Introduction
Immature brains are known to be exceedingly
sensitive to oxidative stress, due to high levels of unsaturated
fatty acids, a high rate of oxygen consumption, low concentrations
of antioxidants, a high content of metals catalyzing free radical
formation and a large proportion of sensitive immature cells
(1–3). This sensitivity undoubtedly
contributes to the severe neuronal damage observed following
hypoxic-ischemic encephalopathy (HIE) in newborns (3). HIE occurs in 0.1–0.2% of term or
near-term infants, among whom approximately 20% die and up to 40%
of the survivors often suffer devastating disabilities, such as
cerebral palsy, mental retardation and epilepsy (4–7).
Thus, the reduction of damaging oxidants in neonatal brains
following hypoxic-ischemic (HI) injury is one of the main
therapeutic goals for the effective treatment of HIE.
Compared with adult patients, infants have a poorer
prognosis as HI injury not only leads to injury to neuronal cells,
but it also disrupts ongoing brain development. Insulin-like growth
factor-I (IGF-I) is an essential trophic factor for neuronal
proliferation and differentiation (8,9).
Its potential neuroprotective effects against HI injury in immature
brains have been evaluated in fetal sheep (10) and neonatal rat models (11). In the rat model, although the
exogenous administration of IGF-I was shown to exert
neuroprotective effects and to improve long-term neurological
function (11), only a 40%
reduction in brain injury was achieved at 3 days of recovery when
IGF-I was administered by intraventricular injection immediately
following HI injury (11). On the
other hand, the delayed subcutaneous administration of IGF-I (24
and 48 h post-injury) has been shown to significantly increase the
volume of surviving brain tissue and to improve behavioral
development at 2 months of age (19). These time-dependent
neuroprotective effects may be attributed to the suppression of
IGF-I activity due to increased levels of oxidative stress in the
acute phase of injury. Therefore, we hypothesized that the
reduction of the levels of oxidative stress may enhance the
neuroprotective effects of IGF-I.
Sodium pyruvate (SP) is a substrate of the
tricarboxylic acid cycle and an extracellular antioxidant (12,13). In a previous study, we found that
SP improved the survival of primary cortical neurons under
conditions of oxygen glucose deprivation (OGD) and, when
administered 30 min after HI injury, it reduced HI injury to
neonatal rat brains and improved long-term behavioral recovery
(14). Compared with SP, ethyl
pyruvate (EP) is a more stable lipophilic ester derivative of
pyruvate and has been proven to reduce HI injury to neonatal brains
through anti-cell death and anti-inflammatory mechanisms (15). As EP has a longer half-life and
fewer side-effects, we hypothesized that the early administration
of EP may improve the protective effects of IGF-I against HI in
immature brains. In this study, we examined this hypothesis in
vitro using a model of ODG, as well as in vivo using a
model of neonatal rat HI injury.
Materials and methods
Primary cortical neuron culture
The animal experiments were approved by the Ethics
Committee of Indiana University School of Medicine, Indianapolis,
IN, USA. The cultured primary neurons were derived from newborn
Sprague-Dawley rats (10–12 pups) as previously described (6). Briefly, after removing the meninges,
the cortical tissue was minced and maintained in Dulbecco’s
modified Eagle’s medium (DMEM) at 4°C. An aliquot of 0.25% trypsin
(15050-065; Invitrogen, Carlsbad, CA, USA) and DNase (DN25; Sigma,
St. Louis, MO, USA) was added to the tissue and incubated for 15
min at 37°C to produce a single cell suspension. Following
centrifugation, the cells were resuspended in neurobasal (NB)
medium (10888; Gibco/Life Technologies, Grand Island, NY, USA)
supplemented with 2% B-27 (17504-044; Invitrogen), 0.5 mM glutamine
(25030-081; Gibco), 100 U/ml penicillin and 100 µg/ml
streptomycin. The cells were plated into poly-L-lysine-coated
(P1399; Sigma) dishes at 5×105/ml. The culture medium
was changed at 24 h and 4 days in vitro (DIV) and fibroblast
growth factor (FGF; 5 ng/ml final concentration; F0291; Sigma) was
added to the culture medium. The cells were ready to use at 6–7
DIV.
OGD
On day 7, the cultured primary cortical neurons were
gently washed with phosphate-buffered saline (PBS) and the medium
was then changed to glucose-free DMEM (11965; Gibco) before the
cells were placed in a humidified chamber gassed with 95%
N2/5% CO2 at 37°C. The medium of the control
cells was changed to DMEM with glucose (11966; Gibco) and the cells
were kept in a regular incubator (5% CO2 and 21%
O2, 37°C). After 2.5 h, the cells were removed from the
hypoxic chamber to the regular incubator after changing the medium
back to NB medium and treated with EP (0.5 mM; E47808; Sigma)
and/or IGF-I (25 ng/ml; 100-11; Peprotech, Inc., Rocky Hill, NJ,
USA).
Lactate dehydrogenase (LDH) release and
MTT assay
Cell injury was assessed by measuring the amount of
LDH released into the culture medium using a cytotoxicity detection
kit (G1780; Promega Corp., Madison, WI, USA) according to the
manufacturer’s instructions. Briefly, 50 µl of culture
medium were mixed with 50 µl substrate followed by
incubation in the dark at 37°C for 30 min. Subsequenlty, 50
µl stop solution were added and the absorbance was measured
at 490 nm using a microplate reader (M2003; Sigma).
Cell viability was monitored by MTT colorimetric
assay (M2003; Sigma). A total of 10 µl MTT was added to 500
µl of cell culture medium followed by incubation at 37°C for
4 h. After discarding the medium, 500 µl dimethyl sulfoxide
were added and the absorbance at 570 nm was recorded using a
microplate reader (M2003; Sigma).
