApoE(−/−)/IKKɛ(−/−) murine
groups. However, after exposure for 12 weeks to HFD, no distinct
difference in the level of either serum TC or TG was observed
between the ApoE(−/−) and ApoE(−/−)/IKKɛ(−/−) murine groups
(Fig. 2C, Table II). Consistent with these data,
the LDL measurements also showed the same trends as that of TC and
TG, whereas the HDL levels were totally reversed (Fig. 2C, Table II). Collectively, these data
suggest that IKKɛ deficiency leads to decreased weight gain, but
not plasma lipid levels.
IKKɛ deficiency attenuates chronic,
high-fat diet-induced inflammation in the heart after HFD
feeding
To assess in more detail whether the impacts of IKKɛ
deficiency could be seen in the adult mammalian heart, we measured
the expression of key metabolic and inflammatory markers followed
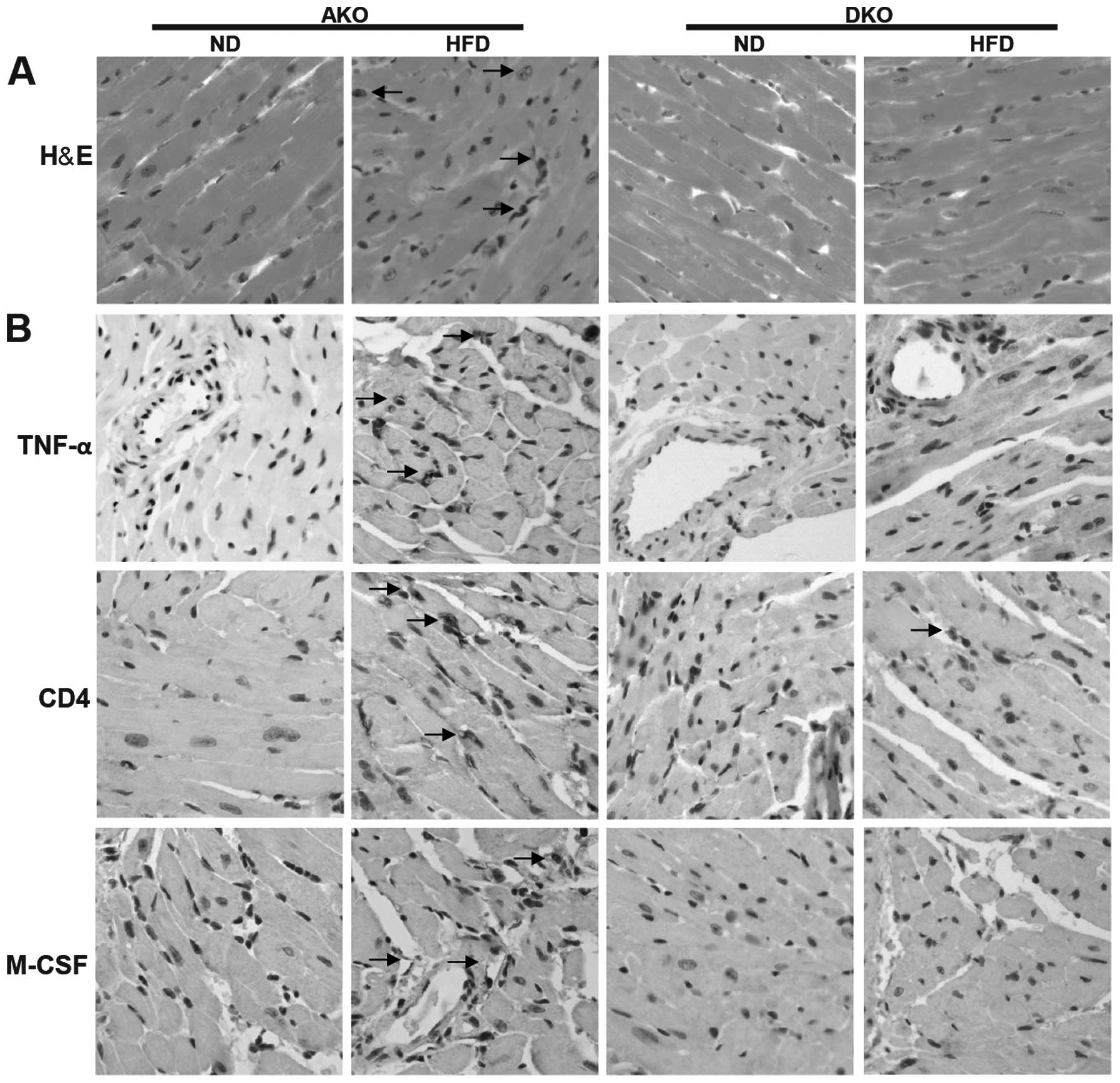
by histopathological analysis. H&E staining of myocardial
nuclei exhibited slightly but significantly higher heterogeneity,
disarray and uneven distribution in the ApoE(−/−)/IKKɛ(−/−) group,
while no significant changes were detected in the other groups
(Fig. 3A). However, Oil Red O
staining revealed that lipid deposition was not different between
the ApoE(−/−) and ApoE(−/−)/IKKɛ(−/−) murine groups mice (data not
shown).
