1. Introduction
Oxygen (O2) constitutes 20.8% of the
atmospheric air, and is the third-most abundant element in the
universe, after hydrogen and helium. It is not only a key component
of all major biomolecules of living organisms, but also a key
constituent of inorganic compounds. Oxygen homeostasis is crucially
important to maintain the survival of all vertebrate species
(1). Therefore, organisms
developed a way to coordinate the oxygen levels in the
intracellular compartments in order to maintain homeostasis. When
these mechanisms fail, and the intracellular concentration of
oxygen decreases, a stress condition called hypoxia is created.
Hypoxia can be defined as a condition lacking the necessary oxygen
to meet metabolic requirements. The level at which this is reached
will vary depending on the metabolic requirements of the cell.
Hypoxia is a relevant physiological stress associated with many
processes, such as adaptation to high altitudes or human diseases
(e.g, cancer) (2). The
hypoxia-inducible factors (HIFs) are a family of transcription
factors whose levels are regulated in response to hypoxic stimuli,
and when active can enact a transcriptional program that allows the
cell to respond to the hypoxic environment.
Another important physiological stress is
inflammation. Inflammation represents a protective attempt to
eliminate pathogens and initiate the healing process of a wound. As
in hypoxia, cells have evolved sophisticated mechanisms to control
the inflammatory response to pathogens. A key element of these
mechanisms is a family of transcription factors termed nuclear
factor κ-light-chain-enhancer of activated B cells (NF-κB). NF-κB
is composed of several family members that activate signalling
pathways in response to a variety of stimuli (such as virus,
bacteria or cytokines) which ultimately engage a complex
transcriptional program, allowing the cell to respond to this
environmental stress (3).
Several diseases, including rheumatoid arthritis
(RA), inflammatory bowel disease and colorectal cancer (CRC) result
from the deregulation of the hypoxia and inflammation pathways
(4–6). Consequently, recent scientific
research has been focussed on attempting to understand how these
pathways are regulated, crosstalk and respond in disease. In this
review, we describe the current understanding of the role of the
HIF and NF-κB transcription factor families in response to hypoxia
and inflammation and discuss their crosstalk in RA, inflammatory
bowel disease and CRC, with relevance for future therapies for the
management of these conditions.
2. The HIF system
The HIFs are a family of transcription factors that
sense changes in environmental oxygen and orchestrate a
transcriptional program, which forms an important part of the
cellular response to the hypoxic environment. HIF-1 was first
identified over 20 years ago through studies of erythropoietin gene
expression (7). HIF is a
heterodimeric transcription factor that consists of a
constitutively expressed HIF-1β subunit and an
O2-regulated HIF-α subunit (8). Three isoforms of HIF-α have been
identified since these initial studies (HIF-1α, -2α and -3α)
(Fig. 1A). The HIF-α isoforms are
all characterized by the presence of basic helix-loop-helix
(bHLH)-Per/ARNT/Sim (PAS) and oxygen-dependent degradation (ODD)
domains (Fig. 1A). Both HIF-1α
and HIF-2α have important cellular functions as transcription
factors with some redundancy in their targets (9,10).
HIF-2α protein shares sequence similarity and functional overlap
with HIF-1α, but its distribution is restricted to certain cell
types, and in some cases, it mediates distinct biological functions
(11). HIF-3α is the most
recently discovered isoform. The regulation of HIF-3α expression is
complex in comparison to HIF-1α and HIF-2α with several splice
variants that can function as a competitive inhibitors of the
HIF-α-HIF-1β interaction (12,13), or by directly activating target
genes in hypoxia that mediate both hypoxia dependent and
independent functions. The role of HIF-3α in the cellular response
to hypoxia remains an active area of study (14). Several splice variants of HIF-1β
[also known as aryl hydrocarbon receptor nuclear translocator
(ARNT)] have been identified (15,16). Though their exact functions are
not known, at least one splice variant has been associated with
poor prognosis in oestrogen receptor-negative breast cancer
(17).
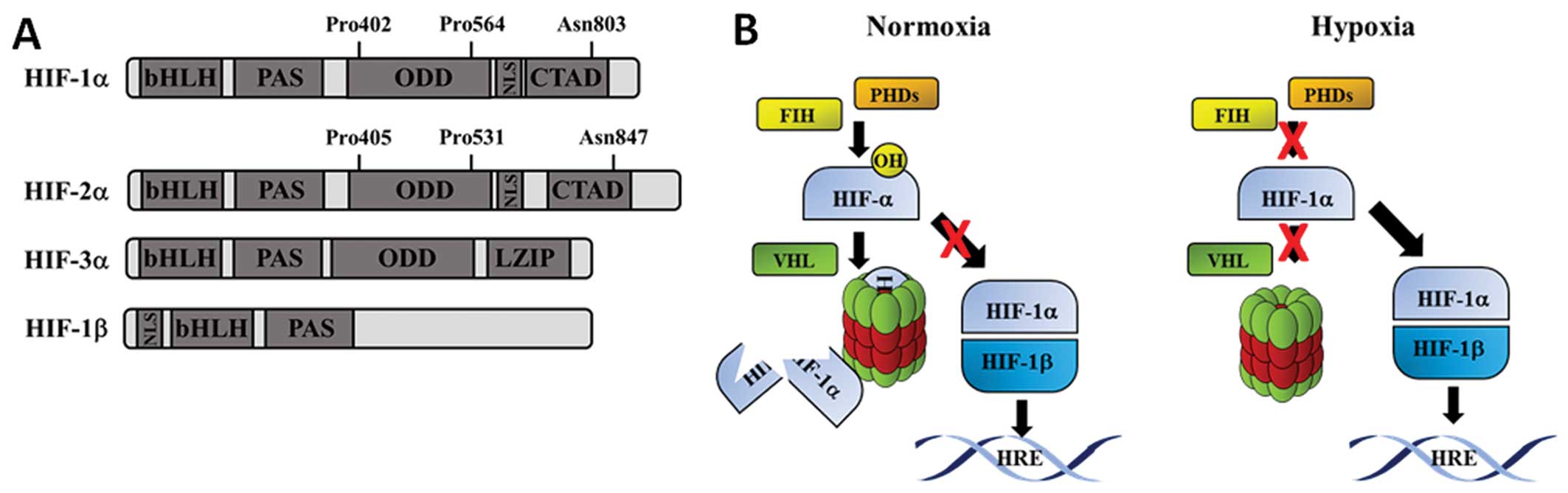 | Figure 1(A) Schematic diagram of HIF
proteins. Boxes represent different protein domains. The
hydroxylation sites for HIF-1α and HIF-2α are noted above the
schematic structure. (B) Schematic diagram of HIF pathway. In the
presence of oxygen (normoxia), c bind to HIF-1α and catalyse the
hydroxylation of proline residues. Once hydroxylated, HIF-1α binds
rapidly to the vHL, which results in its polyubiquitination. This
targets HIF-1α for proteasome-mediated degradation. In the presence
of low oxygen (hypoxia), HIF-1α is stabilized and can translocate
to the nucleus. HIF-1α dimerises with its partner HIF-1β and
transactivates target genes containing hypoxia response elements
(HREs). HIF, hypoxia-inducible factor; vHL, von Hippel Lindau;
bHLH, basic helix-loop-helix; CTAD, C-terminal transactivation
domain; LZIP, leucine zipper; NLS, nuclear localisation signal;
ODD, oxygen-dependent domain; PAS, Per/ARNT/Sim domain. |
3. HIF regulation by οxygen
The regulation of the HIF-α subunits by oxygen
occurs mainly at the post-transcriptional level, and is mediated by
hydroxylation-dependent proteasomal degradation (Fig. 1B). In well-oxygenated cells, HIF-α
is hydroxylated in its ODD. For HIF-1α this is at prolines (Pro)402
and Pro564 (18), whereas HIF-2α
is hydroxylated at Pro405 and Pro531 (Fig. 1B) (19). Proline hydroxylation is catalysed
by a class of dioxygenase enzymes called prolyl hydroxylases
(PHDs). There are three known PHDs, 1–3, all of which have been
shown to hydroxylate HIF-1α. PHD2 has a higher affinity for HIF1α,
whereas PHD1 and PHD3 have higher affinity for HIF-2α (20,21). All PHDs require Fe2+
and α-ketoglutarate (α-KG) as co-factors for their catalytic
activity and have an absolute requirement for molecular oxygen as a
co-substrate, making their activity reduced in hypoxia (22–25).
Prolyl-hydroxylation of HIF-α attracts the von
Hippel-Lindau (vHL) tumour suppressor protein, which recruits the
Elongin C-Elongin B-Cullin 2-E3-ubiquitin-ligase complex, leading
to the Lys48-linked poly-ubiquitination and proteasomal degradation
of HIF-1α (Fig. 1B) (26–28). Interestingly, PHDs have also been
shown to be able to sense amino acid availability through α-KG
oscillations (29), and the
centrosomal protein Cep192 has been described as a hydroxylation
target for PHD1 (30), indicating
an additional function for these enzymes as nutrient sensors and
regulators of cell cycle progression. Both PKM2 and HCLK2 have also
both recently been described as new hydroxylation targets for PHD3
(31,32).
In hypoxia the PHDs are inactive, or have reduced
activity, since they require molecular oxygen as a cofactor. Under
these conditions HIF-α is stabilized, can form a heterodimer with
HIF-1β in the nucleus and bind to the consensus cis-acting hypoxia
response element (HRE) nucleotide sequence 5′-RCGTG-3′, which is
present within the enhancers and/or promoters of HIF target genes
(Fig. 1B) (33–35). HIF-α stabilisation therefore
allows the cell to enact a transcriptional programme that is
appropriate to the hypoxic environment (18) (Fig.
1B).
4. HIF target genes
The HIF heterodimer can regulate the expression of
over 100 target genes involved in a broad range of physiological
functions including: angiogenesis, erythropoiesis, metabolism,
autophagy, apoptosis and other physiological responses to hypoxia
(36). Canonical HIF signalling
is based on the recognition of a putative HRE in the promoter or
enhancer of the target gene that results in the recruitment of the
HIF heterodimer and machinery required for transcription.