Model of neonatal HI injury
Briefly, 7-day-old Sprague-Dawley rat pups (8 per
litter, weighing 13–18 g) were anesthetized with a mixture of
isoflurane (3–4% for induction and 2% for maintenance) and 30%
O2/70% N2. The left carotid artery of each
pup was exposed and ligated with 6-0 surgical silk. After a 2-h
recovery period, the pups were placed in 2-litre airtight and
watertight glass flasks, submerged in a 37.0°C water bath, and
exposed to a humidified mixture of 8% O2 and 92%
N2. After 2.5 h of hypoxia, the pups were then returned
to their dams and received the different treatments. The
environmental temperature following HI injury was 25°C. EP was
administered 30 min after HI injury by intraperitoneal (i.p.)
injection (25 mg/kg; E47808; Sigma) and IGF-I was administered 24 h
after HI injury by subcutaneous (s.c.) injection (3 mg/kg; 100-11;
Peprotech, Inc.). The same volume of normal saline was injected and
served as the vehicle. The sham-operated group underwent the same
surgical procedure apart from carotid artery ligaton and exposure
to hypoxia.
Foot fault test
Foot-fault tests were performed at 4 weeks of
recovery as previously described (14). The rats were placed on an elevated
stainless steel wire (diameter, 0.4 cm) grid (1 m above the floor
with 3 cm2 holes). Each pup was placed on the grid and
the number of foot faults was counted out of 50 steps for forelimbs
or hindlimbs. A foot fault was defined as when the animal misplaced
a forelimb or hindlimb and the paw fell between the grid bars. The
examiners were blinded from the study design.
2,3,5-Triphenyltetrazolium chloride
monohydrate (TTC) staining
TTC staining was performed as previously described
(16). At 48 h after HI injury,
the rat brains were removed after sacrifice and immediately
sectioned coronally into 6 slices (2-mm-thick) in a brain matrix
(RBM-4000C Rodent Brain Matrix, Adult Rat, Coronal Sections; ASI
Instruments, Inc. Warren, MI, USA). The brain slices were incubated
in TTC (T8877; 1%; Sigma) at 37°C for 10 min and fixed in 10%
buffered formalin. Images of the sections were acquired using a
digital camera and the survival area was measured using ImageJ
software. For each brain, the surviving area of brain tissue was
calculated as the ratio of the area of ipsilateral TTC-stained
tissue (non-ischemic) to the area of contralateral TTC-stained
tissue.
Sample preparation
At 3, 6, 12, 24, 48, 72 h, 7 days or 4 weeks after
HI injury, the animals were deeply anesthetized with an overdose of
sodium pentobarbital and then perfused transcardially with cold
0.9% saline, followed by 4% paraformaldehyde (PFA) in PBS. The
brains were removed and post-fixed in PFA overnight, then
cryoprotected with 30% sucrose for 48 h. Serial coronal sections
(30 µm) were cut and stored at −20°C.
Cresyl violet staining
For cresyl violet staining, the sections obtained on
day 7 were incubated with cresyl violet for 30 min at 37°C. Ethanol
solution differentiated the stain. The sections were then rinsed
with distilled water and fully air dried.
Fluoro-Jade B (FJB) staining
FJB staining was performed as previously described
(16). For FJB staining, the
sections obtained at 3–72 h were incubated with a solution of 0.06%
potassium permanganate for 30 min, and then incubated with a
0.0004% solution of FJB (AG310; Millipore, Billerica, MA, USA) and
4′,6-diamidino-2-phenylindole (DAPI; Sigma) for 20 min. The
sections were then rinsed with distilled water and fully air dried.
The number of FJB-positive neurons in 3 sections was determined in
the ipsilateral hippocampus that was affected by HI injury.
3-Bromodeoxyuridine (BrdU) staining
For BrdU labeling, the rat pups were administered
daily i.p. injections (50 µg/g in 0.9% saline; Sigma) from
day 1 to day 7 post-HI injury. BrdU+ cells were detected
at 72 h (P10) or 4 weeks (P35) after surgery using an antibody
against BrdU (OBT0030, rat; 1:400; Accurate Chemical &
Scientific Corp., Westbury, NY, USA). Briefly, the brain sections
were incubated with blocking solution (0.1% Triton X-100, 1% bovine
serum albumin, and 5% normal goat serum in PBS) for 1 h at room
temperature, followed by an overnight incubation with primary
antibody (anti-rat BrdU; 1:400, OBT0030; Accurate Chemical &
Scientific Corp.) at 4°C. The sections were then washed with PBS
and incubated with a secondary antibody (Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) at room temperature for 1
h. After being treated with DAPI for 2 min, the sections were
washed with PBS and mounted on slides with Fluoromount G (Cat. no.
17984-25; Electron Microscopy Sciences, Hatfield, PA, USA).
Immunofluorescence staining
The differentiation states of the BrdU+
neurons were determined by co-labeling with an antibody against
doublecortin (DCX) (AB2253, guinea pig; 1:1,500; Millipore) at 72 h
following HI injury (P10) or with an antibody against neuronal
nuclei antigen (NeuN) (MAB377, mouse; 1:100; Millipore) at 4 weeks
following HI injury (P35).
Following immunostaining, the sections were analyzed
by light microscopy at a magnification, ×20 using an inverted
microscope (Zeiss Axiovert 200M; Carl Zeiss, Göttingen, Germany)
interfaced with a digital camera (Zeiss Axion Cam MRc5; Carl Zeiss)
controlled by a computer.
Statistical analysis
Data are presented as the means ± standard error of
the mean. Statistical differences between >2 groups were
analyzed by using a one-way analysis of variance followed by the
Turkey multiple comparison test. A value of P<0.05 was
considered to indicate a statistically significant difference.
Results
Neuroprotective effects of the
combination of EP and IGF-I in cultured neurons under conditions of
OGD
To examine the neuroprotective effects of EP and/or
IGF-I against HI injury, we first examined the degree of neuronal
injury by LDH assay and cell viability (by MTT assay) in the
absence or presence of increasing concentrations of EP or IGF-I
under conditions of OGD (17).
Under conditions of OGD, the LDH levels were increased in the
culture medium and the MTT levels were decreased in the cell lysate
at 24 h of reoxygenation, suggesting neuronal injury under
oxidative stress (Fig. 1A and B).
