Introduction
The misfolding and aggregation of specific proteins
is a common hallmark of a number of neurodegenerative disorders,
including highly prevalent illnesses, such as Alzheimer’s and
Parkinson’s disease, as well as more rare disorders, such as prion
diseases (1). Prion diseases,
also known as transmissible spongiform encephalopathies (TSEs), are
a group of fatal neurodegenerative disorders caused by the
misfolding of normal cellular prion (PrPC) into an
abnormal form of scrapie isoform of prion protein
(PrPSc) (2). The basic
mechanism of prion infectivity is the ability to self-reproduce
through the conversion of PrPC into PrPSc, a
transformed protease-resistant protein that is believed to be the
infectious agent in the transmissible form of prion disease
(3). Although it is well
established that PrPSc plays a major role in the origin
and transmission of TSEs, the mechanisms through which the
disease-specific PrP exerts its harmful effects on neurons remain
unknown (4).
PrP(106-126), a peptide fragment of
PrPSc, was used in this study for the experimental
induction of PrPSc infection. PrP(106-126) possesses
several of the pathogenic and physiological properties of
PrPSc, including the ability to induce apoptosis in
hippocampal neurons and to induce the proliferation of astrocytes
(5). It also catalyzes the
aggregation of endogenous cellular PrP (PrPC) into an
amyloidogenic form that shares several characteristics with
PrPSc (6). Therefore,
this peptide is useful for in vitro studies of prion-induced
neuronal apoptosis.
The term ‘autophagy’, which means ‘eating of self’,
was first coined by Deter and De Duve several decades ago and was
primarily based on the observed degradation of the mitochondria and
cellular components within lysosomes (7). Autophagy encompasses the different
routes that cells use to deliver cytoplasmic substrates to
lysosomes for degradation (8).
There are three defined types of autophagy: macroautophagy,
microautophagy and chaperone-mediated autophagy (CMA), all of which
promote the proteolytic degradation of the intracellular components
at the lysosome (9). In the
present study, we focused on macroautophagy. Upon induction, the
phagophore expands and encloses a portion of the cytoplasm,
resulting in the formation of a double-membraned structure termed
the autophagosome, which fuses with a lysosome for degradation
(10). Autophagy, a common
morphological feature in dying cells, was often erroneously
presumed to be a cell death pathway. However, it now seems clear
that one of its major functions is to keep cells alive under
stressful ‘life-threatening’ conditions (11). The suppression of autophagy is
associated with certain diseases, including a subset of cancers,
neurodegenerative disorders, infectious diseases and inflammatory
bowel disorders, and a decline in autophagy function is a common
feature of aging (10). The
neuroprotective effects mediated by the inducers of autophagy are
related to an increase in the mitochondrial turnover as a result of
autophagy activation (12,13).
In addition, autophagy reduces the amount of abnormal PrP (14).
Caffeine is a widely used psychoactive drug that has
been long used to increase alertness and energy. It is also known
to exert neuroprotective effects against Parkinson’s disease, one
of the many neurodegenerative disorders (15). However, little is known about the
effects of caffeine on autophagy and cellular homeostasis in prion
diseases.
In this study, we investigated the protective
effects of caffeine against the neurotoxicity that is associated
with prions using the PrP(106-126) peptide in a SH-SY5Y
neuroblastoma cell line. Our results demonstrate that caffeine
enhances autophagy in a dose-dependent manner in neuroblastoma
cells. Furthermore, we reveal that caffeine-induced autophagy
protects these cells against PrP(106-126)-induced apoptosis.
Materials and methods
Cell culture
The human neuroblastoma cell line, SH-SY5Y, was
obtained from the American Type Culture Collection (ATCC;
Rockville, MD, USA). The cells were cultured in minimum essential
medium (Invitrogen-Gibco, Carlsbad, CA, USA) supplemented with 10%
fetal bovine serum (Invitrogen-Gibco), 100 U/ml penicillin and 0.1
mg/ml gentamycin in a humidified incubator maintained at 37°C and
5% CO2. The cells were treated with various doses of
caffeine (Sigma-Aldrich, St. Louis, MO, USA) for 1 h and were then
exposed to 50 μM PrP(106-126) for 24 h with or without the
autophagy inhibitors, 3-methyladenine (3-MA; 200 μM) and wortmannin
(50 nM) (Sigma-Aldrich).