Proteomics approaches have been used to identify protein changes in
response to hypoxia in comparison with gene changes. Changes in
just over 100 proteins in response to hypoxia have been identified
(37,38). However, proteins identified
represent both known and undescribed HIF targets, raising the
possibility of HIF action outside of the conventional canonical
pathway. Indeed, in addition to canonical signalling, there are
various described mechanisms by which the stabilised HIF isoforms
can influence the activity of other signalling pathways independent
of the HIF heterodimer or a HRE. Non-canonical HIF signalling has
been demonstrated to regulate aspects of Notch (39), c-Myc (40) and p53 (41) signalling.
5. Inflammation and the NF-κB pathway
Inflammation is a complex physiological process
characterised by the activation of several coordinated signalling
pathways in response to stress. Generally, the inflammatory
response involves both anti- and pro-inflammatory mediators, given
by the expression of small peptides (e.g., cytokines),
glycoproteins (e.g., cluster of differentiation (CD)], and
transcription factors, such as NF-κB.
NF-κB is considered the main pro-inflammatory family
of transcription factors (42–44). In mammalians, it is characterised
as a family of five Rel-domain proteins; RelA, RelB, cRel, p100/p52
and p105/p50 (Fig. 2A).
Interestingly, it has been shown that almost all combinations of
homo- or hetero-dimers between the five NF-κB subunits are possible
(45). This is important, not
only because it gives an extra layer of complexity to the NF-κB
system, but also because it gives specificity according to cellular
context, stimuli or DNA sequences that are bound to the subunits
(44,46). All the NF-κB subunits are
characterised by a conserved 300-amino acid domain, the Rel
homology domain (RHD), which is located in the N-terminus of the
protein (Fig. 2A), and is
responsible for dimerisation, and DNA binding. While RelA, RelB and
cRel contain a C-terminal transactivation domain (TAD) (Fig. 2A), p105 and p100 contain
Ankyrin-repeats motifs in their C-terminus (ANK) (Fig. 2A), responsible for the
dimerisation with other subunits, and subsequent
sequestration/inactivation in the cytoplasm (Fig. 2B).
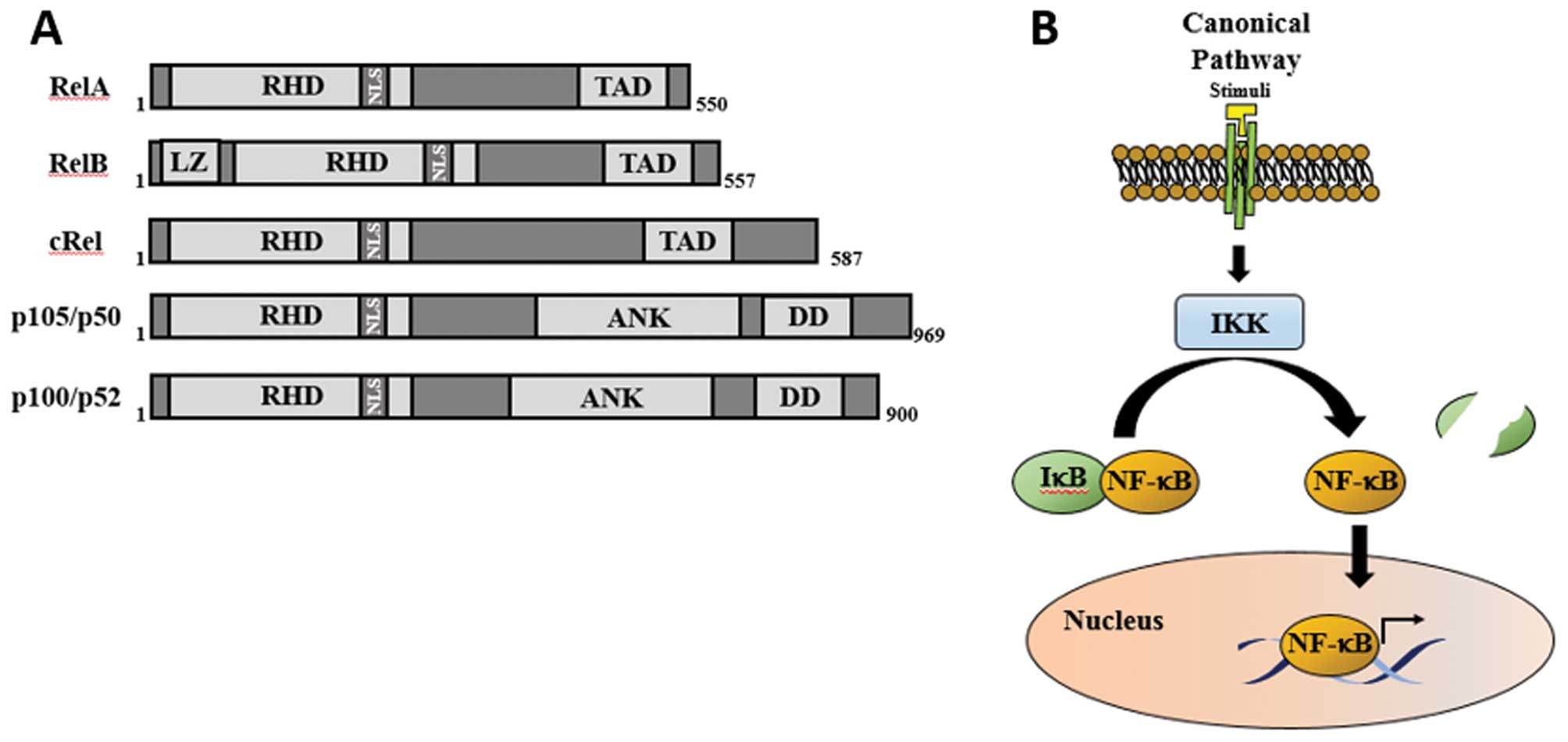 | Figure 2(A) Schematic diagram of NF-κB
subunits. p50 and p52 are not shown, and they are derived from p105
and p100, respectively. Boxes represent different protein domains.
(B) NF-κB canonical pathway. The presence of a stimuli results in
the activation of the IKK complex, which mediates the
phosphorylation of IκB protein, which signals it for proteasomal
degradation. This results in NF-κB dimer release and translocation
into the nucleus. NF-κB, nuclear factor-κB; IKK, inhibitor of κB
kinase; RHD, Rel homology domain; TAD, C-terminal transactivation
domain; LZ, leucine zipper motif; NLS, nuclear localisation signal;
ANK, ankyrin-repeat motifs; DD, death domain. |
There are distinct pathways for the activation of
NF-κB, according to the stimulus, as well as the kinases and NF-κB
subunits involved (3). The most
common, and most well studied is the classical or canonical NF-κB
pathway (Fig. 2B). In
unstimulated cells, the NF-κB dimers remain inactive in the
cytosol, bound to an inhibitory protein, inhibitor of NF-κB (IκB)
(47). Upon stimulation, for
example by the pro-inflammatory cytokine, tumour necrosis factor-α
(TNF-α), the inhibitor of κB kinase (IκB kinase; IKK), is
activated, and phosphorylates IκB. This leads to the degradation of
IκB and the release/translocation of the NF-κB complex into the
nucleus (48). In the nucleus,
the activated NF-κB complex binds to specific 9–10 base pair DNA
sequences (κB sites) to activate a complex regulatory network in
response to a specific stimulus (49). The combination of different
possible homo- and heterodimers, stimuli and cellular context leads
to a myriad of possible outcomes, namely the activation or
inhibition of apoptosis, cellular growth and carcinogenesis
(50).
The NF-κB system is complex and is involved in
multiple biological roles; it is thus expected that it is
deregulated in many different diseases. NF-κB abnormal activation
has been associated with several human diseases, such as
inflammation-related diseases (inflammatory bowel disease and
asthma), cancer (apoptosis suppression), viral infections (HIV) and
genetic diseases (incontinentia pigmenti) (51).
6. Crosstalk between hypoxia and
inflammation in disease
Hypoxia and inflammation are intimately linked. It
has been reported that individuals with mountain sickness presented
with increased inflammatory cytokines circulating in the blood
(52). Additionally, healthy
volunteers who have been exposed to a hypoxic environment for three
nights in high altitudes (>3,400 meters), presented with high
levels of the inflammatory cytokine, interleukin (IL)-6, in the
blood (53). On the other hand,
several inflammatory diseases, such as RA and inflammatory bowel
disease, also exhibit areas of combined hypoxia and inflammation,
which are usually associated with a poor prognosis of the disease
(54–57).
Hypoxia and inflammation are also connected at the
molecular level (48,58,59). HIF (hypoxia) and NF-κB
(inflammation) have been shown to have several common target genes,
common regulators, and importantly, common stimuli (48). NF-κB activation has been shown to
stabilise HIF-1α in hypoxia, and, together with HIF-1β, in
inflammation (60,61). On the other hand, HIF-1α has been
shown to repress NF-κB in vivo and in vitro under
inflammatory conditions (59,62,63). The complexity of the combined
response of HIF and NF-κB in hypoxia makes the crosstalk of these
two pathways more intricate, and difficult to study. However, by
developing a suitable inflammatory model, where the pathways can be
controlled, as well as the conditions of the stimuli, these studies
could provide very useful information that ultimately should be
used to uncover new therapeutic strategies in a diverse range of
diseases where hypoxia and inflammation are predominant features.
In this review, the crosstalk between the main players induced in
both inflammation and hypoxia in three clinical settings is
addressed.
Hypoxia and inflammation crosstalk in
RA
RA is a systemic autoimmune disorder characterised
by chronic inflammation of the synovial membranes of joint tissues
at multiple anatomical sites which ultimately leads to localised
destruction and debilitating deformity (64,65). The RA joint synovium is
characterised by both inflammatory and hypoxic regions (Fig. 3), which are highly infiltrated
with lymphocytes (CD4+ T cells, and B cells),
macrophages and macrophage-like and fibroblast-like synoviocytes
(66). The molecular basis of RA
is still poorly understood, mainly because RA is a heterogeneous
disease composed of several possible treatment responses, and
clinical manifestations (67–69). These differences make RA difficult
to treat, and further studies on the crosstalk between pathways
involved in the disease are required.