As the concentration of EP or IGF-I increased in the culture
medium, the LDH levels decreased and the MTT levels increased
(Fig. 1A and B), both in a
dose-dependent manner, indicating a decrease in neuronal injury and
an increase in neuronal cell viability. The neuroprotective effects
were the most prominent when EP (0.5 mM) was combined with IGF-I
(25 ng/ml) as compared to treatment with EP or IGF-I alone
(Fig. 1C).
EP and IGF-I promote long-term behavioral
development following HI injury
We then evaluated the neuroprotective effects of EP
and/or IGF-I in a commonly used neonatal rat model of HI injury
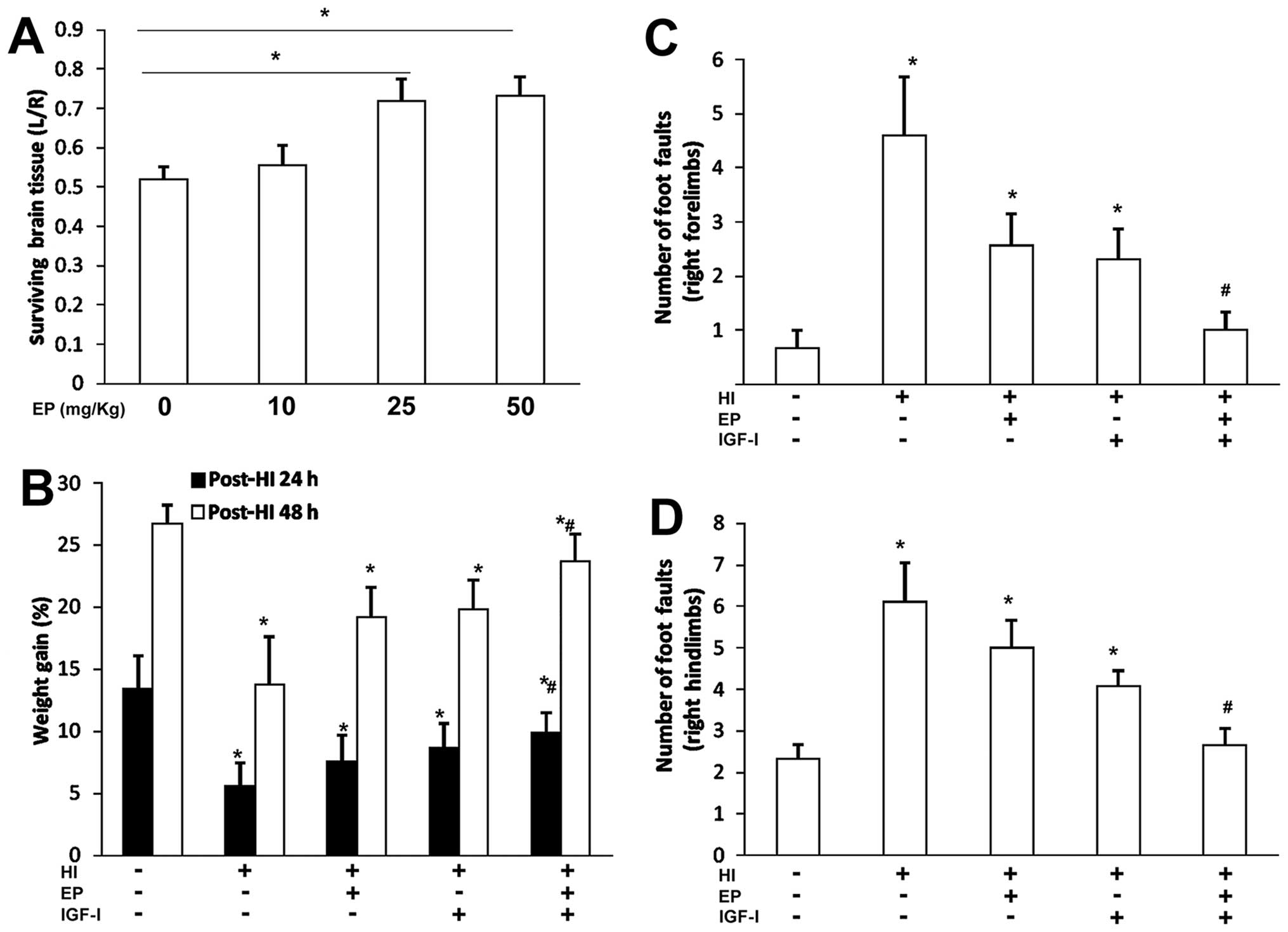
(18). At 48 h of recovery, EP
began to show protective effects against HI injury at the dose of
25 mg/kg (administered at 30 min after HI injury), indicated by an
increase in the volume area of surviving brain tissue (Fig. 2A). We selected a previously
published IGF-I (3 mg/kg, 24 h post HI) treatment dose and schedule
(19) to evaluate the
neuroprotective effects of the two treatments.
Compared with the sham-operated rats, HI injury to
the ipisilateral section of the brain resulted in weight loss
within 24 h (Fig. 2B). While the
rats in the sham-operated group gained approximately 13% of body
weight the following day, the rats in the treated groups gained
more weight over the same time period, with the combined treatment
group gaining the most weight (Fig.
2B).
We employed a foot fault test to compare the
sensorimotor behavior of the rats in the different treatment groups
with that of the rats in the sham-operated group at 4 weeks of
recovery (Fig. 2C and D). When
the total number of foot faults per 50 steps was recorded within 5
min, the rats in the vehicle-treated group showed a significantly
increased number of foot faults compared with the rats in the
sham-operated group (right forelimb, Fig. 2C; right hindlimb, Fig. 2D). However, treatment with EP or
IGF-I decreased the number of foot faults, and combination
treatment with EP and IGF-I resulted in a further reduction in the
number of both right forelimb and hindlimb foot faults compared
with either treatment alone.