Treatment with PrP(106-126)
Synthetic PrP(106-126) peptides (sequence,
Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly)
were synthesized by Peptron (Seoul, Korea). The peptides were
dissolved in sterile dimethyl sulfoxide at a stock concentration of
10 mM and stored at −4°C.
Terminal deoxynucleotidyl transferase
dUTP nick end-labeling (TUNEL) assay
TUNEL assay was performed to measure the degree of
cellular apoptosis using an In Situ ApoBrdU DNA fragmentation assay
kit (BioVision, Mountain View, CA, USA), according to the
manufacturer’s instructions. The cells were counterstained with
propidium iodide (PI) to show cell nuclei.
Annexin V assay
Apoptosis in the detached cells was assessed using
an Annexin V assay kit (Santa Cruz Biotechnology, Santa Cruz, CA,
USA) according to the manufacturer’s instructions. The Annexin V
levels were determined by measuring fluorescence at an excitation
of 488 nm and an emission of 525/30 using a Guava EasyCyte HT
System (Millipore, Bedford, MA, USA).
Trypan blue exclusion assay
The number of viable cells was determined by trypan
blue dye exclusion (Sigma-Aldrich) using a hemocytometer. The
result was expressed as a percentage relative to the control.
Western blot analysis
The SH-SY5Y cells were lysed in lysis buffer [25 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4,
100 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 5 mM
MgCl2, 0.1 mM dithiothreitol (DTT), and a protease
inhibitor mixture] and whole cell proteins were electrophoretically
resolved on a 10–15% sodium dodecyl sulfate polyacrylamide gel and
transferred onto a nitrocellulose membrane. Immunoreactivity was
detected through sequential incubation with primary antibodies,
horseradish peroxidase-conjugated secondary antibodies and enhanced
chemiluminescence reagents using the horseradish peroxidase