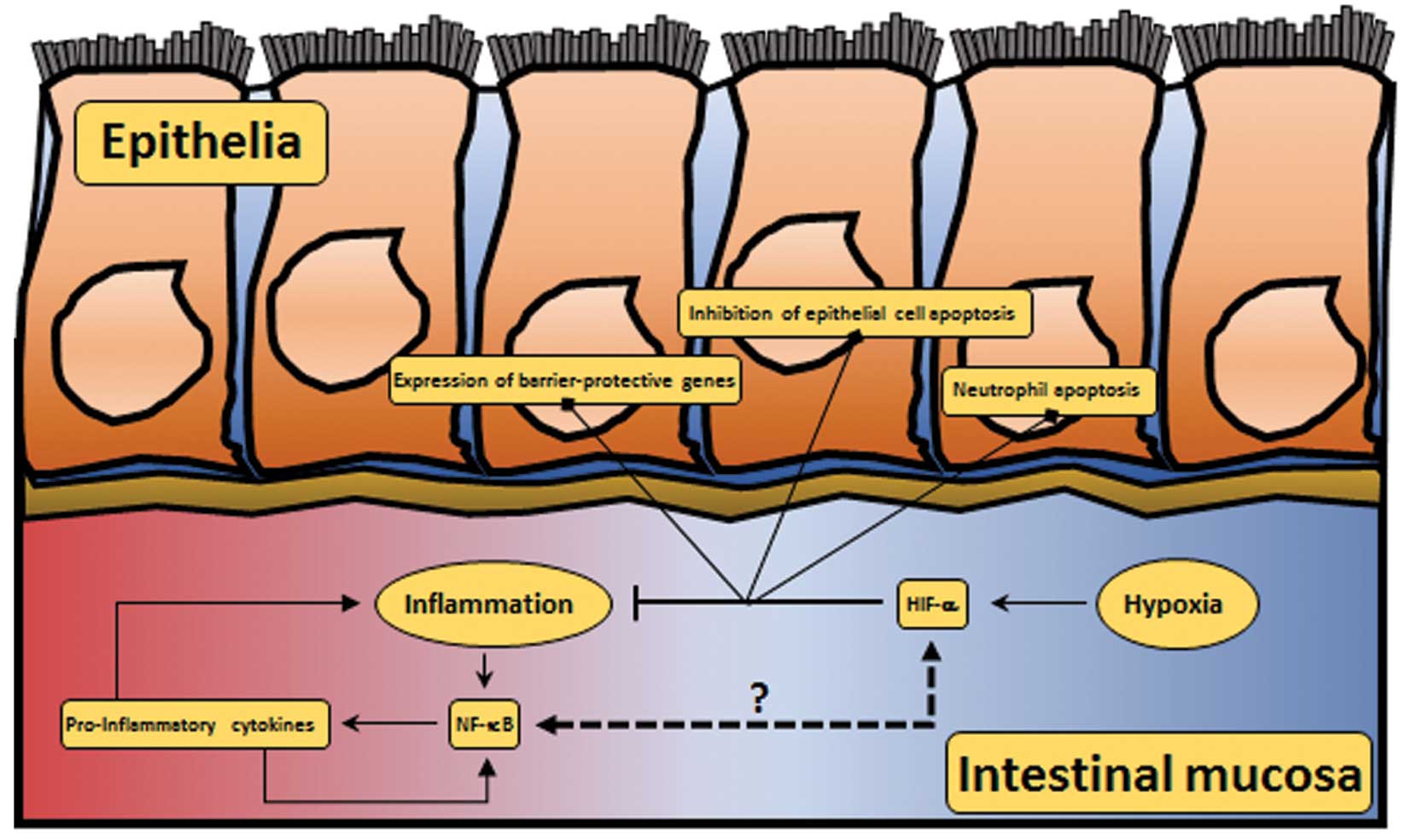
 | Figure 3HIF and NF-κB crosstalk in RA. In RA,
the synovial join is characterised by hypoxic and inflammatory
regions (in blue and red, respectively). Hypoxia leads to the
activation of HIF-1α, which is involved in several cellular
processes (such as apoptosis, vasomotor control, energy metabolism
and angiogenesis). Additionally, hypoxia leads to the activation of
HIF-2α, which is involved in the activation of pro-inflammatory
cytokines. In RA, inflammation leads to the activation of NF-κB,
which activates a pro-inflammatory programme, including
pro-inflammatory cytokines, chemokines, metalloproteases and
metabolic proteins. While HIF-1α has been implicated in the
repression of the NF-κB pathway (59), HIF-2α has been shown to increase
inflammation (78). In RA, this
crosstalk remains poorly understood. RA, rheumatoid arthritis; HIF,
hypoxia-inducible factor; NF-κB, nuclear factor-κB. |
The role of NF-κB in RA
The deregulation of several transcription factors,
such as NF-κB, activator protein-1 (AP-1), and signal transducer
and activator of transcription (STATs), has been strongly
associated with the inflammatory setting of RA (70–72). NF-κB, in particular, has been
shown to be highly activated in the RA synovium (73,74). This is exceptionally important due
to the major role of NF-κB in activating inflammatory responses,
such as through the activation of the pro-inflammatory cytokine,
TNF-α, or the chemokine, IL-8 (75). The activation of a coordinated and
complex network of pro-inflammatory cytokines, chemokines,
metalloproteases (MPPs) and metabolic proteins by NF-κB, leads to
the activation of a positive feedback loop, enhancing the
activation of more pro-inflammatory signals that ultimately results
in chronic and persistent inflammation (Fig. 3) (75,76).
The role of HIF in RA
The HIF family of proteins are additional
transcription factors with direct relevance to RA (77,78). Recently, HIF-1α was identified as
a key player in RA, and therefore as a potential therapeutic target
(79). HIF is important to
coordinate the hypoxia response in the synovial tissue, and the
deregulation or failure of that response leads to cellular
dysfunction, and can ultimately lead to cell death (80). Furthermore, the intense hypoxic
region in the synovial tissue (2–4%), activates a hypoxic response
through HIF, which is involved in regulating several genes involved
in apoptosis, vasomotor control, energy metabolism, and
importantly, angiogenesis (Fig.
3) (16,48,81–83).
Even though the role of HIF in RA has been firmly
established, the contribution of each α-subunit remains poorly
understood. Recently, HIF-2α was implicated as the essential
catabolic regulator of inflammation in RA (78). In that study, the authors
demonstrated that the overexpression of HIF-2α in joint tissues,
but not HIF-1α, was sufficient to induce RA pathogenesis (78). The full contribution of the
α-subunits to RA remains elusive. However, it seems clear that each
α-subunit contributes differently to the progression of RA. HIF-1α
plays a more anti-inflammatory role, whereas HIF-2α acts in a
pro-inflammatory manner. What regulates this differential
expression of the isoforms is still unknown. However, taking into
consideration that NF-κB is the main activator of the HIF
transcription factors, it would be interesting to understand
whether NF-κB has any role in this HIF-1α to HIF-2α switch, and
whether that would be dependent of the presence of hypoxia,
inflammation, or both combined.
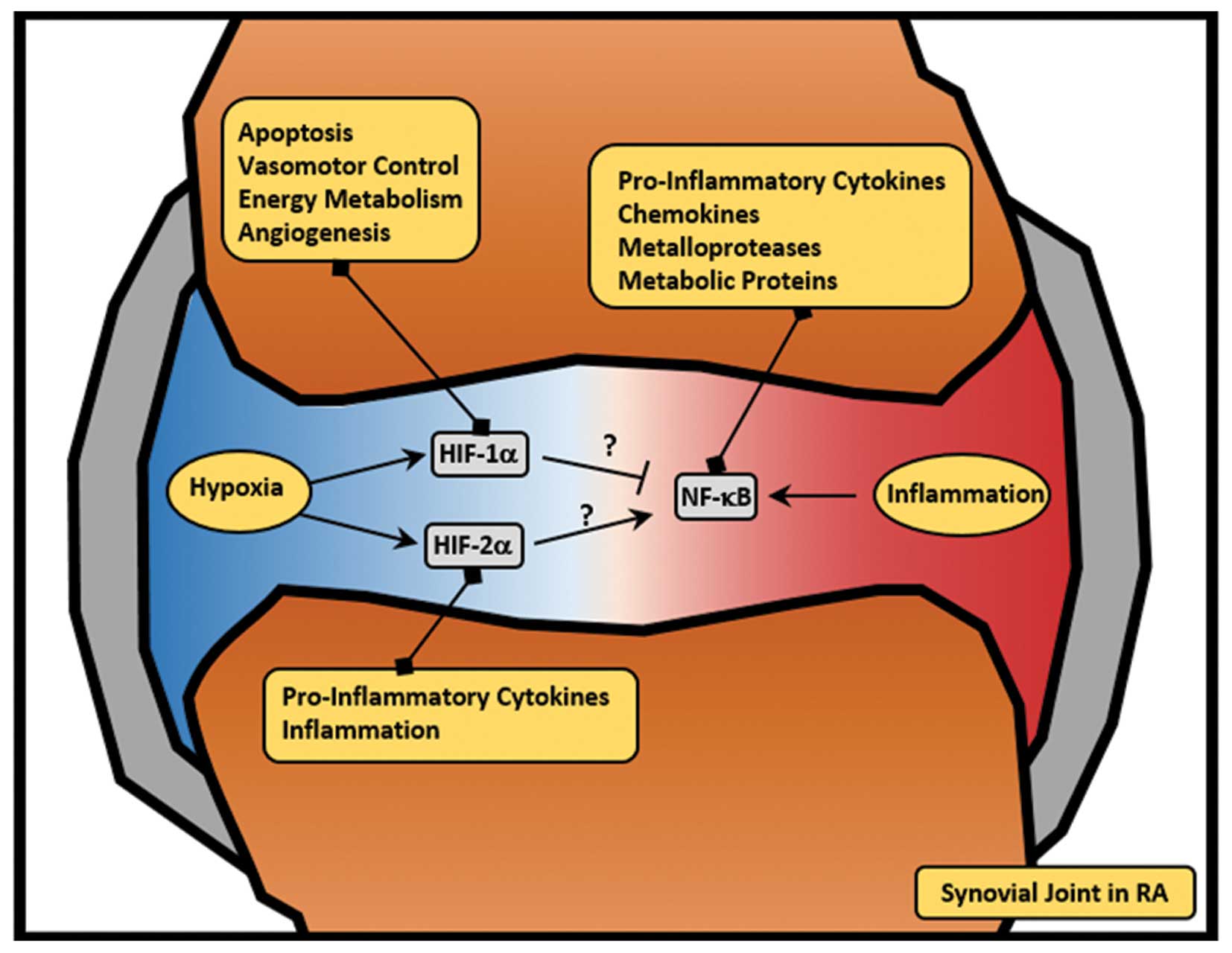
Inflammatory bowel disease (IBD)
The intestinal mucosa is exposed to steep hypoxic
gradients (63) and is in a
constant state of controlled inflammation, which is necessary to
allow tolerance to otherwise harmless ingested dietary antigens
(Fig. 4) (84). This fine balance is pathologically
disturbed in inflammatory bowel disease (IBD); a
relapsing-remitting progressive disorder of the gastrointestinal
tract that comprises both Crohn’s and ulcerative colitis. The
symptoms of IBD can range from mild to severe and include abdominal
pain, intestinal bleeding, weight loss, fever and diarrhoea
(85). The two IBD sub-types have
different distribution patterns: ulcerative colitis is restricted
to the colon, whereas Crohn’s colitis can affect any part of the GI
tract. Both are thought to occur when inappropriate immunological
activity in the intestinal mucosa results in epithelial barrier
dysfunction leading to exposure of the mucosal immune system to
luminal antigenic material and further cycles of inflammation and
barrier dysfunction that underlie disease progression (86,87).