Comparison of ApoE(−/−) and ApoE(−/−)/IKKɛ(−/−)
animals on ND revealed similar inflammatory mediators including
TNF-α, CD4 and M-CSF (Fig. 3B).
Following HFD feeding, the inflammatory markers were increased in
the two groups, and showed a trend towards higher values compared
to the ApoE(−/−)/IKKɛ(−/−) mice, suggesting suppressive
inflammatory activity in the heart of ApoE(−/−)/IKKɛ(−/−) mice.
Thus, IKKɛ may contribute to the generation of low-grade, sustained
inflammation in the heart after HFD feeding.
Ablation of IKKɛ blocks NF-κB activation
induced by HFD
To investigate the regulatory mechanisms of IKKɛ in
animal models, we examined the levels of p50, p65, IκB-α and the
NF-κB pathway downstream factor (TGF-β, IL-β) proteins in hearts.
By western blot analysis, the expression levels of total-p50
(T-p50) and total-p65 (T-p65), two important components that
usually form the most common heterodimers of NF-κB, were shown not
to be different between ApoE(−/−)/IKKɛ(−/−) and ApoE(−/−) murine
groups (Fig. 4A). However,
phosphorylated p50 (Pi-p50) and phosphorylated p65 (Pi-p65),
comprising the functionally active form of the transcription factor
in the nucleus, and IκB-α, which binds with NF-κB to inhibit its
activation, were also detected (Fig.
4A). The results showed that the expression of Pi-p50 and
Pi-p65 (Fig. 4A) were
significantly higher in the ApoE(−/−) group compared to the
ApoE(−/−)/IKKɛ(−/−) group after 12 weeks HFD feeding, while the
expression pattern of IκB-α (Fig.
4A) was reversed among the four groups of mice. The protein
expression of IκB-α was highly increased in the HFD-fed
ApoE(−/−)/IKKɛ(−/−) group compared to the ApoE(−/−) group, while
that of the other two groups of mice remained at the same level. In
addition, the protein expression of the NF-κB pathway downstream
factors, TGF-β and IL-β, were also downregulated in the HFD-fed
ApoE(−/−)/IKKɛ(−/−) group compared with the ApoE(−/−) group
(Fig. 4B).
 | Figure 4Ablation of IκB kinase (IKK) ɛ blocks
nuclear factor-κB (NF-κB) activation induced by high-fat diet
(HFD). (A) Protein levels of T-p50, T-p65, Pi-p50, Pi-p65, IκB-α,
TGF-β and IL1-β were measured by western blot analysis. (B)
Quantitative results of the total-p50 (T-p50), total-p65 (T-p65),
phosphorylated p50 (Pi-p50) and phosphorylated p65 (Pi-p65), IκB-α,
TGF-β and IL1-β in AKO and DKO murine hearts at 12 weeks fed with
normal diet (ND) or HFD. Values are means ± SEM, n=4 per group.