detection kit (Westsave Gold;,AbFrontier Inc., Seoul, Korea). The
primary antibodies used for immunoblotting were as follows:
anti-LC3 (Novus Biologicals, Littleton, CO, USA),
anti-phospho-SAPK/JNK (Cell Signaling Technology Inc., Danvers, MA,
USA), anti-phospho-Akt (Epitomics, Burlingame, CA, USA) and
anti-β-actin (Sigma-Aldrich). Images were captured using a Fusion
FX7 imaging system (Vilber Lourmat, ZI Sud Torcy, France). The
densitometry of the signal bands was analyzed using Bio-1D software
(Vilber Lourmat, Marne la Vallée, France).
Results
Caffeine inhibits PrP(106-126)-induced
apoptosis
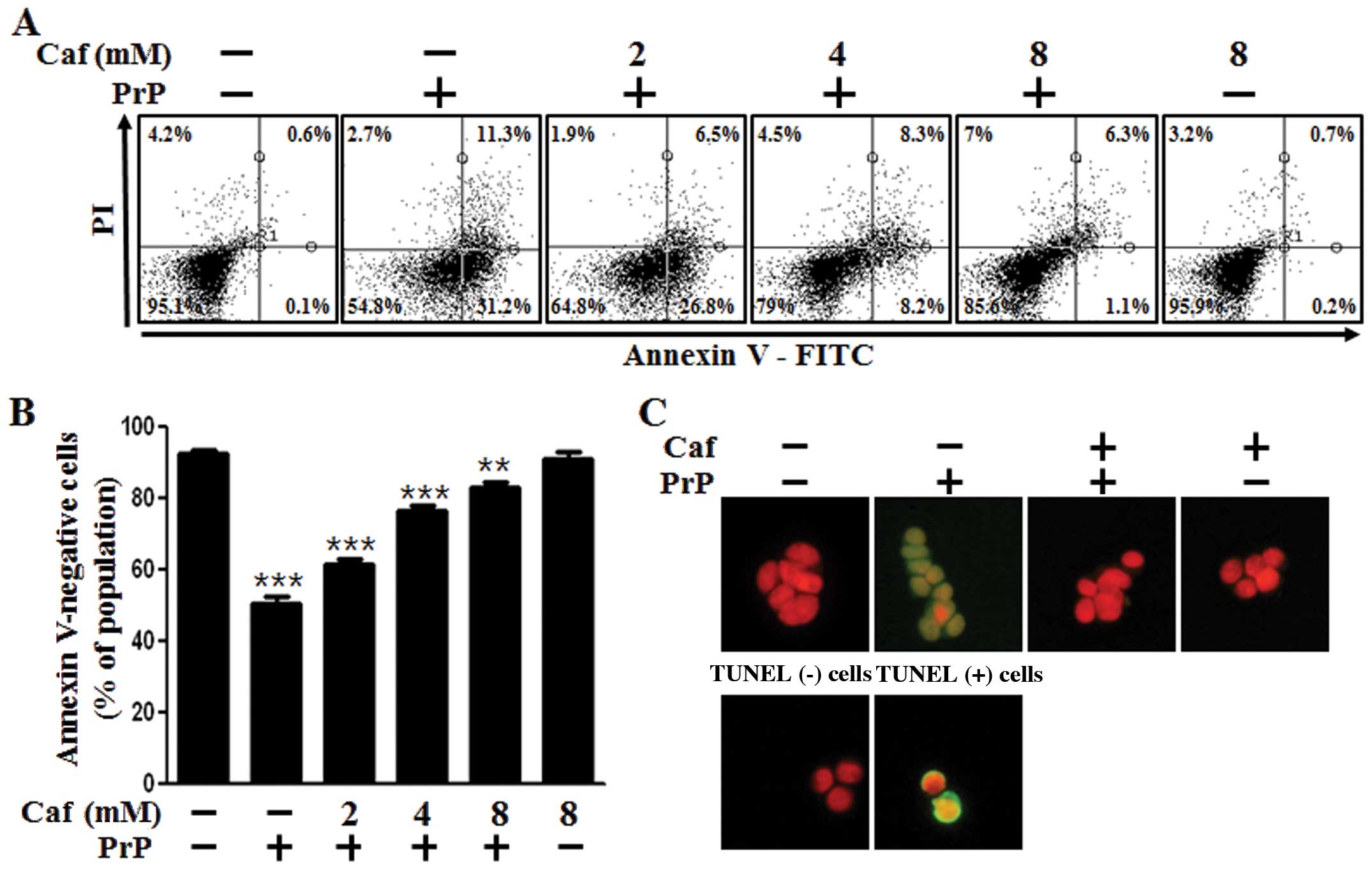
We examined whether caffeine protects neuronal cells
against prion-mediated neurotoxicity and whether these effects are
associated with the induction of autophagy. First, we investigated
the effects of caffeine on PrP(106-126)-mediated neurotoxicity in
the SH-SY5Y cells using an Annexin V assay. The SH-SY5Y cells were
exposed to caffeine with or without PrP(106-126). The cell
viability of the PrP(106-126)-treated cells was decreased by
approximately 50% compared with the untreated controls. The cell
viability of the cells treated with caffeine only was comparable to
that of the untreated controls. Importantly, treatment with
caffeine inhibited PrP(106-126)-induced neurotoxicity in the
SH-SY5Y cells (Fig. 1A and B). We
also used a TUNEL assay (Fig.
1C), microscopic imaging (Fig.
1D) and a trypan blue exclusion assay (Fig. 1E). As shown in Fig. 1C, the apoptotic process in the
PrP(106-126)-treated cells emitted green fluorescence, indicating
DNA strand breakage. As shown in Fig.
1E, treatment with caffeine reversed the cell death induced by
treatment with PrP(106-126) in a dose-dependent manner. These
results indicated that caffeine was effective in preventing
PrP(106-126)-induced apoptosis in the SH-SY5Y cells.