Role of the HIF system in IBD
Hypoxia has been found to play a role in IBD. Lower
resting oxygen levels have been demonstrated in sections of IBD
tissue compared to the controls using a 2-nitroimidazole based
approach (63). In keeping with
these observations, HIF-1α and HIF-2α activation has been
associated with disease and increased vascular density in human
specimens (88). The increased
vascular density was subsequently demonstrated to be effected by
vascular endothelial growth factor (VEGF), an established target of
the HIF system (89).
Compartmental analyses of the effects of hypoxia have been possible
in murine models, where hypoxia has been shown to affect the
epithelium, primarily during periods of inflammation (63). The colonic epithelium is the most
hypoxic and HIF-active tissue layer because it is physically
farthest away from the colonic vascular plexus and closest to the
anoxic bowel lumen. This effect is exacerbated by oxygen
consumption by luminal bacteria (90), and the presence of inflammatory
mediators and lipopolysaccharide (LPS), which have been shown to
regulate HIF activity (48).
In the context of IBD, HIF system activity is
thought to be protective, acting through three mechanisms: i)
inhibition of epithelial cell apoptosis; ii) enhanced expression of
barrier-protective genes; and the iii) promotion of neutrophil
apoptosis (Fig. 4) (86). Evidence of the anti-apoptotic
effects of HIF has been demonstrated indirectly through experiments
to investigate the role of the hydroxylase inhibitor,
dimethyloxaloylglycine (DMOG), in colitis. Using a murine model of
dextran sodium sulfate (DSS)-induced colitis, HIF stabilisation
following treatment with DMOG has been shown to prevent apoptosis
in a mechanism thought to be mediated by the anti-apoptotic
protein, cIAP-2 (91). Recently,
this effect has been specifically attributed to PHD1, since the
homozygous loss of PHD1, but not PHD2 or PHD3, has been shown to be
protective in the same murine model of DSS-induced colitis
(92). This effect is most likely
HIF-dependent, since the conditional knockout of HIF-1α in mouse
intestinal epithelial cells has been shown to result in an enhanced
susceptibility to the development of colitis (63).
In addition to its anti-apoptotic effects, the HIF
system can protect against colitis through the expression of
barrier-protective genes. Several HIF-dependent target genes have
been proposed as mediators of this effect: CD55 (93), ecto-50 nucleotidase (94), A2B receptor (95) MUC-3 (96), intestinal trefoil factor (97), and P-glycoprotein (98) all play a role in the regulation of
the intestinal mucosa barrier and have all been demonstrated to be
regulated in a hypoxia-dependent manner.
There is also evidence of the differential effects
of the HIF-α isoform in IBD. HIF-2α expression has been shown to be
increased in colon tissues of mice after the induction of colitis.
This was also observed in patients with ulcerative colitis or
Crohn’s disease (62).
Interestingly, in that study, while the loss of HIF-2α was
associated with attenuated colonic inflammation, the overexpression
of HIF-2α led to spontaneous colitis and increased
inflammation.
Role of NF-κB in IBD
Other transcriptional programs are active in IBD in
addition to those enacted by the HIF system. IBD is primarily an
inflammatory pathology and NF-κB activity has been linked to its
progression (5). A high degree of
NF-κB induction has been demonstrated in intestinal macrophages and
epithelial cells (99). In IBD,
inflammatory cytokines can drive NF-κB activation, leading to the
production of more inflammatory cytokines and potentiating further
NF-κB activation (Fig. 4).
NF-κB-induced TNF-α expression is one example of this type of
positive feedback loop (100).
Interestingly, NF-κB can have a dual role in IBD, potentiating
inflammation in intestinal macrophages while protecting from
inflammation in mucosal epithelial cells. Sharing interesting
similarities to the effects of HIF activation, NF-κB signalling in
intestinal epithelial cells has been shown to be protective against
the development of colitis (101). Deletion of the NF-κB pathway in
intestinal epithelial cells results in decreased expression of
anti-apoptotic genes, such as Bcl-xL, and leads to reduced
epithelial barrier function and increased susceptibility to colitis
(102). Conditional knockout of
NEMO and subsequent NF-κB inhibition has been shown to result in
severe epithelial inflammation in a murine model (102). Similarly, epithelial
cell-specific IKKβ deletion has been shown to result in the
sustained production of pro-inflammatory Th1 cytokines and
increased intestinal inflammation (103). Several treatments have been
proposed to target NF-κB activity in IBD, including proteasome
blockade, the administration of non-coding RNAs to interfere with
NF-κB-DNA binding and anti-TNF-α immunotherapy. However, all have
been met with significant systemic toxicity due to the broad role
of NF-κB in multiple organs.
NF-κB-HIF crosstalk in IBD
Sharing similarities with the microenvironment of
RA, in IBD both inflammation and hypoxia are present in the
intestinal epithelium and contribute to disease progression
(104). It is generally [but not
universally (105)] understood
that both NF-κB and HIF activity are protective in episodes of
colitis (101). Significant
crosstalk between these pathways has already been established, and
it has been proposed that both pathways may act in concert to
contribute to the epithelial barrier function of the colon in a
process that is deregulated in IBD.
One example of this crosstalk is the regulation of
apoptosis by both pathways (106,107). The caspase recruitment domain
family, member 9 (CARD9) is understood to function as a molecular
scaffold for the assembly of a BCL10 signalling complex that
activates NF-κB (106), and has
also been shown to be involved in the regulation of
hypoxia-sensitive pathways (107). CARD9 therefore represents one
point of crosstalk that may be important in the development of IBD
as a promising target for further investigation.
Our laboratory and others have demonstrated
NF-κB-dependent HIF-1α mRNA regulation (61,108). NF-κB can also regulate HIF
signalling through IKKγ and HIF-2α, which increases HIF-2α
transcriptional activity through interaction with cAMP response
element-binding (CREB) binding protein (CBP)/p300 (109). Negative feedback through the
NF-κB-dependent induction of the micro-RNA, miR-155, in
response to LPS has been shown to target HIF-1α for silencing
(110). Furthermore, our
laboratory have recently demonstrated an evolutionarily conserved
negative feedback mechanism through which HIF can regulate NF-κB in
a mechanism that is dependent on the kinases, TAK-IKK and CDK6
(59).
CRC
CRC is a lethal disease affecting over 500,000
individuals annually (111). In
contrast to the protective effects of HIF and NF-κB activity in
IBD, both can play important roles in the development of colorectal
malignancy. In CRC, the hypoxic milieu is similar to that of IBD
but, critically, the cells are transformed to allow them to react
differently to the activation of either system. In addition,
chronic inflammation is a hallmark of cancer (112). The role of the HIF system and
the role of NF-κB activity are considered below, and the
significance of their crosstalk with respect to the development of
CRC is examined.
Role of the HIF system in CRC
The role of the HIFs in cancer progression has long
been appreciated due to their ability to promote angiogenesis
through one of the principally identified HIF-1α target genes,
VEGFA (113). However, it is
becoming more evident that hypoxia and the HIF system can affect
tumour growth through modulation of proliferation, apoptosis and
epithelial to mesenchymal transition (EMT) (Fig. 5). Hypoxia and the subsequent HIF
activation are generally understood to be prognostically bad and
lead to tumour progression (114). In CRC, HIF-1α stabilisation has
been shown to lead to a poor disease outcome. Shay et al
(114) demonstrated that the
inhibition of HIF signalling using acriflavine halted the
progression of an autochthonous model of established
colitis-associated colon cancer in immuno-competent mice. In their
model, treatment with acriflavine was shown to decrease tumour
number, size and advancement, in an effect thought to be mediated
through the inhibition of HIF-dependent targets, such as VEGFA.
