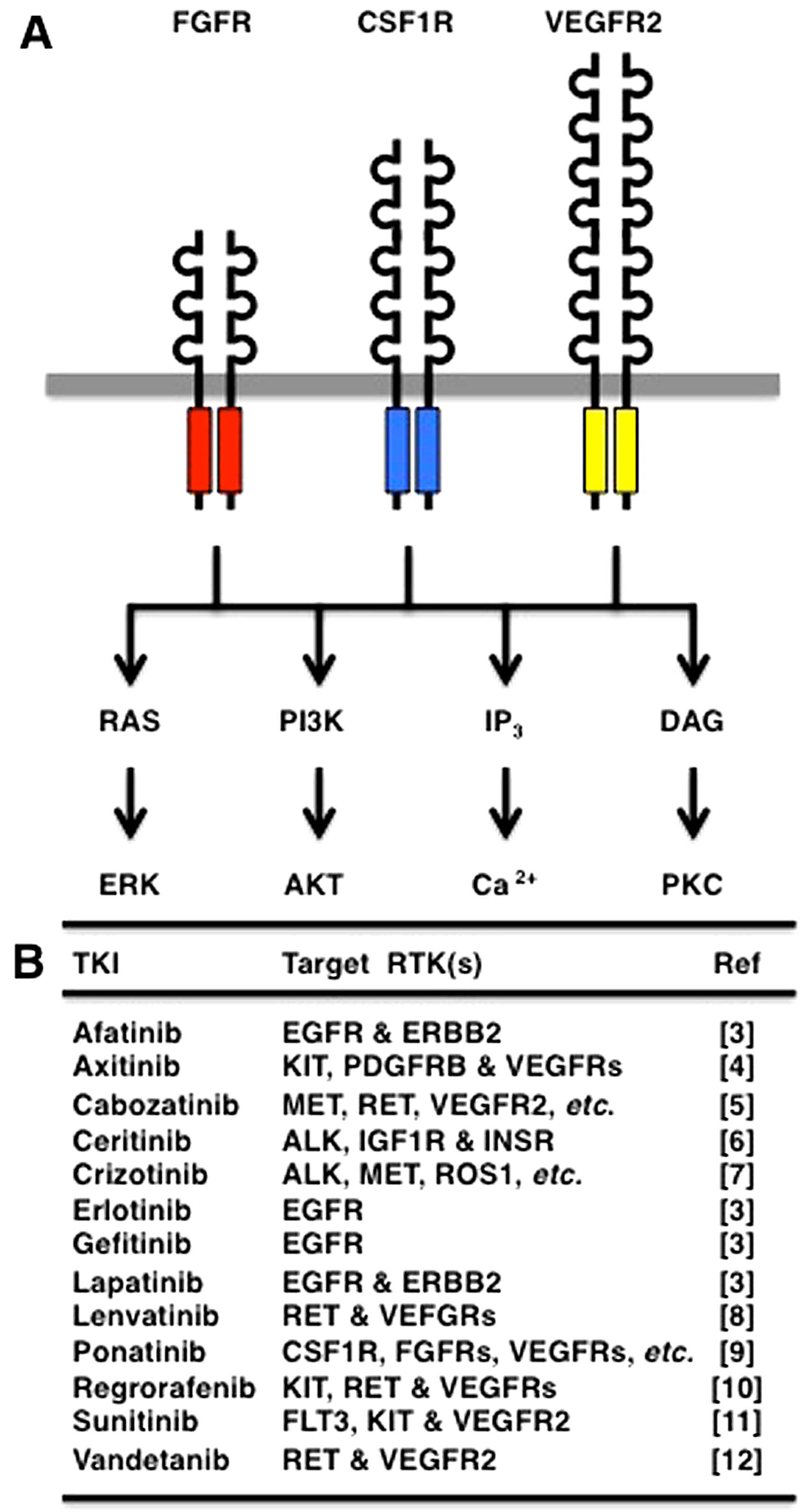
Receptor tyrosine kinases (RTKs) are
transmembrane-type receptors with cytoplasmic tyrosine kinase
domains (1), which transduce
extracellular signals to a variety of intracellular signaling
cascades, such as RAS-ERK, PI3K-AKT, IP3-Ca2+
and DAG-PKC (Fig. 1A).