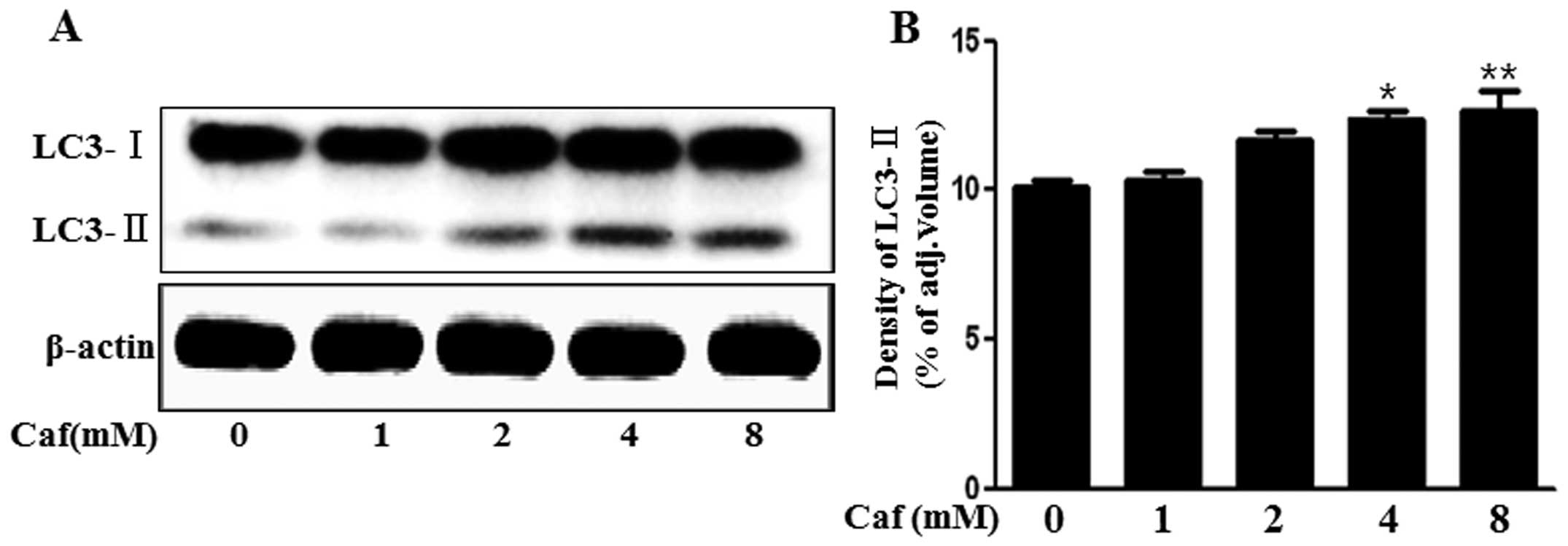
Caffeine induces the production of the
autophagosomal marker, LC3-II
The LC3 protein is localized and aggregated on the
autophagosome and is therefore considered to be a marker for
autophagy. LC3 transforms from LC3-I into LC3-II during
autophagosome formation (16).
Thus, in the present study, we investigated whether caffeine
induces autophagy by measuring LC3 transformation. As shown in
Fig. 2, we found that the levels
of the late autophagosomal marker, LC3-II, increased in the
caffeine-treated group in a dose-dependent manner compared with the
control, as shown by western blot and densitometric analyses
(Fig. 2A and B). These results
suggested that caffeine induced autophagy in a dose-dependent
manner.
Caffeine inhibits PrP(106-126)-induced
apoptosis by activating autophagy
Accumulating evidence suggests a neuroprotective
role of autophagy in the control of cell death (17–19). In this study, to investigate the
effects of caffeine on the activation of autophagy, we analyzed the
levels of LC3-II and cell viability using autophagy inhibitors. We
found that the caffeine-induced increase in the LC3-II levels was
inhibited by the autophagy inhibitors, 3-MA and wortmannin, as
shown by western blot and densitometryic analyses (Fig. 3A and B). We also found that the
effects of caffeine on cell viability were reversed by treatment
with 3-MA and wortmannin, as shown by an Annexin-V assay and graph
analysis (Fig. 3C and D). In
Fig. 3C, which shows the results
procuced by the Annexin V assay, the dots represent fluorescent
cells in the plot. Green fluorescence (x-axis) indicates apoptosis
and red fluorescence (y-axis) indicates necrosis. Cells in the
first quadrant signify the late apoptotic condition and cells in
the fourth quadrant signify the early apoptotic condition. We found
that recovery in cell viability induced by caffeine was reversed by
treatment with 3-MA and wortmannin. These results strongly suggest
that caffeine exerts neuroprotective effects through the induction
of autophagy.
Effects of caffeine on Akt and JNK
signaling
We then investigated whether caffeine has an effect
on growth and death signals. It is well known that Akt promotes
growth factor-mediated cell survival both directly and indirectly.
Nakaso et al demonstrated that caffeine prevents the
apoptosis of neurons through Akt activation (20). JNK activation has been shown to be
induced by PrP(106-126)-induced apoptosis (21). Therefore, we examined Akt and JNK
activation following treatment with caffeine and PrP(106-126). Our
results demonstrated that treatment with caffeine reversed the
suppressive effects on Akt activation induced by PrP(106-126) and
suppressed the activation of JNK induced by PrP(106-126). These
results suggest that caffeine exerts protective effects on
intracellular signaling. Moreover, our resuts suggest that caffeine
plays a role similar to that of a JNK inhibitor in cell apoptosis.