Neuroprotective effects of the
combination of EP and IGF-I in vivo
At 48 h of recovery, the amount of surviving brain
tissue in the EP + IGF-I group was 84.2±9.0%, significantly higher
than that with EP treatment alone (71.8±14.2%) or IGF-I treatment
alone (68.8±10.3%). These neuroprotective effects were additive and
not transient (Fig. 3A and B). At
7 days of recovery, the amount of surviving brain cortex tissue in
the EP + IGF-I group was 89.7±6.8%, significantly higher than that
with EP treatment alone (73.2±1.4%) or IGF-I treatment alone
(70.0±8.6%). The amount of surviving tissue in the hippocampus in
the EP + IGF-I group was 78.0±9.6%, significantly higher than that
with EP treatment alone (58.3±1.2%) or IGF-I treatment alone
(58.5±9.3%) (Fig. 3C and D).
Therefore, treatment with EP (25 mg/g, 30 min post-HI injury) in
combination with IGF-I (3 mg/kg, 24 h post-HI injury) exerted
neuroprotective effects against HI injury, as indicated by the
increase in the volume of surviving brain tissue at 48 h (Fig. 3A and B) and 7 days (Fig. 3C and D) of recovery.
Treatment with EP and IGF-I decreases
apoptosis in the damaged hippocampus following HI injury
The increase in the volume of surviving tissue in
the brain may have been a result of a reduced number of injured
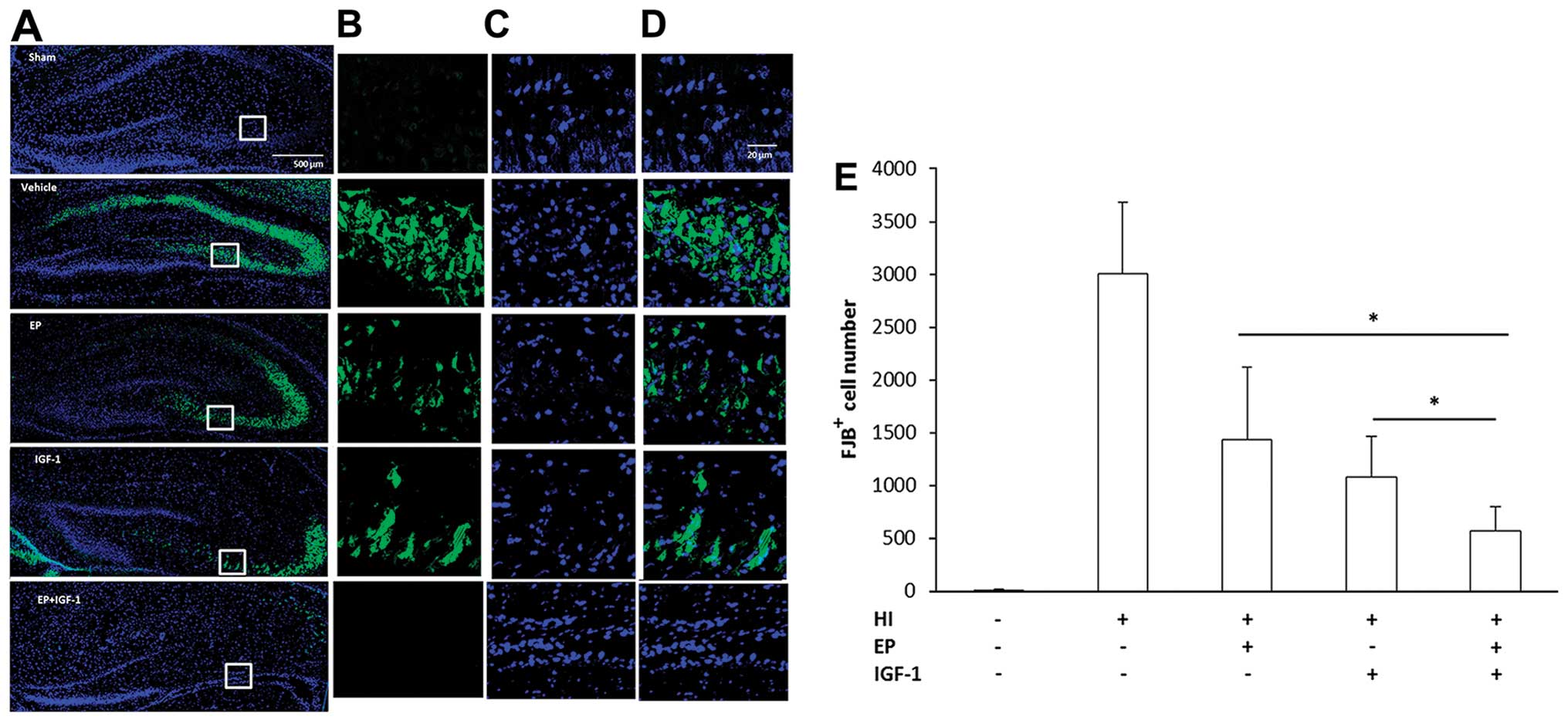
neurons. To clarify this, we labeled the injured neurons with FJB
beginning at 3 h after injury and for up to at least 7 days.
Fig. 4 illustrates the
distribution of FJB+ cells in the hippocampus. The
FJB+ neurons were detected at 3 h of recovery and
reached peak numbers from 48–72 h (data not shown). At 72 h of
recovery, HI injury to the neonatal brain increased the number of
FJB+ cells both in the dentate gyrus (DG) and cornu
ammonis 3 (CA3) region of the hippocampus (Fig. 4A–D). At a higher magnification,
the FJB+ neurons displayed distinct apoptotic nuclei
with either a condensed or fragmented morphology. Compared with the
vehicle-treated group, the numbers of FJB+ neurons were
significantly decreased by EP treatment or IGF-I treatment, whereas
combined treatment with EP and IGF-I led to a further decrease in
the number of FJB+ neurons compared to the groups
treated with EP or IGF-I alone (Fig.
4E).
HI injury promotes neuronal cell
proliferation
To determine the effect of HI injury on
neurogenesis, we labeled proliferating cells with BrdU during the
first 7 days following HI injury. Within the hippocampus of the
rats in the sham-operated group, the majority of BrdU+
cells was distributed in the subgranular zone (SGZ), where neural
stem cells or immature neurons reside (Fig. 5). In comparison, the
BrdU+ cells in the rats in the group subjected to HI
injury were not confined to the SGZ, but were scattered around the
entire DG, suggesting that newborn cells had migrated to other
locations. Overall, the number of BrdU+ cells in the DG
of the rats in the group subjected to HI injury was double that of
the cells in the DG of rats in the sham-operated group (Fig. 5C).