These data provide a direct link between HIF-1α expression and
tumour progression. However, HIF isoform activation can be
antagonistic in the context of tumour progression. In contrast to
the effect of high HIF-1α expression, high HIF-2α expression has
recently been reported to prevent CRC progression (115). The antagonistic effects of
HIF-1α and HIF-2α are important for the regulation of proliferation
and apoptosis in cancer biology (40,82,116). The HIF system can affect
proliferation through the regulation of cMyc. HIF-1α can promote
cell cycle arrest by the direct opposition of c-Myc activity and
the induction of p21 in CRC (116). Conversely, HIF-2α has been shown
to promote proliferation in through its augmentation of cMyc
function (40).
 | Figure 5HIF and NF-κB crosstalk in CRC. In
CRC, the intestinal lumen is characterised by hypoxic and
inflammatory regions (in blue and red, respectively). HIF-1α is
activated in hypoxia, and is involved in the modulation of tumour
growth, apoptosis, and EMT. Inflammation leads to the activation of
NF-κB, which is involved in the expression of pro-inflammatory
cytokines, ROS production, and tumour survival. In CRC, there are
some points of crosstalk between NF-κB and HIF, namely in the
regulation of p53, APC, and cMyc. HIF, hypoxia-inducible factor;
NF-κB, nuclear factor-κB; CRC, colorectal cancer; EMT, epithelial
to mesenchymal transition; ROS, reactive oxygen species; APC,
adenomatous polyposis coli. |
The HIF system can also affect apoptosis through the
regulation of p53. p53 stability leads to apoptosis in somatic
cells and it is frequently mutated in cancers in pursuit of
immortality. HIF-1α has been shown to stabilise wild-type p53 via
physical interaction through its ODD (41,117). As a form of negative feedback,
p53 can promote the degradation of HIF-1α (118). The negative feedback of
wild-type p53 on HIF-1α could explain the increased stability of
HIF-1α in tumours that express mutant p53 which is incapable of
degrading HIF-1α. The net result of the p53-HIF-1α interaction is
increased apoptosis in damaged cells that are exposed to hypoxia
(119). HIF-2α can inhibit p53
phosphorylation, resulting in a reduction in p53 pathway activity
and the prevention of apoptosis in response to damaging stimuli
(120). In addition to its role
in the regulation of p53, HIF has been linked to the positive
regulation of apoptosis through the control of several
pro-apoptotic factors, including caspase-3, Fas and Fas ligand
(121).
Hypoxia is a critical determinant of the motile and
invasive phenotype of cancer cells. HIF activation is also
important in the regulation of genes involved in EMT, including the
direct regulation of the EMT-promoting transcription factors, Snail
and Twist, which have both been described as direct targets of the
HIF system (122–124). EMT is a critical event in the
induction of tumour metastasis (125). Notch has also been shown to
mediate HIF-1α-dependent EMT (126).
Role of NF-κB in CRC
The role of NF-κB in CRC is an active area of study
(101,127–129). Inflammation is an important
trigger in the establishment and development of CRC. Patients with
long-standing IBD have an increased risk of developing CRC
(127,128). In this context, NF-κB activation
can promote tumourigenesis and CRC progression. In CRC, chronic
inflammation results in sustained reactive oxygen species (ROS)
production, leading to DNA damage (Fig. 5) (130). Treatment with non-steroidal
anti-inflammatory drugs (NSAIDs) reduces the development of CRC in
patients with IBD and hereditary CRC (131,132), and the inactivation of NF-κB
signalling reduces the formation of inflammation-associated tumours
(101,129). IL-6 has been shown to be
important for the number and size of tumours formed in mice
(133), and IKKβ conditional
knockout mice have been shown to develop more numerous tumours
(134).
As with the HIF system, the mechanism of
NF-κB-induced tumourigenesis and progression can be multifactorial.
The activation of the NF-κB pathway confers survival,
proliferation, angiogenic and migratory advantages (Fig. 5) (112,135–138); all of which are hallmarks of
cancer (112). NF-κB activation
can block apoptosis by regulating the anti-apoptosis proteins, such
as inhibitor of apoptotic proteins (IAPs) (139), or by the inhibition of prolonged
c-Jun N-terminal kinase (JNK) signalling, modulating the
accumulation of ROS (140).
Alternatively, NF-κB activation can enhance IL-2 production, which
can activate Janus kinase 3 (Jak3) by autophosphorylation (141). Jak3 can activate STAT3. Jak3 and
STAT3 over-activation has been observed in human colon cancer in
vivo and in vitro, and shown to prevent apoptosis,
leading to poor prognosis (142,143). In addition, NF-κB activation can
affect proliferation and cell growth through the regulation of its
target genes, cyclin D1 and cMyc (144–146), and promote angiogenesis through
the regulation of VEGF and IL-8 (136). Finally, NF-κB activation has
been shown to affect the expression of matrix metalloprotease-9
(MMP-9), in murine colon adenocarcinoma cells (147), an important protein in the
regulation of migration and invasion.
NF-κB-HIF crosstalk in CRC
The data presented above demonstrate a clear overlap
between the effectors of the HIF and NF-κB systems in the
establishment and development of CRC. Solid tumours are
characterised by the presence of hypoxia, as well as inflammation
(6). Potential points for
crosstalk include the regulation of cMyc and p53 (Fig. 5). NF-κB interacts with the
co-activators, p300 and CREB-binding protein, to inhibit p53
function. This effect is reinforced by the NF-κB-dependent
upregulation of the p53 inhibitor, mouse double minute 2 (MDM2)
(3,148) and is similar to that exerted on
p53 by HIF-1α. The expression of NF-κB, HIF, VEGF and Bcl-3 has
been shown to correlate with proliferation, angiogenesis, decreased
survival and a poor clinical outcome (149,150). In addition, TNF-α has been shown
to stabilise Snail and β-catenin in a process that requires the
downregulation of glycogen synthase kinase-3β (GSK3β) by NF-κB and
the activation of Akt cascades, resulting in the promotion of EMT
(151). These data are
clinically important since NF-κB and Twist have been associated
with lymph node metastasis in patients with CRC (152). Interestingly, HIF has been shown
to interact with both Snail and Twist, making this another
potential point for crosstalk between the pathways (153).
In addition to the mechanisms outlined above, there
is a complex interplay between HIF, NF-κB and adenomatous polyposis
coli (APC), that appears to be important in CRC (Fig. 5). One of the earliest events in
the development of CRC is loss of the APC gene. Our laboratory has
recently reported a functional crosstalk between HIF-1α and APC at
the transcriptional level (154). HIF activation represses APC
expression, acting at its promoter to result in positive activation
and proliferation through the Wnt/β-catenin signalling and the
TCF-LEF pathway (155),
reduction in genetic and microtubule stability and reductions in
cell migration (6,156).
The repression of APC by HIF-1α is complicated by
the fact that medium levels of β-catenin can induce NF-κB,
resulting in positive feedback, and high levels of β-catenin
inhibit NF-κB, resulting in negative feedback (6). Further studies are required to
determine the functional significance of this interaction in
vivo. However, it represents another exciting point of
crosstalk with importance for CRC disease progression.
7. Conclusion
In this review, the current understanding of the
mechanisms of the HIF and NF-κB systems has been discussed with
specific reference to the crosstalk between these two
stress-responsive pathways. This crosstalk is significant for many
disease processes and its role in RA, inflammatory bowel disease
and CRC has been discussed in detail. It is important to note that
the crosstalk between these pathways has significance beyond
pathological processes. For example, in healthy individuals who
live at a high altitude, prolonged HIF activation can lead to
reduced NF-κB activity, effectively dampening the immune response.
Further studies in this area is required; however, it is
interesting that the anecdotal evidence of increased H.
pylori infection in Tibetan monks exists (157). Individuals with mountain
sickness have presented with increased levels of inflammatory
cytokines circulating in the blood (52). Another study demonstrated that
healthy volunteers who spent three nights at high altitudes
(>3,400 meters), presented with high levels of the inflammatory
cytokine, IL-6 (53). This
hypoxia-inflammation crosstalk is also relevant in the clinical
context. It was shown that ischemia in organ grafts increased the
risk of inflammation and, consequently, graft failure or organ
rejection (158). Accurate
systematic experimentation is important to determine the mechanisms
of the crosstalk between these pathways since these findings may
have an impact on multiple disease processes, apart from those
discussed herein. These include diabetes and systemic sclerosis,
where limb perfusion is not optimal, resulting in increased tissue
breakdown in the absence of an appropriate inflammatory response,
leading to an increased infection rate.
Acknowledgments
We would like to thank members of the S.R.
laboratory for their helpful discussions. J.B. is a CR-UK clinical
fellow, D.B. is funded by a Wellcome Trust ISSF award, the S.R.
laboratory is funded by a CR-UK Senior Research Fellowship
(C99667/A12918). This study was also supported by a Wellcome Trust
Strategic Award (097945/B/11/Z).
Abbreviations:
|
IKK
|
inhibitor of κB kinase
|
|
NF-κB
|
nuclear factor-κB
|
|
HIF
|
hypoxia-inducible factor
|
|
ARNT
|
aryl hydrocarbon nuclear
translocator
|
|
PHD
|
prolyl hydroxylase
|
|
vHL
|
von Hippel Lindau
|
|
TNF
|
tumour necrosis factor
|
References
|
1
|
Semenza GL: Regulation of oxygen
homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda).
24:97–106. 2009. View Article : Google Scholar
|
|
2
|
Semenza GL: HIF-1 and human disease: One
highly involved factor. Genes Dev. 14:1983–1991. 2000.PubMed/NCBI
|
|
3
|
Perkins ND: The diverse and complex roles
of NF-κB subunits in cancer. Nat Rev Cancer. 12:121–132.
2012.PubMed/NCBI
|
|
4
|
Thornton RD, Lane P, Borghaei RC, Pease
EA, Caro J and Mochan E: Interleukin 1 induces hypoxia-inducible
factor 1 in human gingival and synovial fibroblasts. Biochem J.
350:307–312. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taylor CT: Interdependent roles for
hypoxia inducible factor and nuclear factor-kappaB in hypoxic
inflammation. J Physiol. 586:4055–4059. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Näthke I and Rocha S: Antagonistic
crosstalk between APC and HIF-1α. Cell Cycle. 10:1545–1547. 2011.
View Article : Google Scholar
|
|
7
|
Semenza GL and Wang GL: A nuclear factor
induced by hypoxia via de novo protein synthesis binds to the human
erythropoietin gene enhancer at a site required for transcriptional
activation. Mol Cell Biol. 12:5447–5454. 1992.PubMed/NCBI
|
|
8
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl
Acad Sci USA. 92:5510–5514. 1995. View Article : Google Scholar
|
|
9
|
Carroll VA and Ashcroft M: Role of
hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the
regulation of HIF target genes in response to hypoxia, insulin-like
growth factor-I, or loss of von Hippel-Lindau function:
Implications for targeting the HIF pathway. Cancer Res.
66:6264–6270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou J, Schmid T, Schnitzer S and Brüne B:
Tumor hypoxia and cancer progression. Cancer Lett. 237:10–21. 2006.