Based on these results, we suggest that treatment with caffeine
inhibits the PrP(106-126)-mediated neuronal apoptotic pathway by
regulating autophagy.
Discussion
The aim of this study was to investigate the role of
caffeine-induced autophagy and the regulation of
PrP(106-126)-induced apoptosis by caffeine in neuronal cells. Our
results suggest that the increase in autophagy induced by caffeine
and the resultant reduction in PrP(106-126)-induced neurotoxicity
may be the key mechanisms underlying the observed neuroprotective
effects of caffeine.
Caffeine is widely used in modern society and is
classified as safe by the food and drug administration. Beverages
containing caffeine, such as coffee, tea, soft drinks and energy
drinks, enjoy great popularity. Caffeine is a psychoactive drug and
functions as a central nervous system stimulant, temporarily
relieving drowsiness and restoring alertness. A recent study
suggested that caffeine induces neuronal GSH synthesis by promoting
cysteine uptake, thus leading to neuroprotection (22). Caffeine has also been shown to
exert protective effects against Alzheimer’s and Parkinson’s
disease in human and animal studies (23).
Certain studies have suggested that caffeine induces
autophagy (24,25). Several other studies have
suggested that autophagy is a double-edged sword, with both a
beneficial and a harmful potential in cancer (26) and neurodegeneration (27). On the positive side, autophagy is
the cellular pathway that mediates the lysosomal degradation of
intracellular long-lived macromolecules or organelles for
subsequent reuse under physiological conditions of differentiation,
starvation, or stress, such as oxidative stress, endoplasmic
reticulum stress and the accumulation of abnormal protein (9,28).
Saiki et al reported that caffeine at concentrations >2.5
mM induced apoptosis through the enhancement of autophagy and that
the levels of LC3-II were markedly increased at concentrations
>10 mM (25). However, in this
study, 8 mM caffeine did not decrease cell viability compared with
the control, although it clearly increased the levels of the
autophagosomal marker, LC3-II. We also found that caffeine induced
apoptosis at concentrations >10 mM (data not shown). We suggest
that the caffeine used in the study by Saiki et al (25) was slightly different from the one
we used, with respect to certain characteristics, such as purity or
grade. Based on our findings, we suggest that caffeine exerts
neuroprotective effects through the induction of autophagy.
Certain studies have shown that caffeine inhibits
the PI3K/Akt pathway (29,30).
By contrast, our experimental data indicated that Akt activation
was increased by treatment with caffeine. Nakaso et al also
suggested that caffeine activated the PI3K/Akt pathway in SH-SY5Y
cells (20). As shown in Fig. 4A and C, the activation of JNK by
PrP (106-12) was decreased by treatment with caffeine. This
indicates that caffeine suppresses apoptosis-related signaling in
neuronal cells, as JNK is closely associated with apoptosis in
neurons (31). These findings
support our hypothesis that caffeine exerts protective effects
against PrP(106-126)-induced apoptosis in SH-SY5Y cells.
The cross-talk between apoptosis and autophagy is
complex and sometimes contradictory; however, it is a critical
determinant of the overall fate of the cell. To the best of our
knowledge, this is the first study indicating that
caffeine-mediated autophagy may play a critical role in the
neuroprotection against PrP(106-126)-induced neurotoxicity. We
therefore suggest that caffeine may be a valid therapeutic agent
for prion-related neurodegenerative diseases.
Acknowledgements
This study was supported by a grant from the
National Research Foundation of Korea (NRF), funded by the Korean
government (MISP) (2013R1A4A1069486).
References
|
1
|
Costanzo M and Zurzolo C: The cell biology
of prion-like spread of protein aggregates: mechanisms and
implication in neurodegeneration. Biochem J. 452:1–17.
2013.PubMed/NCBI
|
|
2
|
Prusiner SB: Novel proteinaceous
infectious particles cause scrapie. Science. 216:136–144. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prusiner SB: Prions. Proc Natl Acad Sci
USA. 95:13363–13383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aguzzi A: Staining, straining and
restraining prions. Nat Neurosci. 11:1239–1240. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Forloni G, Angeretti N, Chiesa R, et al:
Neurotoxicity of a prion protein fragment. Nature. 362:543–546.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Selvaggini C, De Gioia L, Cantu L, et al:
Molecular characteristics of a protease-resistant, amyloidogenic
and neurotoxic peptide homologous to residues 106-126 of the prion
protein. Biochem Biophys Res Commun. 194:1380–1386. 1993.