Treatment with EP and IGF-I promotes
neurogenesis in the damaged hippocampus following HI injury
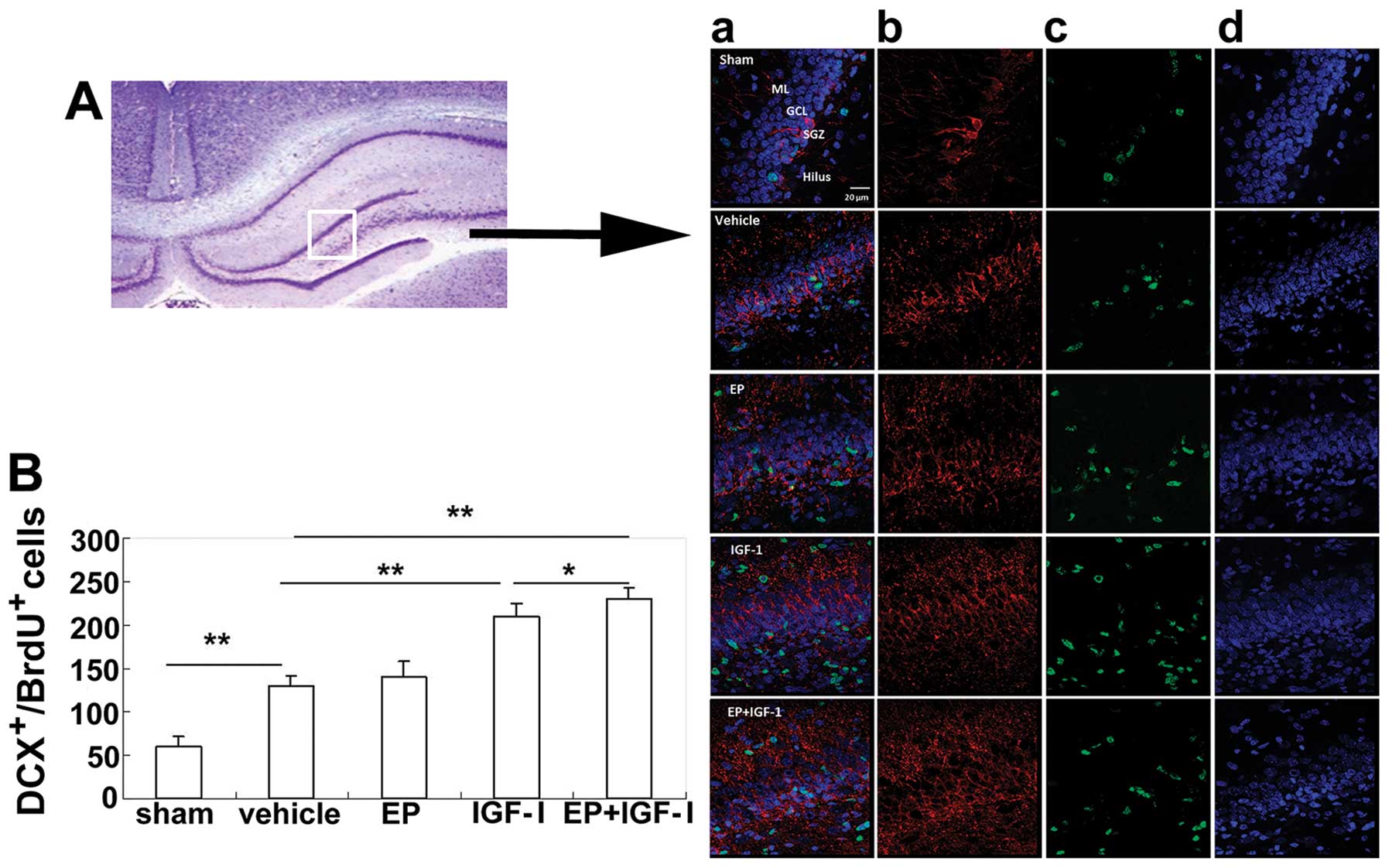
To examine the neuronal differentiation of
BrdU+ cells, we double labeled BrdU+ cells
with either DCX, a marker for immature neurons, or NeuN, a marker
for mature neurons. At 72 h post-HI injury, the number of
BrdU+DCX+ cells in the group subjected to HI
injury was increased compared to that of the rats in the
sham-operated group (Fig. 6).