View Article : Google Scholar
|
|
11
|
Patel SA and Simon MC: Biology of
hypoxia-inducible factor-2alpha in development and disease. Cell
Death Differ. 15:628–634. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Makino Y, Cao R, Svensson K, Bertilsson G,
Asman M, Tanaka H, Cao Y, Berkenstam A and Poellinger L: Inhibitory
PAS domain protein is a negative regulator of hypoxia-inducible
gene expression. Nature. 414:550–554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamashita T, Ohneda O, Nagano M, et al:
Abnormal heart development and lung remodeling in mice lacking the
hypoxia-inducible factor-related basic helix-loop-helix PAS protein
NEPAS. Mol Cell Biol. 28:1285–1297. 2008. View Article : Google Scholar :
|
|
14
|
Zhang P, Yao Q, Lu L, Li Y, Chen PJ and
Duan C: Hypoxia-inducible factor 3 is an oxygen-dependent
transcription activator and regulates a distinct transcriptional
response to hypoxia. Cell Rep. 6:1110–1121. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bárdos JI and Ashcroft M: Negative and
positive regulation of HIF-1: A complex network. Biochim Biophys
Acta. 1755:107–120. 2005.PubMed/NCBI
|
|
16
|
Rocha S: Gene regulation under low oxygen:
Holding your breath for transcription. Trends Biochem Sci.
32:389–397. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qin C, Wilson C, Blancher C, Taylor M,
Safe S and Harris AL: Association of ARNT splice variants with
estrogen receptor-negative breast cancer, poor induction of
vascular endothelial growth factor under hypoxia, and poor
prognosis. Clin Cancer Res. 7:818–823. 2001.PubMed/NCBI
|
|
18
|
Kaelin WG Jr and Ratcliffe PJ: Oxygen
sensing by metazoans: The central role of the HIF hydroxylase
pathway. Mol Cell. 30:393–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haase VH: Renal cancer: Oxygen meets
metabolism. Exp Cell Res. 318:1057–1067. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Berra E, Benizri E, Ginouvès A, Volmat V,
Roux D and Pouysségur J: HIF prolyl-hydroxylase 2 is the key oxygen
sensor setting low steady-state levels of HIF-1alpha in normoxia.
EMBO J. 22:4082–4090. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Appelhoff RJ, Tian YM, Raval RR, Turley H,
Harris AL, Pugh CW, Ratcliffe PJ and Gleadle JM: Differential
function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the
regulation of hypoxia-inducible factor. J Biol Chem.
279:38458–38465. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Epstein AC, Gleadle JM, McNeill LA, et al:
C. elegans EGL-9 and mammalian homologs define a family of
dioxygenases that regulate HIF by prolyl hydroxylation. Cell.
107:43–54. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fandrey J, Gorr TA and Gassmann M:
Regulating cellular oxygen sensing by hydroxylation. Cardiovasc
Res. 71:642–651. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bruegge K, Jelkmann W and Metzen E:
Hydroxylation of hypoxia-inducible transcription factors and
chemical compounds targeting the HIF-alpha hydroxylases. Curr Med
Chem. 14:1853–1862. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Frede S, Stockmann C, Freitag P and
Fandrey J: Bacterial lipopolysaccharide induces HIF-1 activation in
human monocytes via p44/42 MAPK and NF-kappaB. Biochem J.
396:517–527. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ivan M, Kondo K, Yang H, Kim W, Valiando
J, Ohh M, Salic A, Asara JM, Lane WS and Kaelin WG Jr: HIFalpha
targeted for VHL-mediated destruction by proline hydroxylation:
Implications for O2 sensing. Science. 292:464–468. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jaakkola P, Mole DR, Tian YM, et al:
Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation
complex by O2-regulated prolyl hydroxylation. Science.
292:468–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu F, White SB, Zhao Q and Lee FS:
HIF-1alpha binding to VHL is regulated by stimulus-sensitive
proline hydroxylation. Proc Natl Acad Sci USA. 98:9630–9635. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Durán RV, MacKenzie ED, Boulahbel H,
Frezza C, Heiserich L, Tardito S, Bussolati O, Rocha S, Hall MN and
Gottlieb E: HIF-independent role of prolyl hydroxylases in the
cellular response to amino acids. Oncogene. 32:4549–4556. 2013.
View Article : Google Scholar :
|
|
30
|
Moser SC, Bensaddek D, Ortmann B, Maure
JF, Mudie S, Blow JJ, Lamond AI, Swedlow JR and Rocha S: PHD1 links
cell-cycle progression to oxygen sensing through hydroxylation of
the centrosomal protein Cep192. Dev Cell. 26:381–392. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo W, Hu H, Chang R, Zhong J, Knabel M,
O’Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is
a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell.
145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie L, Pi X, Mishra A, Fong G, Peng J and
Patterson C: PHD3-dependent hydroxylation of HCLK2 promotes the DNA
damage response. J Clin Invest. 122:2827–2836. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pugh CW, Tan CC, Jones RW and Ratcliffe
PJ: Functional analysis of an oxygen-regulated transcriptional
enhancer lying 3′ to the mouse erythropoietin gene. Proc Natl Acad
Sci USA. 88:10553–10557. 1991. View Article : Google Scholar
|
|
34
|
Semenza GL, Jiang BH, Leung SW, Passantino
R, Concordet JP, Maire P and Giallongo A: Hypoxia response elements
in the aldolase A, enolase 1, and lactate dehydrogenase A gene
promoters contain essential binding sites for hypoxia-inducible
factor 1. J Biol Chem. 271:32529–32537. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schödel J, Oikonomopoulos S, Ragoussis J,
Pugh CW, Ratcliffe PJ and Mole DR: High-resolution genome-wide
mapping of HIF-binding sites by ChIP-seq. Blood. 117:e207–e217.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Semenza GL: Regulation of cancer cell
metabolism by hypoxia-inducible factor 1. Semin Cancer Biol.
19:12–16. 2009. View Article : Google Scholar
|
|
37
|
Han YH, Xia L, Song LP, Zheng Y, Chen WL,
Zhang L, Huang Y, Chen GQ and Wang LS: Comparative proteomic
analysis of hypoxia-treated and untreated human leukemic U937
cells. Proteomics. 6:3262–3274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Djidja MC, Chang J, Hadjiprocopis A, et
al: Identification of hypoxia-regulated proteins using MALDI-mass
spectrometry imaging combined with quantitative proteomics. J
Proteome Res. 13:2297–2313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gustafsson MV, Zheng X, Pereira T, Gradin
K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U and
Bondesson M: Hypoxia requires notch signaling to maintain the
undifferentiated cell state. Dev Cell. 9:617–628. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gordan JD, Bertout JA, Hu CJ, Diehl JA and
Simon MC: HIF-2alpha promotes hypoxic cell proliferation by
enhancing c-myc transcriptional activity. Cancer Cell. 11:335–347.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
An WG, Kanekal M, Simon MC, Maltepe E,
Blagosklonny MV and Neckers LM: Stabilization of wild-type p53 by
hypoxia-inducible factor 1alpha. Nature. 392:405–408. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View Article : Google Scholar
|
|
43
|
No authors listed. Celebrating 25 years of
NF-κB. Nat Immunol. 12:6812011. View Article : Google Scholar
|
|
44
|
Campbell KJ and Perkins ND: Regulation of
NF-kappaB function. Biochem Soc Symp. 73:165–180. 2006.PubMed/NCBI
|
|
45
|
Wong D, Teixeira A, Oikonomopoulos S, et
al: Extensive characterization of NF-κB binding uncovers
non-canonical motifs and advances the interpretation of genetic
functional traits. Genome Biol. 12:R702011. View Article : Google Scholar
|
|
46
|
Gilmore TD: The Rel/NF-kappaB signal
transduction pathway: Introduction. Oncogene. 18:6842–6844. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen F, Castranova V, Shi X and Demers LM:
New insights into the role of nuclear factor-kappaB, a ubiquitous
transcription factor in the initiation of diseases. Clin Chem.
45:7–17. 1999.PubMed/NCBI
|
|
48
|
Bandarra DR and Rocha S: A tale of two
transcription factors: NF-κB and HIF crosstalk. OA Mol Cell Biol.
1:62013. View Article : Google Scholar
|
|
49
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Perkins ND and Gilmore TD: Good cop, bad
cop: The different faces of NF-kappaB. Cell Death Differ.
13:759–772. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aggarwal BB, Takada Y, Shishodia S,
Gutierrez AM, Oommen OV, Ichikawa H, Baba Y and Kumar A: Nuclear
transcription factor NF-kappa B: Role in biology and medicine.
Indian J Exp Biol. 42:341–353. 2004.PubMed/NCBI
|
|
52
|
Hackett PH and Roach RC: High-altitude
illness. N Engl J Med. 345:107–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hartmann G, Tschöp M, Fischer R,
Bidlingmaier C, Riepl R, Tschöp K, Hautmann H, Endres S and Toepfer
M: High altitude increases circulating interleukin-6, interleukin-1
receptor antagonist and C-reactive protein. Cytokine. 12:246–252.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim HL, Cho YS, Choi H, Chun YS, Lee ZH
and Park JW: Hypoxia-inducible factor 1alpha is deregulated by the
serum of rats with adjuvant-induced arthritis. Biochem Biophys Res
Commun. 378:123–128. 2009. View Article : Google Scholar
|
|
55
|
Boyd HK, Lappin TR and Bell AL: Evidence
for impaired erythropoietin response to anaemia in rheumatoid
disease. Br J Rheumatol. 30:255–259. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Grenz A, Clambey E and Eltzschig HK:
Hypoxia signaling during intestinal ischemia and inflammation. Curr
Opin Crit Care. 18:178–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Eltzschig HK, Sitkovsky MV and Robson SC:
Purinergic signaling during inflammation. N Engl J Med.