Phylogenetic analyses of 518 protein kinases revealed that RTKs are
clustered with non-receptor-type tyrosine kinases (2), and analyses of 54 human RTKs using
the Clustal Omega program revealed that RTKs are classified into
the epidermal growth factor receptor (EGFR) group (EGFR, ERBB2,
MET, RYK, etc.), the fibroblast growth factor receptor (FGFR) group
[FGFRs, colony stimulating factor 1 receptor (CSF1R), vascular
endothelial growth factor (VEGF)R2, etc.], the insulin receptor
(INSR) group (INSR, IGF1R, ALK, ROS1, etc.), the RAR-related orphan
receptor (ROR) group (ROR1, ROR2, DDR2, NTRK1, etc.) and the EPH
receptor (EPH) group (EPHA1, EPHB1, PTK7, etc.) (Fig. 2).
Since the aberrant activation of RTKs is a driving
force of human carcinogenesis, small-molecule inhibitors targeting
RTKs have been developed for cancer therapy (3–12).
For example, erlotinib and gefetinib target EGFR; afatinib and
lapatinib target EGFR and ERBB2; and ponatinib (AP24534) targets
multiple RTKs, such as CSF1R, FGFRs, PDGFRs, RET and VEGFRs
(Fig. 1B).
FGFR1, FGFR2, FGFR3 and FGFR4 constitute the FGFR
family of RTKs with three immunoglobulin-like domains in the
extracellular region (13–16).
FGF1 (acidic FGF), FGF2 (basic FGF), FGF3-FGF10, FGF16, FGF17,
FGF18, FGF20 and FGF22 bind to heparin-sulfate proteoglycan for
paracrine signaling through FGFRs, whereas FGF19, FGF21 and FGF23
bind to Klotho proteins for endocrine signaling through FGFRs.
FGFRs are involved in the regulation of cell survival,
proliferation, differentiation and motility during embryogenesis,
adult-tissue homeostasis and carcinogenesis (17–20).
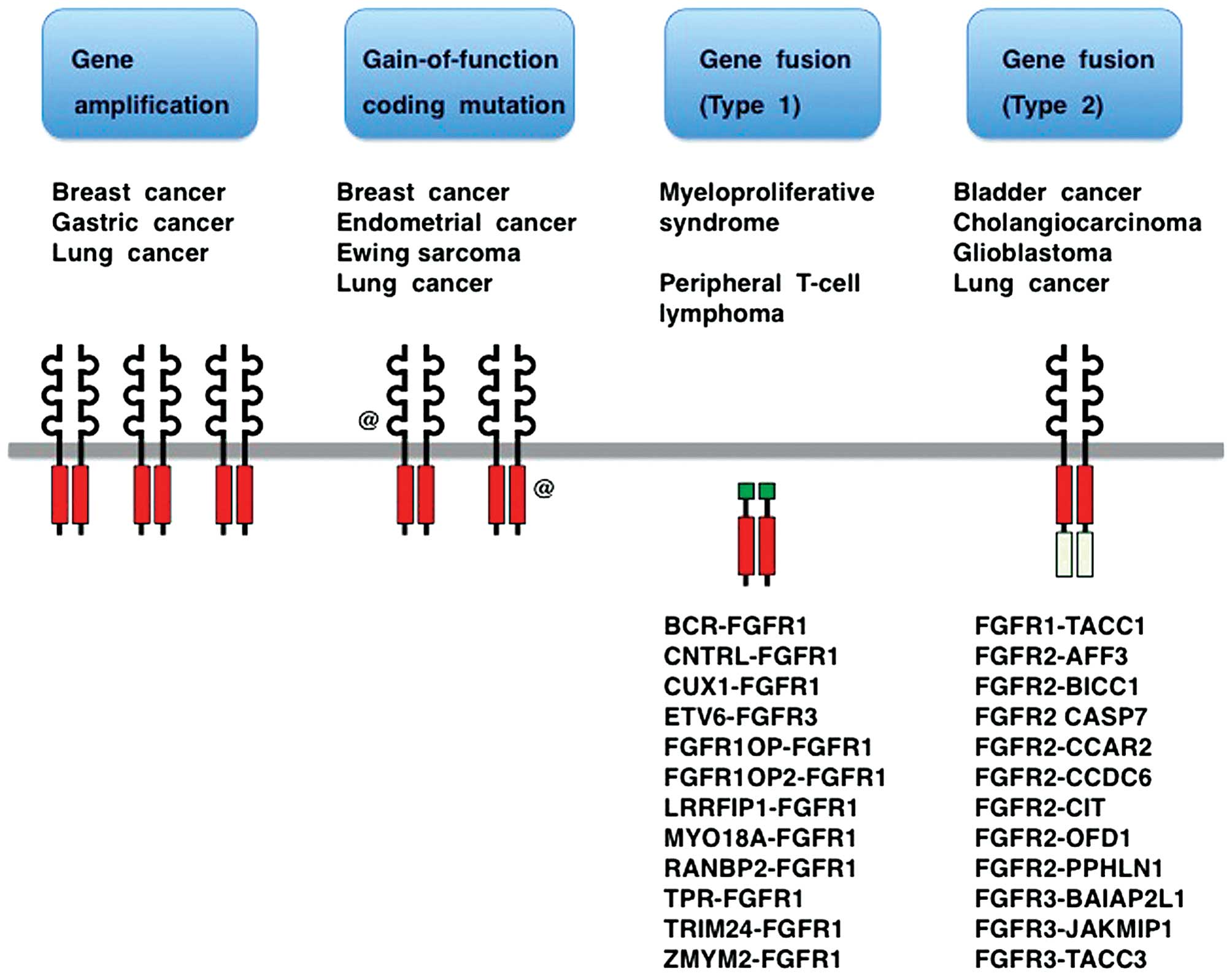
Gene amplification, gain-of-function coding mutation
and gene fusion are three major classes of FGFR alterations
in human cancer (Fig. 3)
(14,21–24). Clinical trials of several tyrosine
kinase inhibitors (TKIs) targeting FGFRs are ongoing (25–28), while TKI resistance and
tumor-stromal interaction related to FGFRs are hot issues (29–32). Knowledge of FGFRs has been
exponentially growing as a result of the advancement of massively
parallel sequencing technology combined with the global trend
toward translational medicine. In this review, recent progress in
the field of FGFR medicine is reviewed with emphases on FGFR
alterations in human cancer, the classification of small-molecule