Although mainly distributed in the SGZ of the rats in the
sham-operated group, the BrdU+DCX+ cells were
also distributed in the granule cell layer (GCL) in the group
subjected to HI (Fig. 6A, panel
d), indicating that more BrdU+DCX+ cells may
have migrated. Although the numbers of the
BrdU+DCX+ cells in the group treated with EP
were similar to those of the vehicle-treated group, the numbers
were markedly increased in the IGF-I-treated group and the combined
treatment group, indicating that IGF-I, but not EP, stimulated
neurogenesis (Fig. 6B). At 4
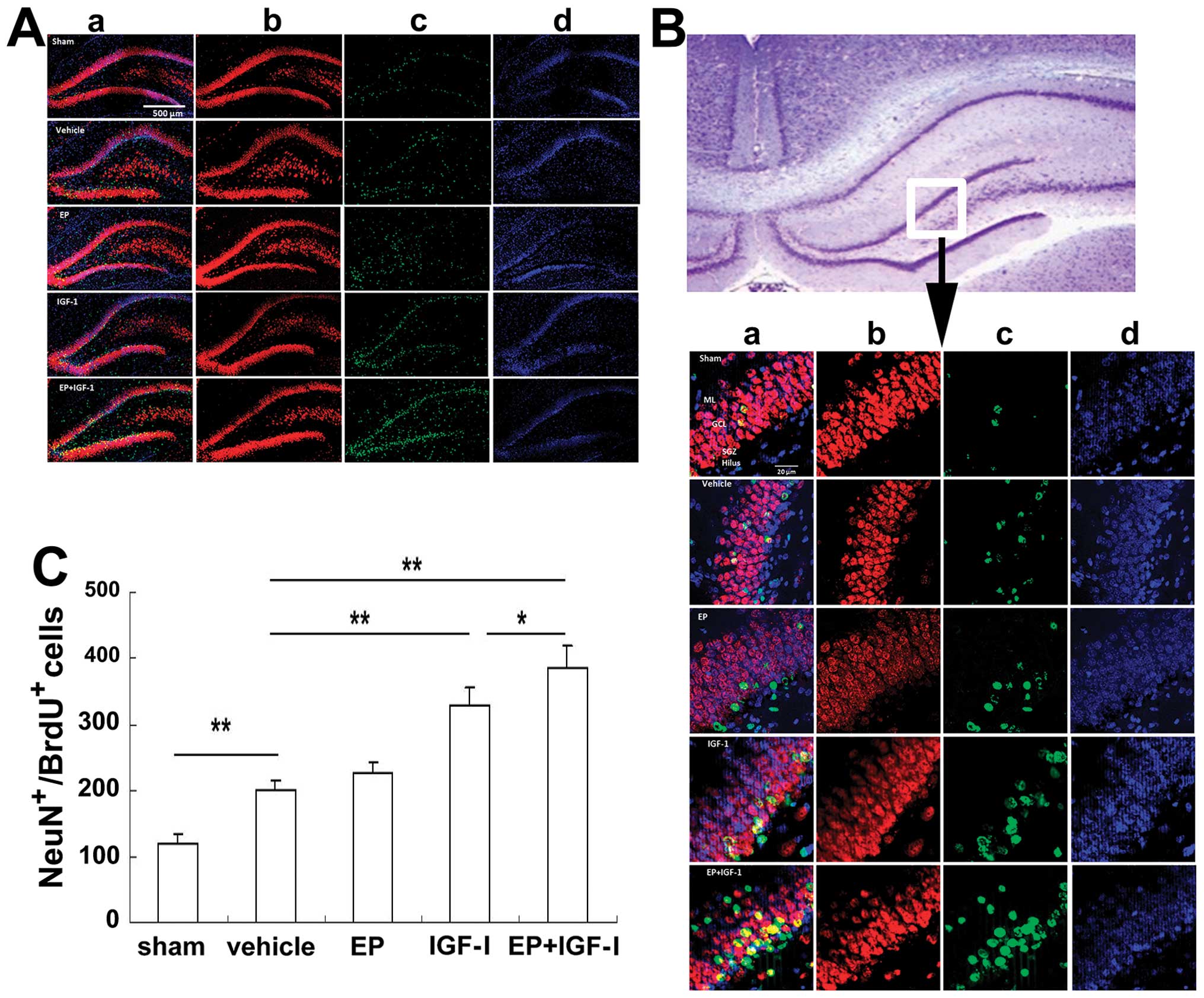
weeks post-injury, the number of NeuN+BrdU+
neurons was also elevated (Fig.
7) in the group subjected to HI compared to the sham-operated
group. Of note, the increase in the number of DCX+ cells
(Fig. 6B) and NeuN+
cells (Fig. 7C), showing a
similar pattern among the treatment groups, suggesting that IGF-I
not only stimulated neurogenesis, but also neuronal survival and
differentiation.
Discussion
HI injury results in severe damage to immature
brains as it disturbs multiple developmental events in addition to
normal brain functions. Compared with the adult brain, neonatal
brains often display different forms of cell death (20) and different responses to
therapeutic interventions (21,22). One of the underlying causes for
these differences is the inability of the immature brains to handle
the rapidly accumulating levels of oxidative stress immediately
following HI injury (1–3). In fact, treatments with antioxidants
provide beneficial effects as has been shown in animal models of HI
injury (14,23,24). While the removal of oxidants only
offers temporary relief, resumption of the normal developmental
process is equally urgent for the young brain. In contination of
our previous study on the beneficial effects of SP (14), in this study, we focused on the
neuroprotective effects of EP and IGF-I.
Our choice of this combination was based on the
following considerations: first, SP provided long-term
neuroprotective effects to immature brains following HI, but it is
not suitable for clinical use due to its short half-life,
instability and side-effects in adults (25). It has been shown that EP, a more
stable and lipophilic derivative of pyruvic acid (25), exerts neuroprotective effects on
cortical neurons equal to those of SP under conditions of OGD. More
importantly, EP (25 mg/kg) reduced brain injury to the same extent
(~20%) as SP (500 mg/kg), but at a much lower dose (data not
shown). Due to its anti-inflammatory effects (26), EP is evidently the better choice
for further investigation. These positive results showing the
neuroprotective effects of EP, are contrast to those of another
study demonstrating the negative effects of neuroprotectants
(27). The results in that case
showed no reduction in the severity of infarction in an in
vivo rat model, despite the fact that the model used was
similar to the one we used. The main differences between the
studies were that the rats were a different strain (Wistar compared
to Sprague-Dawley), the doses were lower than our doses (10 and 40
mg/kg compared to our most effective doses at 25 and 50 mg/kg) and
the exposure to hypoxia was shorter (50 min compared to 150 min).
The outcome was scored only in terms of macroscopic brain injury so
there is a possibility that some effects were missed. The exact
reasons for these different results warrant further investigation,
as it is important that these effects be reproducible in different
model systems if their potential for clinical treatment is to be
fulfilled (27).
Second, IGF-I is a pleiotrophic factor essential to
the survival of immature neurons and maintenance of the high
metabolism needed to fulfill developmental needs, such as neuronal
migration, axon extension and synaptogenesis (28). Serum IGF-I levels have been shown
to be decreased in human newborns suffering from HIE (29) and neuronal IGF-I mRNA (30) and IGF-I serum levels have been
shown to be decreased in neonatal rat brains following HI injury
(19,31). However, immediate intraventricular
(32) and intranasal delivery
(33) was less effective than
delayed subcutaneous delivery (24 and 48 h) in terms of the
reduction of the voluem of injured tissue and long-term behavioral
recovery. This improvement with delayed treatment indicates that
IGF-I is less effective during the acute phase when oxidative
stress is at its highest. A similar result was observed in a study
on rat oligodendrocyte progenitors, where delayed cell death caused
by glutamate was prevented by both the immediate and late (16 h
post-exposure) administration of IGF-I, while cell proliferation
was promoted (34). In addition
IGF-I reverse the loss in oligodendrocyte transcription
factor-positive cells in white matter following HI injury (34). Therefore, combining IGF-I with
early EP treatment to reduce oxidative stress may protect immature
neurons against injury and assist their normal development.