View Article : Google Scholar
|
|
7
|
Deter RL and De Duve C: Influence of
glucagon, an inducer of cellular autophagy, on some physical
properties of rat liver lysosomes. J Cell Biol. 33:437–449. 1967.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rubinsztein DC, Marino G and Kroemer G:
Autophagy and aging. Cell. 146:682–695. 2011. View Article : Google Scholar
|
|
9
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Filomeni G, Graziani I, De Zio D, et al:
Neuroprotection of kaempferol by autophagy in models of
rotenone-mediated acute toxicity: possible implications for
Parkinson’s disease. Neurobiol Aging. 33:767–785. 2012.PubMed/NCBI
|
|
13
|
Garcia JJ, Pinol-Ripoll G,
Martinez-Ballarin E, et al: Melatonin reduces membrane rigidity and
oxidative damage in the brain of SAMP8 mice. Neurobiol Aging.
32:2045–2054. 2011. View Article : Google Scholar
|
|
14
|
Nakagaki T, Satoh K, Ishibashi D, et al:
FK506 reduces abnormal prion protein through the activation of
autolysosomal degradation and prolongs survival in prion-infected
mice. Autophagy. 9:2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu K, Xu YH, Chen JF and Schwarzschild MA:
Neuroprotection by caffeine: time course and role of its
metabolites in the MPTP model of Parkinson’s disease. Neuroscience.
167:475–481. 2010.PubMed/NCBI
|
|
16
|
Rubinsztein DC, Cuervo AM, Ravikumar B, et
al: In search of an “autophagomometer”. Autophagy. 5:585–589.
2009.
|
|
17
|
Park JH, Lee JE, Lee SJ, et al: Potential
autophagy enhancers protect against fipronil-induced apoptosis in
SH-SY5Y cells. Toxicol Lett. 223:25–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xia DY, Li W, Qian HR, Yao S, Liu JG and
Qi XK: Ischemia preconditioning is neuroprotective in a rat
cerebral ischemic injury model through autophagy activation and
apoptosis inhibition. Braz J Med Biol Res. 46:580–588. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim J, Kim TY, Cho KS, Kim HN and Koh JY:
Autophagy activation and neuroprotection by progesterone in the
G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis.
Neurobiol Dis. 59:80–85. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakaso K, Ito S and Nakashima K: Caffeine
activates the PI3K/Akt pathway and prevents apoptotic cell death in
a Parkinson’s disease model of SH-SY5Y cells. Neurosci Lett.
432:146–150. 2008.PubMed/NCBI
|
|
21
|
Carimalo J, Cronier S, Petit G, et al:
Activation of the JNK-c-Jun pathway during the early phase of
neuronal apoptosis induced by PrP106-126 and prion infection. Eur J
Neurosci. 21:2311–2319. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aoyama K, Matsumura N, Watabe M, Wang F,
Kikuchi-Utsumi K and Nakaki T: Caffeine and uric acid mediate
glutathione synthesis for neuroprotection. Neuroscience.
181:206–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Ghribi O and Geiger JD: Caffeine
protects against disruptions of the blood-brain barrier in animal
models of Alzheimer’s and Parkinson’s diseases. J Alzheimers Dis.
20(Suppl 1): S127–S141. 2010.
|
|
24
|
Winter G, Hazan R, Bakalinsky AT and
Abeliovich H: Caffeine induces macroautophagy and confers a
cytocidal effect on food spoilage yeast in combination with benzoic
acid. Autophagy. 4:28–36. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saiki S, Sasazawa Y, Imamichi Y, et al:
Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wei K, Wang P and Miao CY: A double-edged
sword with therapeutic potential: an updated role of autophagy in
ischemic cerebral injury. CNS Neurosci Ther. 18:879–886. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar
|
|
29
|
Foukas LC, Daniele N, Ktori C, Anderson
KE, Jensen J and Shepherd PR: Direct effects of caffeine and
theophylline on p110 delta and other phosphoinositide 3-kinases.
Differential effects on lipid kinase and protein kinase activities.
J Biol Chem. 277:37124–37130. 2002. View Article : Google Scholar
|
|
30
|
Sarkaria JN, Busby EC, Tibbetts RS, et al:
Inhibition of ATM and ATR kinase activities by the radiosensitizing
agent, caffeine. Cancer Res. 59:4375–4382. 1999.PubMed/NCBI
|
|
31
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|