*P<0.05 vs. AKO ND; §P<0.05 vs. DKO ND;
#P<0.05 vs. AKO HFD; Densitometric data are from one
representative experiment of three separate experiments. (C)
Representative merged images show the protein expression of NF-κB
cascade components Pi-p50 and Pi-p65 as determined by
immunofluorescent staining. Nuclei (Hoechst, blue), positive
staining (red), n=4 per group; magnification, ×400. (D)
Localization of Pi-p50 and Pi-p65 was widely distributed throughout
the nucleus of the myocardium. |
Consistent with results of the western blot
analysis, we also observed that the expression levels of Pi-p50 and
Pi-p65 were lower in the myocardium of the HFD-fed
ApoE(−/−)/IKKɛ(−/−) group than the ApoE(−/−) group by
immunofluorescent analysis (Fig.
4C). Localization of Pi-p50 and Pi-p65 was widely distributed
throughout the nucleus of the myocardium. The localization of these
phosphorylated proteins transferred to the nuclei, suggesting that
most of the activated forms of the nuclear factor shifted to the
nuclei of the myocardium to function as a downstream target gene
transcriptional regulator in the ApoE(−/−) group (Fig. 4D). Thus, these results confirm the
inhibitory effect of IKKɛ ablation on NF-κB pathway.
IKKɛ-mediated inflammation is associated
with the NF-κB pathway
To identify the molecular mechanisms underlying the
positive effects of IKKɛ on inflammatory response, we determined
whether IKKɛ directly interacted with the T-p50/p65 heterodimer by
immunoprecipitation experiments. The results showed that IKKɛ
co-precipitated along with T-p50 and T-p65 (Fig. 5), which map IKKɛ. Additionally,
the NF-κB pathway mediated the interaction during inflammation.
This interaction suggested that expression levels of the NF-κB
pathway were significantly correlated with IKKɛ activation.
Discussion
Findings of previous studies suggest that obesity
generates a state of chronic, low-grade inflammation (15,22,23). Transcription factor NF-κB is a
strong transcriptional activator that may play a crucial role in
this response, although involvement of the liver and adipose tissue
or whether the heart is affected and the processes involved, remain
uncertain. The results of this study identify and delineate one
pathway by which the non-canonical IKKɛ leads to the activation of
canonical NF-κB signaling, and plays a significant role in
regulating the expression of genes involved in chronic, low-grade
inflammatory response in hearts of obese mice.
In the present study, we first examined the mRNA
levels of the inducible IKK family members induced by HFD in the
ApoE(−/−) murine hearts. Notably, IKKɛ only was upregulated in the
ApoE(−/−) mice-fed HFD. Consistent with this finding, the protein
expression levels of IKKɛ in the hearts from the ApoE(−/−) mice
after 12-week HFD also showed the same trends by western blot
analyses. Furthermore, the monitoring of body weight showed that
upregulation of IKKɛ may be correlated with weight gain, whereas
the increased weight gain was completely blocked in the
ApoE(−/−)/IKKɛ(−/−) group. Thus, these results suggest an
inhibitory role for IKKɛ in HFD-induced obesity. Notably, under
HFD, ablation of IKKɛ in mice did not significantly alter the total
levels of plasma lipid levels, including TC, TG, LDL and HDL, which
excluded the potential role for lipid alteration in the HFD-induced
morphological and molecular adaptation.