The above hypothesis was supported by the results of
the present study: early EP treatment combined with the delayed
s.c. injection of IGF-I led to reduced neuronal cell death under
conditions of OGD, and reduced damage to the neonatal brain due to
HI injury, both in short-term and long-term experiments. In the
developing hippocampus, the DG is known to be more vulnerable to HI
injury than the CA3 region (36).
In this study, we found that neuronal cell death occurred both in
the DG and CA3 regions, which may result from a longer exposure to
hypoxia (2.5 h) than in other studies (usually <2 h) (35–37). At 72 h of recovery, treatment with
EP and IGF-I alone reduced the number of injured neurons, while
combined treatment further reduced the number of FJB+
neurons. These neuroprotective effects may be a result of their
complimentary neuroprotective actions. IGF-I promotes neuronal cell
survival by activating the key survival signaling kinase, Akt
(38), whereas EP scavenges free
radicals and reduces inflammatory reactions (15).
Apart from promoting neuronal cell survival, IGF-I
also stimulates neurogenesis (39,40). Unlike adult brains, to which
traumatic brain injury mainly induces the proliferation of reactive
astrocytes (41), HI injury to
neonatal mouse brains promotes the proliferation of multiple cell
types, including microglia, endothelial cells, oligodendrocytes and
neurons (42). Following HI
injury, we found that the BrdU+ cells were mainly
distributed in the granular and molecular neuronal cell layers of
the hippocampus, which contain neurons from secondary neurogenesis
(43). To characterize
neurogenesis and differentiation following HI injury, we double
stained BrdU+ cells with DCX, a marker for immature
neurons, or NeuN, a marker for mature neurons. At 3 days and 4
weeks following HI injury, a parallel trend emerged: while EP alone
did not have much of an effect, IGF-I or combined treatment with
both EP and IGF-I resulted in a similar increase in the number of
BrdU+DCX+ immature neurons, as well as in the
number of BrdU+NeuN+ mature neurons. In
addition, a greater number of BrdU+DCX+ cells
had migrated into the GCL and had likely become incorporated into
the neuronal network, as demonstrated by improved motor
coordination at 4 weeks of recovery.
In conclusion, the findings of this study
demonstrate that combining early EP administration with the delayed
s.c. injection of IGF-I exerts neuroprotective effects on immature
neurons under conditions of OGD and following HI injury. These
effects were likely the result of a reduction in neuronal cell
death and an increase in neurogenesis/differentiation following HI
injury.
Acknowledgments
This study was supported by a grant from the Eunice
Kennedy Shriver National Institute of Child Health and Human
Development (no. 1R01HD059979 to W.L.) and by the Indiana Spinal
Cord Brain Injury Fund.
References
|
1
|
Ikonomidou C, Mosinger JL, Salles KS,
Labruyere J and Olney JW: Sensitivity of the developing rat brain
to hypobaric/ischemic damage parallels sensitivity to
N-methyl-aspartate neurotoxicity. J Neurosci. 9:2809–2818.
1989.PubMed/NCBI
|
|
2
|
Jiang X, Mu D, Manabat C, et al:
Differential vulnerability of immature murine neurons to
oxygen-glucose deprivation. Exp Neurol. 190:224–232. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ikonomidou C and Kaindl AM: Neuronal death
and oxidative stress in the developing brain. Antioxid Redox
Signal. 14:1535–1550. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Berger R and Garnier Y: Perinatal brain
injury. J Perinat Med. 28:261–285. 2000.PubMed/NCBI
|
|
5
|
du Plessis AJ and Volpe JJ: Perinatal
brain injury in the preterm and term newborn. Curr Opin Neurol.
15:151–157. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Logitharajah P, Rutherford MA and Cowan
FM: Hypoxic-ischemic encephalopathy in preterm infants: antecedent
factors, brain imaging, and outcome. Pediatr Res. 66:222–229. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vannucci RC: Hypoxic-ischemic
encephalopathy. Am J Perinatol. 17:113–120. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Russo VC, Gluckman PD, Feldman EL and
Werther GA: The insulin-like growth factor system and its
pleiotropic functions in brain. Endocr Rev. 26:916–943. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aberg ND, Brywe KG and Isgaard J: Aspects
of growth hormone and insulin-like growth factor-I related to
neuroprotection, regeneration, and functional plasticity in the
adult brain. ScientificWorldJournal. 6:53–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guan J, Bennet L, George S, et al:
Insulin-like growth factor-1 reduces postischemic white matter
injury in fetal sheep. J Cereb Blood Flow Metab. 21:493–502. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guan J: Insulin-like growth factor-1 and
its derivatives: potential pharmaceutical application for ischemic
brain injury. Recent Pat CNS Drug Discov. 3:112–127. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Desagher S, Glowinski J and Premont J:
Pyruvate protects neurons against hydrogen peroxide-induced
toxicity. J Neurosci. 17:9060–9067. 1997.PubMed/NCBI
|
|
13
|
Mazzio E and Soliman KF: Pyruvic acid
cytoprotection against 1-methyl-4-phenylpyridinium,
6-hydroxydopamine and hydrogen peroxide toxicities in vitro.
Neurosci Lett. 337:77–80. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pan R, Rong Z, She Y, Cao Y, Chang LW and
Lee WH: Sodium pyruvate reduces hypoxic-ischemic injury to neonatal
rat brain. Pediatr Res. 72:479–489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen H, Hu X, Liu C, et al: Ethyl pyruvate
protects against hypoxic-ischemic brain injury via anti-cell death
and anti-inflammatory mechanisms. Neurobiol Dis. 37:711–722. 2010.