367:2322–2333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bandarra D, Biddlestone J, Mudie S, Muller
HA and Rocha S: Hypoxia activates IKK-NF-κB and the immune response
in Drosophila melanogaster. Biosci Rep. 34:342014. View Article : Google Scholar
|
|
59
|
Bandarra D, Biddlestone J, Mudie S, Muller
HA and Rocha S: HIF-1α restricts NF-κB dependent gene expression to
control innate immunity signals. Dis Model Mech. Dec 15–2014.Epub
ahead of print.
|
|
60
|
van Uden P, Kenneth NS, Webster R, Müller
HA, Mudie S and Rocha S: Evolutionary conserved regulation of
HIF-1β by NF-κB. PLoS Genet. 7:e10012852011. View Article : Google Scholar
|
|
61
|
van Uden P, Kenneth NS and Rocha S:
Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem
J. 412:477–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Xue X, Ramakrishnan S, Anderson E, Taylor
M, Zimmermann EM, Spence JR, Huang S, Greenson JK and Shah YM:
Endothelial PAS domain protein 1 activates the inflammatory
response in the intestinal epithelium to promote colitis in mice.
Gastroenterology. 145:831–841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Karhausen J, Furuta GT, Tomaszewski JE,
Johnson RS, Colgan SP and Haase VH: Epithelial hypoxia-inducible
factor-1 is protective in murine experimental colitis. J Clin
Invest. 114:1098–1106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sewell KL and Trentham DE: Pathogenesis of
rheumatoid arthritis. Lancet. 341:283–286. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Al-Shukaili AK and Al-Jabri AA: Rheumatoid
arthritis, cytokines and hypoxia. What is the link. Saudi Med J.
27:1642–1649. 2006.PubMed/NCBI
|
|
66
|
Gaber T, Dziurla R, Tripmacher R,
Burmester GR and Buttgereit F: Hypoxia inducible factor (HIF) in
rheumatology: Low O2! See what HIF can do. Ann Rheum
Dis. 64:971–980. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hueber W, Kidd BA, Tomooka BH, et al:
Antigen microarray profiling of autoantibodies in rheumatoid
arthritis. Arthritis Rheum. 52:2645–2655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
van Baarsen LG, Wijbrandts CA, Rustenburg
F, Cantaert T, van der Pouw Kraan TC, Baeten DL, Dijkmans BA, Tak
PP and Verweij CL: Regulation of IFN response gene activity during
infliximab treatment in rheumatoid arthritis is associated with
clinical response to treatment. Arthritis Res Ther. 12:R112010.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
van Wietmarschen HA, Dai W, van der Kooij
AJ, et al: Characterization of rheumatoid arthritis subtypes using
symptom profiles, clinical chemistry and metabolomics measurements.
PLoS One. 7:e443312012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sweeney SE and Firestein GS: Signal
transduction in rheumatoid arthritis. Curr Opin Rheumatol.
16:231–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Morel J and Berenbaum F: Signal
transduction pathways: new targets for treating rheumatoid
arthritis. Joint Bone Spine. 71:503–510. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Firestein GS and Manning AM: Signal
transduction and transcription factors in rheumatic disease.
Arthritis Rheum. 42:609–621. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Benito MJ, Murphy E, Murphy EP, van den
Berg WB, FitzGerald O and Bresnihan B: Increased synovial tissue
NF-kappa B1 expression at sites adjacent to the cartilage-pannus
junction in rheumatoid arthritis. Arthritis Rheum. 50:1781–1787.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Handel ML, McMorrow LB and Gravallese EM:
Nuclear factor-kappa B in rheumatoid synovium. Localization of p50
and p65. Arthritis Rheum. 38:1762–1770. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Müller-Ladner U, Pap T, Gay RE, Neidhart M
and Gay S: Mechanisms of disease: The molecular and cellular basis
of joint destruction in rheumatoid arthritis. Nat Clin Pract
Rheumatol. 1:102–110. 2005. View Article : Google Scholar
|
|
76
|
Simmonds RE and Foxwell BM: Signalling,
inflammation and arthritis: NF-kappaB and its relevance to
arthritis and inflammation. Rheumatology (Oxford). 47:584–590.
2008. View Article : Google Scholar
|
|
77
|
Westra J, Molema G and Kallenberg CG:
Hypoxia-inducible factor-1 as regulator of angiogenesis in
rheumatoid arthritis -therapeutic implications. Curr Med Chem.
17:254–263. 2010. View Article : Google Scholar
|
|
78
|
Ryu JH, Chae CS, Kwak JS, et al:
Hypoxia-inducible factor-2α is an essential catabolic regulator of
inflammatory rheumatoid arthritis. PLoS Biol. 12:e10018812014.
View Article : Google Scholar
|
|
79
|
Hu F, Shi L, Mu R, et al:
Hypoxia-inducible factor-1α and interleukin 33 form a regulatory
circuit to perpetuate the inflammation in rheumatoid arthritis.
PLoS One. 8:e726502013. View Article : Google Scholar
|
|
80
|
Brouwer E, Gouw AS, Posthumus MD, van
Leeuwen MA, Boerboom AL, Bijzet J, Bos R, Limburg PC, Kallenberg CG
and Westra J: Hypoxia inducible factor-1-alpha (HIF-1alpha) is
related to both angiogenesis and inflammation in rheumatoid
arthritis. Clin Exp Rheumatol. 27:945–951. 2009.
|
|
81
|
Muz B, Khan MN, Kiriakidis S and Paleolog
EM: Hypoxia. The role of hypoxia and HIF-dependent signalling
events in rheumatoid arthritis. Arthritis Res Ther. 11:2012009.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Moniz S, Biddlestone J and Rocha S:
Grow2: The HIF system, energy homeostasis and the cell
cycle. Histol Histopathol. 29:589–600. 2014.PubMed/NCBI
|
|
83
|
Kenneth NS and Rocha S: Regulation of gene
expression by hypoxia. Biochem J. 414:19–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Poonam P: The biology of oral tolerance
and issues related to oral vaccine design. Curr Pharm Des.
13:2001–2007. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Podolsky DK: Inflammatory bowel disease. N
Engl J Med. 347:417–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Cummins EP, Doherty GA and Taylor CT:
Hydroxylases as therapeutic targets in inflammatory bowel disease.
Lab Invest. 93:378–383. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Abraham C and Cho JH: Inflammatory bowel
disease. N Engl J Med. 361:2066–2078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Giatromanolaki A, Sivridis E, Maltezos E,
Papazoglou D, Simopoulos C, Gatter KC, Harris AL and Koukourakis
MI: Hypoxia inducible factor 1alpha and 2alpha overexpression in
inflammatory bowel disease. J Clin Pathol. 56:209–213. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Danese S, Dejana E and Fiocchi C: Immune
regulation by microvascular endothelial cells: Directing innate and
adaptive immunity, coagulation, and inflammation. J Immunol.
178:6017–6022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Werth N, Beerlage C, Rosenberger C, et al:
Activation of hypoxia inducible factor 1 is a general phenomenon in
infections with human pathogens. PLoS One. 5:e115762010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Cummins EP, Seeballuck F, Keely SJ, Mangan
NE, Callanan JJ, Fallon PG and Taylor CT: The hydroxylase inhibitor
dimethyloxalylglycine is protective in a murine model of colitis.
Gastroenterology. 134:156–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tambuwala MM, Cummins EP, Lenihan CR, et
al: Loss of prolyl hydroxylase-1 protects against colitis through
reduced epithelial cell apoptosis and increased barrier function.
Gastroenterology. 139:2093–2101. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Louis NA, Hamilton KE, Kong T and Colgan
SP: HIF-dependent induction of apical CD55 coordinates epithelial
clearance of neutrophils. FASEB J. 19:950–959. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Synnestvedt K, Furuta GT, Comerford KM,
Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF and
Colgan SP: Ecto-5′-nucleotidase (CD73) regulation by
hypoxia-inducible factor-1 mediates permeability changes in
intestinal epithelia. J Clin Invest. 110:993–1002. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kong T, Westerman KA, Faigle M, Eltzschig
HK and Colgan SP: HIF-dependent induction of adenosine A2B receptor
in hypoxia. FASEB J. 20:2242–2250. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Louis NA, Hamilton KE, Canny G, Shekels
LL, Ho SB and Colgan SP: Selective induction of mucin-3 by hypoxia
in intestinal epithelia. J Cell Biochem. 99:1616–1627. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Furuta GT, Turner JR, Taylor CT, Hershberg
RM, Comerford K, Narravula S, Podolsky DK and Colgan SP:
Hypoxia-inducible factor 1-dependent induction of intestinal
trefoil factor protects barrier function during hypoxia. J Exp Med.
193:1027–1034. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Comerford KM, Wallace TJ, Karhausen J,
Louis NA, Montalto MC and Colgan SP: Hypoxia-inducible
factor-1-dependent regulation of the multidrug resistance (MDR1)
gene. Cancer Res. 62:3387–3394. 2002.PubMed/NCBI
|
|
99
|
Neurath MF, Pettersson S, Meyer zum
Büschenfelde KH and Strober W: Local administration of antisense
phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B
abrogates established experimental colitis in mice. Nat Med.
2:998–1004. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Holtmann MH and Neurath MF: Differential
TNF-signaling in chronic inflammatory disorders. Curr Mol Med.
4:439–444. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Greten FR, Eckmann L, Greten TF, Park JM,
Li ZW, Egan LJ, Kagnoff MF and Karin M: IKKbeta links inflammation
and tumorigenesis in a mouse model of colitis-associated cancer.
Cell. 118:285–296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Pasparakis M: IKK/NF-kappaB signaling in
intestinal epithelial cells controls immune homeostasis in the gut.
Mucosal Immunol. 1(Suppl 1): S54–S57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zaph C, Troy AE, Taylor BC, et al:
Epithelial-cell-intrinsic IKK-beta expression regulates intestinal
immune homeostasis. Nature. 446:552–556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hauser CJ, Locke RR, Kao HW, Patterson J
and Zipser RD: Visceral surface oxygen tension in experimental
colitis in the rabbit. J Lab Clin Med. 112:68–71. 1988.PubMed/NCBI
|
|
105
|
Shah YM, Ito S, Morimura K, et al:
Hypoxia-inducible factor augments experimental colitis through an
MIF-dependent inflammatory signaling cascade. Gastroenterology.