To understand the effect of IKKɛ, which contributes
to obesity-associated disorders, we examined the morphological and
molecular alteration in hearts. A series of histopathological
changes were evidenced by the application of H&E staining, but
not Oil Red O staining. In addition, under the HFD feeding regimen
used here, we observed that the hearts of the ApoE(−/−) mice had a
low-grade, chronic inflammation/inflammatory cell infiltration, and
certain inflammatory markers (TNF-α, CD4, M-CSF) were elevated,
which could be reversed by knockout of IKKɛ. However, whether IKKɛ
is capable of facilitating obesity-induced inflammation remains
controversial. At present, studies conducted by Scheja et al
suggest that IKKɛ knockout had no effect on inflammation in major
metabolic tissues and on insulin resistance when the anti-obesity
effect of the IKKɛ knockout was overridden by a more aggressive HFD
regimen (24). Thus, the
inflammatory effects of IKKɛ in metabolic disease are apparently
dependent on its obesogenic effects. Reilly et al study have
also shown that the anti-obesogenic effects of IKKɛ and TBK1
inhibitors, amlexanox, suppress inflammation and improve metabolic
homeostasis in murine models of obesity (25). Thus, there is controversy
regarding the effect of IKKɛ knockout on body weight regulation,
inflammation and insulin resistance. As discussed above, it seems
that multiple mechanisms are involved, but one of the first and
potentially most important control mechanisms characterized for
inflammation is its regulation of weight gain. The most significant
metabolic effect of IKKɛ knockout in our study was also the
prevention of weight gain on a HFD, which is consistent with recent
advancements in the development of atherosclerosis from our
laboratory (21). Thus,
limitations of this study include the influence of the prevention
of weight gain on inflammation. Concerning the cell, Olefsky et
al showed that the protein kinase IKKɛ is an important
connection between obesity and inflammation (26). As such, findings of this study
show that IKKɛ is a bridge between obesity and inflammation
(21,26). Thus, the direct regulatory
mechanism involving IKKɛ and inflammation in heart should be
further investigated.
An essential component of the NF-κB signaling
mechanism is the canonical IκB kinase complex (IKKα, IKKβ), which
is activated in response to many signals. The upstream activators
and downstream target of this complex are well characterized
(27). By contrast, relatively
little is known concerning the related kinase IKKɛ. Another
hypothesis in the present study was that the expression of IKKɛ
would, at least partially, lead to HFD-inducing the activation of
NF-κB cascade. The expression levels of Pi-p65/p50 in the murine
hearts were all downregulated in the ApoE(−/−)/IKKɛ(−/−) group
compared with the ApoE(−/−) group. Unlike the other components of
the NF-κB cascade, the decrease in expression of IκB-α in the
ApoE(−/−) group was reversed. This result may be explained by HFD
inducing the NF-κB cascade through the phosphorylation and
ubiquitination of IκB-α to release NF-κB for its translocation to
the nucleus. Since there was more degradation of IκB-α, the
expression of the IκB-α protein was likely to decrease, as
confirmed by a previous study (27). Furthermore, consistent with the
upstream NF-κB components, our assessment of the protein levels of
the two downstream inflammatory cytokine markers (IL1-β, TGF-β)
exhibited the same trends in hearts. In this study, the expression
and localization of the NF-κB cascade components were also
investigated by immunofluorescence, which revealed that Pi-p65 and
p50 were upregulated in the murine heart in the ApoE(−/−) group fed
with a HFD, and suppressed its phosphorylation level and NF-κB
signaling in the ApoE(−/−)/IKKɛ(−/−) mice. Furthermore, we observed
the translocation of resting NF-κB from the cytoplasm to the nuclei
in the myocyte, consistent with the manner of NF-κB activation.
Additionally, in the IP experiments, the inducible IκB kinase of
IKKɛ binds to the NF-κB cascade components total-p50 and total-p65,
thereby providing a complete overview of the pathway, starting from
the upstream IKKɛ to the downstream transcription factors. On the
basis of our findings and those of previous studies suggesting this
non-canonical IκB kinase IKKɛ in NF-κB activation, we suggeset that
IKKɛ has a pivotal role in the activities of NF-κB, which in turn
regulates the expression of genes involved in the inflammatory
response in hearts.
In conclusion, we have found a novel role for IKKɛ
in regulating obesity-associated low-grade, chronic inflammation in
murine hearts after HFD feeding, while in the absence of IKKɛ, the
murine hearts are significantly prevented from sustaining an
obesity-associated inflammatory response. These observations
provide new insights that IKKɛ is dispensable for NF-κB activation
and that IKKɛ may have anti-inflammatory properties in obesity.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81070180, 81370259) and the
Ministry of Human Resources and Social Security of China
(2010-412).