View Article : Google Scholar :
|
|
16
|
Rong Z, Pan R, Xu Y, Zhang C, Cao Y and
Liu D: Hesperidin pretreatment protects hypoxia-ischemic brain
injury in neonatal rat. Neuroscience. 255:292–299. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goldberg MP and Choi DW: Combined oxygen
and glucose deprivation in cortical cell culture: calcium-dependent
and calcium-independent mechanisms of neuronal injury. J Neurosci.
13:3510–3524. 1993.PubMed/NCBI
|
|
18
|
Rice JE III, Vannucci RC and Brierley JB:
The influence of immaturity on hypoxic-ischemic brain damage in the
rat. Ann Neurol. 9:131–141. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhong J, Zhao L, Du Y, Wei G, Yao WG and
Lee WH: Delayed IGF-1 treatment reduced long-term
hypoxia-ischemia-induced brain damage and improved behavior
recovery of immature rats. Neurol Res. 31:483–489. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Northington FJ, Chavez-Valdez R and Martin
LJ: Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol.
69:743–758. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perlman JM: Intervention strategies for
neonatal hypoxic-ischemic cerebral injury. Clin Ther. 28:1353–1365.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Savman K and Brown KL: Treating neonatal
brain injury - promise and inherent research challenges. Recent Pat
Inflamm Allergy Drug Discov. 4:16–24. 2010. View Article : Google Scholar
|
|
23
|
Buonocore G and Groenendaal F:
Anti-oxidant strategies. Semin Fetal Neonatal Med. 12:287–295.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hobbs CE and Oorschot DE: Neonatal rat
hypoxia-ischemia: long-term rescue of striatal neurons and motor
skills by combined antioxidant-hypothermia treatment. Brain Pathol.
18:443–454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cruz RJ Jr, Harada T, Sasatomi E and Fink
MP: Effects of ethyl pyruvate and other α-keto carboxylic acid
derivatives in a rat model of multivisceral ischemia and
reperfusion. J Surg Res. 165:151–157. 2011. View Article : Google Scholar
|
|
26
|
Kao KK and Fink MP: The biochemical basis
for the anti-inflammatory and cytoprotective actions of ethyl
pyruvate and related compounds. Biochem Pharmacol. 80:151–159.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gressens P, Le Verche V, Fraser M, Rousset
CI, Schwendimann L, Bennet L, George SA, Wang X, Mallard C, Tilley
BC, et al: Pitfalls in the quest of neuroprotectants for the
perinatal brain. Dev Neurosci. 33:189–198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Werner H and Leroith D: Insulin and
insulin-like growth factor receptors in the brain: Physiological
and pathological aspects. Eur Neuropsychopharmacol. Jan
31–2014.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Satar M, Ozcan K, Yapicioglu H and Narli
N: Serum insulin-like growth factor 1 and growth hormone levels of
hypoxic-ischemic newborns. Biol Neonate. 85:15–20. 2004. View Article : Google Scholar
|
|
30
|
Lee WH, Wang GM, Seaman LB and Vannucci
SJ: Coordinate IGF-I and IGFBP5 gene expression in perinatal rat
brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 16:227–236.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Clawson TF, Vannucci SJ, Wang GM, Seaman
LB, Yang XL and Lee WH: Hypoxia-ischemia-induced apoptotic cell
death correlates with IGF-I mRNA decrease in neonatal rat brain.
Biol Signals Recept. 8:281–293. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brywe KG, Mallard C, Gustavsson M, et al:
IGF-I neuroprotection in the immature brain after hypoxia-ischemia,
involvement of Akt and GSK3beta? Eur J Neurosci. 21:1489–1502.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin S, Fan LW, Rhodes PG and Cai Z:
Intranasal administration of IGF-1 attenuates hypoxic-ischemic
brain injury in neonatal rats. Exp Neurol. 217:361–370. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wood TL, Loladze V, Altieri S, Gangoli N,
Levison SW, Brywe KG, Mallard C and Hagberg H: Delayed IGF-1
administration rescues oligodendrocyte progenitors from
glutamate-induced cell death and hypoxic-ischemic brain damage. Dev
Neurosci. 29:302–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pazos MR, Cinquina V, Gomez A, et al:
Cannabidiol administration after hypoxia-ischemia to newborn rats
reduces long-term brain injury and restores neurobehavioral
function. Neuropharmacology. 63:776–783. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Towfighi J, Mauger D, Vannucci RC and
Vannucci SJ: Influence of age on the cerebral lesions in an
immature rat model of cerebral hypoxia-ischemia: a light
microscopic study. Brain Res Dev Brain Res. 100:149–160. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shrivastava K, Chertoff M, Llovera G,
Recasens M and Acarin L: Short and long-term analysis and
comparison of neurodegeneration and inflammatory cell response in
the ipsilateral and contralateral hemisphere of the neonatal mouse
brain after hypoxia/ischemia. Neurol Res Int. 2012:7815122012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dudek H, Datta SR, Franke TF, et al:
Regulation of neuronal survival by the serine-threonine protein
kinase Akt. Science. 275:661–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aberg MA, Aberg ND, Hedbacker H, Oscarsson
J and Eriksson PS: Peripheral infusion of IGF-I selectively induces
neurogenesis in the adult rat hippocampus. J Neurosci.
20:2896–2903. 2000.
|
|
40
|
D’Ercole AJ, Ye P and O’Kusky JR: Mutant
mouse models of insulin-like growth factor actions in the central
nervous system. Neuropeptides. 36:209–220. 2002. View Article : Google Scholar
|
|
41
|
Gao X, Enikolopov G and Chen J: Moderate
traumatic brain injury promotes proliferation of quiescent neural
progenitors in the adult hippocampus. Exp Neurol. 219:516–523.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bartley J, Soltau T, Wimborne H, et al:
BrdU-positive cells in the neonatal mouse hippocampus following
hypoxic-ischemic brain injury. BMC Neurosci. 6:152005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bayer SA: Development of the hippocampal
region in the rat. I. Neurogenesis examined with 3H-thymidine
autoradiography. J Comp Neurol. 190:87–114. 1980. View Article : Google Scholar : PubMed/NCBI
|