134:2036–2048. 2048 e2031–2033. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Hara H and Saito T: CARD9 versus CARMA1 in
innate and adaptive immunity. Trends Immunol. 30:234–242. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Yang H, Minamishima YA, Yan Q, Schlisio S,
Ebert BL, Zhang X, Zhang L, Kim WY, Olumi AF and Kaelin WG Jr: pVHL
acts as an adaptor to promote the inhibitory phosphorylation of the
NF-kappaB agonist Card9 by CK2. Mol Cell. 28:15–27. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Rius J, Guma M, Schachtrup C, Akassoglou
K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG and Karin M:
NF-kappaB links innate immunity to the hypoxic response through
transcriptional regulation of HIF-1alpha. Nature. 453:807–811.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Bracken CP, Whitelaw ML and Peet DJ:
Activity of hypoxia-inducible factor 2alpha is regulated by
association with the NF-kappaB essential modulator. J Biol Chem.
280:14240–14251. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
O’Connell RM, Rao DS, Chaudhuri AA, Boldin
MP, Taganov KD, Nicoll J, Paquette RL and Baltimore D: Sustained
expression of microRNA-155 in hematopoietic stem cells causes a
myeloproliferative disorder. J Exp Med. 205:585–594. 2008.
View Article : Google Scholar
|
|
111
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Tsuzuki Y, Fukumura D, Oosthuyse B, Koike
C, Carmeliet P and Jain RK: Vascular endothelial growth factor
(VEGF) modulation by targeting hypoxia-inducible
factor-1alpha--> hypoxia response element--> VEGF cascade
differentially regulates vascular response and growth rate in
tumors. Cancer Res. 60:6248–6252. 2000.PubMed/NCBI
|
|
114
|
Shay JE, Imtiyaz HZ, Sivanand S, et al:
Inhibition of hypoxia-inducible factors limits tumor progression in
a mouse model of colorectal cancer. Carcinogenesis. 35:1067–1077.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Rawluszko-Wieczorek AA, Horbacka K,
Krokowicz P, Misztal M and Jagodzinski PP: Prognostic potential of
DNA methylation and transcript levels of HIF1A and EPAS1 in
colorectal cancer. Mol Cancer Res. 12:1112–1127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Koshiji M, Kageyama Y, Pete EA, Horikawa
I, Barrett JC and Huang LE: HIF-1alpha induces cell cycle arrest by
functionally counteracting Myc. EMBO J. 23:1949–1956. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Sánchez-Puig N, Veprintsev DB and Fersht
AR: Binding of natively unfolded HIF-1alpha ODD domain to p53. Mol
Cell. 17:11–21. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter
CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL and Bedi A:
Regulation of tumor angiogenesis by p53-induced degradation of
hypoxia-inducible factor 1alpha. Genes Dev. 14:34–44.
2000.PubMed/NCBI
|
|
119
|
Moeller BJ, Dreher MR, Rabbani ZN,
Schroeder T, Cao Y, Li CY and Dewhirst MW: Pleiotropic effects of
HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 8:99–110.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Bertout JA, Majmundar AJ, Gordan JD, Lam
JC, Ditsworth D, Keith B, Brown EJ, Nathanson KL and Simon MC:
HIF2alpha inhibition promotes p53 pathway activity, tumor cell
death, and radiation responses. Proc Natl Acad Sci USA.
106:14391–14396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Volm M and Koomägi R: Hypoxia-inducible
factor (HIF-1) and its relationship to apoptosis and proliferation
in lung cancer. Anticancer Res. 20:1527–1533. 2000.PubMed/NCBI
|
|
122
|
Evans AJ, Russell RC, Roche O, et al: VHL
promotes E2 box-dependent E-cadherin transcription by HIF-mediated
regulation of SIP1 and snail. Mol Cell Biol. 27:157–169. 2007.
View Article : Google Scholar :
|
|
123
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by
HIF-1alpha promotes metastasis. Nat Cell Biol. 10:295–305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Gort EH, van Haaften G, Verlaan I, et al:
The TWIST1 oncogene is a direct target of hypoxia-inducible
factor-2alpha. Oncogene. 27:1501–1510. 2008. View Article : Google Scholar
|
|
125
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Sahlgren C, Gustafsson MV, Jin S,
Poellinger L and Lendahl U: Notch signaling mediates
hypoxia-induced tumor cell migration and invasion. Proc Natl Acad
Sci USA. 105:6392–6397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Lakatos PL and Lakatos L: Risk for
colorectal cancer in ulcerative colitis: Changes, causes and
management strategies. World J Gastroenterol. 14:3937–3947. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Munkholm P: Review article: The incidence
and prevalence of colorectal cancer in inflammatory bowel disease.
Aliment Pharmacol Ther. 18(Suppl 2): 1–5. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Fernández-Majada V, Aguilera C, Villanueva
A, et al: Nuclear IKK activity leads to dysregulated
notch-dependent gene expression in colorectal cancer. Proc Natl
Acad Sci USA. 104:276–281. 2007. View Article : Google Scholar :
|
|
130
|
Seril DN, Liao J, Yang GY and Yang CS:
Oxidative stress and ulcerative colitis-associated carcinogenesis:
Studies in humans and animal models. Carcinogenesis. 24:353–362.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Sangha S, Yao M and Wolfe MM:
Non-steroidal anti-inflammatory drugs and colorectal cancer
prevention. Postgrad Med J. 81:223–227. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Hoffmeister M, Chang-Claude J and Brenner
H: Do older adults using NSAIDs have a reduced risk of colorectal
cancer. Drugs Aging. 23:513–523. 2006. View Article : Google Scholar
|
|
133
|
Becker C, Fantini MC, Schramm C, et al:
TGF-beta suppresses tumor progression in colon cancer by inhibition
of IL-6 trans-signaling. Immunity. 21:491–501. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Greten FR, Arkan MC, Bollrath J, et al:
NF-kappaB is a negative regulator of IL-1beta secretion as revealed
by genetic and pharmacological inhibition of IKKbeta. Cell.
130:918–931. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
136
|
Richmond A: Nf-kappa B, chemokine gene
transcription and tumour growth. Nat Rev Immunol. 2:664–674. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Schulze-Bergkamen H and Krammer PH:
Apoptosis in cancer-implications for therapy. Semin Oncol.
31:90–119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Kucharczak J, Simmons MJ, Fan Y and
Gélinas C: To be, or not to be: NF-kappaB is the answer - role of
Rel/NF-kappaB in the regulation of apoptosis. Oncogene.
22:8961–8982. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Luo JL, Kamata H and Karin M:
IKK/NF-kappaB signaling: Balancing life and death - a new approach
to cancer therapy. J Clin Invest. 115:2625–2632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Cornejo MG, Boggon TJ and Mercher T: JAK3:
A two-faced player in hematological disorders. Int J Biochem Cell
Biol. 41:2376–2379. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Lin Q, Lai R, Chirieac LR, et al:
Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and
cell lines: Inhibition of JAK3/STAT3 signaling induces apoptosis
and cell cycle arrest of colon carcinoma cells. Am J Pathol.
167:969–980. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Tsareva SA, Moriggl R, Corvinus FM,
Wiederanders B, Schütz A, Kovacic B and Friedrich K: Signal
transducer and activator of transcription 3 activation promotes
invasive growth of colon carcinomas through matrix
metalloproteinase induction. Neoplasia. 9:279–291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS Jr: NF-kappaB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999.PubMed/NCBI
|
|
145
|
Chen C, Edelstein LC and Gélinas C: The
Rel/NF-kappaB family directly activates expression of the apoptosis
inhibitor Bcl-x(L). Mol Cell Biol. 20:2687–2695. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Baldwin AS: Control of oncogenesis and
cancer therapy resistance by the transcription factor NF-kappaB. J
Clin Invest. 107:241–246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Choo MK, Sakurai H, Kim DH and Saiki I: A
ginseng saponin metabolite suppresses tumor necrosis
factor-α-promoted metastasis by suppressing nuclear factor-κB
signaling in murine colon cancer cells. Oncol Rep. 19:595–600.
2008.PubMed/NCBI
|
|
148
|
Thomasova D, Mulay SR, Bruns H and Anders
HJ: p53-independent roles of MDM2 in NF-κB signaling: Implications
for cancer therapy, wound healing, and autoimmune diseases.
Neoplasia. 14:1097–1101. 2012.
|
|
149
|
Puvvada SD, Funkhouser WK, Greene K, Deal
A, Chu H, Baldwin AS, Tepper JE and O’Neil BH: NF-κB and Bcl-3
activation are prognostic in metastatic colorectal cancer.
Oncology. 78:181–188. 2010. View Article : Google Scholar :
|
|
150
|
Kwon HC, Kim SH, Oh SY, et al:
Clinicopathological significance of nuclear factor-kappa B, HIF-1
alpha, and vascular endothelial growth factor expression in stage
III colorectal cancer. Cancer Sci. 101:1557–1561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Wu Y and Zhou BP:
TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and
invasion. Br J Cancer. 102:639–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Schwitalla S, Ziegler PK, Horst D, et al:
Loss of p53 in enterocytes generates an inflammatory
microenvironment enabling invasion and lymph node metastasis of
carcinogen-induced colorectal tumors. Cancer Cell. 23:93–106. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Terzic J, Grivennikov S, Karin E and Karin
M: Inflammation and colon cancer. Gastroenterology. 138:2101–2114.
e21052010. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Newton IP, Kenneth NS, Appleton PL, Näthke
I and Rocha S: Adenomatous polyposis coli and hypoxia-inducible
factor-1{alpha} have an antagonistic connection. Mol Biol Cell.
21:3630–3638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Bienz M and Clevers H: Linking colorectal
cancer to Wnt signaling. Cell. 103:311–320. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
McCartney BM and Näthke IS: Cell
regulation by the Apc protein Apc as master regulator of epithelia.
Curr Opin Cell Biol. 20:186–193. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Wen D, Zhang N, Shan B and Wang S:
Helicobacter pylori infection may be implicated in the topography
and geographic variation of upper gastrointestinal cancers in the
Taihang Mountain high-risk region in northern China. Helicobacter.
15:416–421. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Krüger B, Krick S, Dhillon N, et al: Donor
Toll-like receptor 4 contributes to ischemia and reperfusion injury
following human kidney transplantation. Proc Natl Acad Sci USA.
106:3390–3395. 2009. View Article : Google Scholar : PubMed/NCBI
|