References
|
1
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar
|
|
2
|
Mauro C and Marelli-Berg FM: T cell
immunity and cardiovascular metabolic disorders: does metabolism
fuel inflammation? Front Immunol. 3:1732012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen H: Cellular inflammatory responses:
novel insights for obesity and insulin resistance. Pharmacol Res.
53:469–477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hansson GK and Hermansson A: The immune
system in atherosclerosis. Nat Immunol. 12:204–212. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harrison DG, Vinh A, Lob H and Madhur MS:
Role of the adaptive immune system in hypertension. Curr Opin
Pharmacol. 10:203–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baker RG, Hayden MS and Ghosh S: NF-κB,
inflammation, and metabolic disease. Cell Metab. 13:11–22.
2011.
|
|
7
|
Ghosh S and Hayden MS: New regulators of
NF-kappaB in inflammation. Nat Rev Immunol. 8:837–848. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pasparakis M: Regulation of tissue
homeostasis by NF-kappaB signalling: implications for inflammatory
diseases. Nat Rev Immunol. 9:778–788. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oeckinghaus A, Hayden MS and Ghosh S:
Crosstalk in NF-κB signaling pathways. Nat Immunol. 12:695–708.
2011.
|
|
11
|
Liu F, Xia Y, Parker AS and Verma IM: IKK
biology. Immunol Rev. 246:239–253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang N, Ahmed S and Haqqi TM: Genomic
structure and functional characterization of the promoter region of
human IkappaB kinase-related kinase IKKi/IKKvarepsilon gene. Gene.
353:118–133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Häcker H and Karin M: Regulation and
function of IKK and IKK-related kinases. Sci STKE 2006.
re132006.PubMed/NCBI
|
|
14
|
Chau TL, Gioia R, Gatot JS, et al: Are the
IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly
activated? Trends Biochem Sci. 33:171–180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiang SH, Bazuine M, Lumeng CN, et al:
The protein kinase IKKepsilon regulates energy balance in obese
mice. Cell. 138:961–975. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dai J, Shen DF, Bian ZY, et al: IKKi
deficiency promotes pressure overload-induced cardiac hypertrophy
and fibrosis. PloS One. 8:e534122013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo J, Kim D, Gao J, et al: IKBKE is
induced by STAT3 and tobacco carcinogen and determines
chemosensitivity in non-small cell lung cancer. Oncogene.
32:151–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo JP, Coppola D and Cheng JQ: IKBKE
protein activates Akt independent of phosphatidylinositol
3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain
malignant transformation. J Biol Chem. 286:37389–37398. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tenoever BR, Ng SL, Chua MA, McWhirter SM,
García-Sastre A and Maniatis T: Multiple functions of the
IKK-related kinase IKKepsilon in interferon-mediated antiviral
immunity. Science. 315:1274–1278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ng SL, Friedman BA, Schmid S, et al: IκB
kinase epsilon (IKK(epsilon)) regulates the balance between type I
and type II interferon responses. Proc Natl Acad Sci USA.
108:21170–21175. 2011.
|
|
21
|
Cao C, Zhu Y, Chen W, et al: IKKɛ knockout
prevents high fat diet induced arterial atherosclerosis and NF-κB
signaling in mice. PloS One. 8:e649302013.
|
|
22
|
Shoelson SE, Herrero L and Naaz A:
Obesity, inflammation, and insulin resistance. Gastroenterology.
132:2169–2180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wellen KE and Hotamisligil GS:
Inflammation, stress, and diabetes. J Clin Invest. 115:1111–1119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scheja L, Heese B and Seedorf K:
Beneficial effects of IKKɛ-deficiency on body weight and insulin
sensitivity are lost in high fat diet-induced obesity in mice.
Biochem Biophys Res Commun. 407:288–294. 2011.
|
|
25
|
Reilly SM, Chiang SH, Decker SJ, et al: An
inhibitor of the protein kinases TBK1 and IKK-ɛ improves
obesity-related metabolic dysfunctions in mice. Nat Med.
19:313–321. 2013.
|
|
26
|
Olefsky JM: IKKepsilon: a bridge between
obesity and inflammation. Cell. 138:834–836. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000.PubMed/NCBI
|