FGFR inhibitors and the effects of FGFR inhibitors on the tumor
microenvironment and whole-body homeostasis.
Gastric cancer is the fifth most common malignancy
worldwide, although its incidence and mortality have been
decreasing (33,46). The amplifications of genes
encoding RTKs, such as EGFR, ERBB2, FGFR2 and
MET, occur in gastric cancer (47,48). Gastric cancer with FGFR2
amplification is significantly associated with lymphatic invasion
and a poor prognosis (49,50);
however, the molecular mechanisms through which FGFR2
amplification promotes lymph node metastasis remain unclear.
Preclinical studies using small-molecule FGFR2 inhibitor and
patient-derived cancer xenografts revealed that FGFR2
amplification in human gastric cancer is a promising therapeutic
target (51,52).
Missense mutations of FGFRs involved in congenital
disorders rather than cancer have been well characterized. Gremlin
FGFR1 mutations (P252R and Y372C) and FGFR2 mutations (S252W,
P253R, K526E, N549K, K641R, etc.) in patients with craniosynostosis
and FGFR3 mutations (R248C, S249C, G380R, N540K, K650E, etc.) in
patients with skeletal dysplasia are gain-of-function mutations
(17–20); however, germline FGFR1 mutations
(R254W and V429E) in patients with congenital hypogonadotropic
hypogonadism are loss-of-function mutations (58,59).
These facts clearly indicate that there are
gain-of-function, as well as loss-of-function mutations in FGFRs.
Therefore, validation of gain-of-function based on kinase or
cell-based assay is mandatory before prescribing FGFR inhibitors to
cancer patients with FGFR coding mutations.
Both types of FGFR fusion proteins are endowed with
oncogenic potential through the acquisition of
protein-protein-interaction modules from fusion partners for
ligand-independent dimerization and/or recruitment of aberrant
substrates. Type 1 FGFR fusion proteins acquire oncogenic potential
through altered subcellular localization as a result of the loss of
the extracellular and transmembrane domains of wild-type FGFRs.
Type 2 FGFR fusion proteins lose the PLC-γ-binding tyrosine (Tyr or
Y) residue (Y766 in FGFR1, Y769 in FGFR2 or Y760 in FGFR3) owing to
the C-terminal alterations. To understand the mechanisms of
carcinogenesis caused by the FGFR fusions, substrates and
downstream signaling cascades of FGFR fusion proteins need be
elucidated.
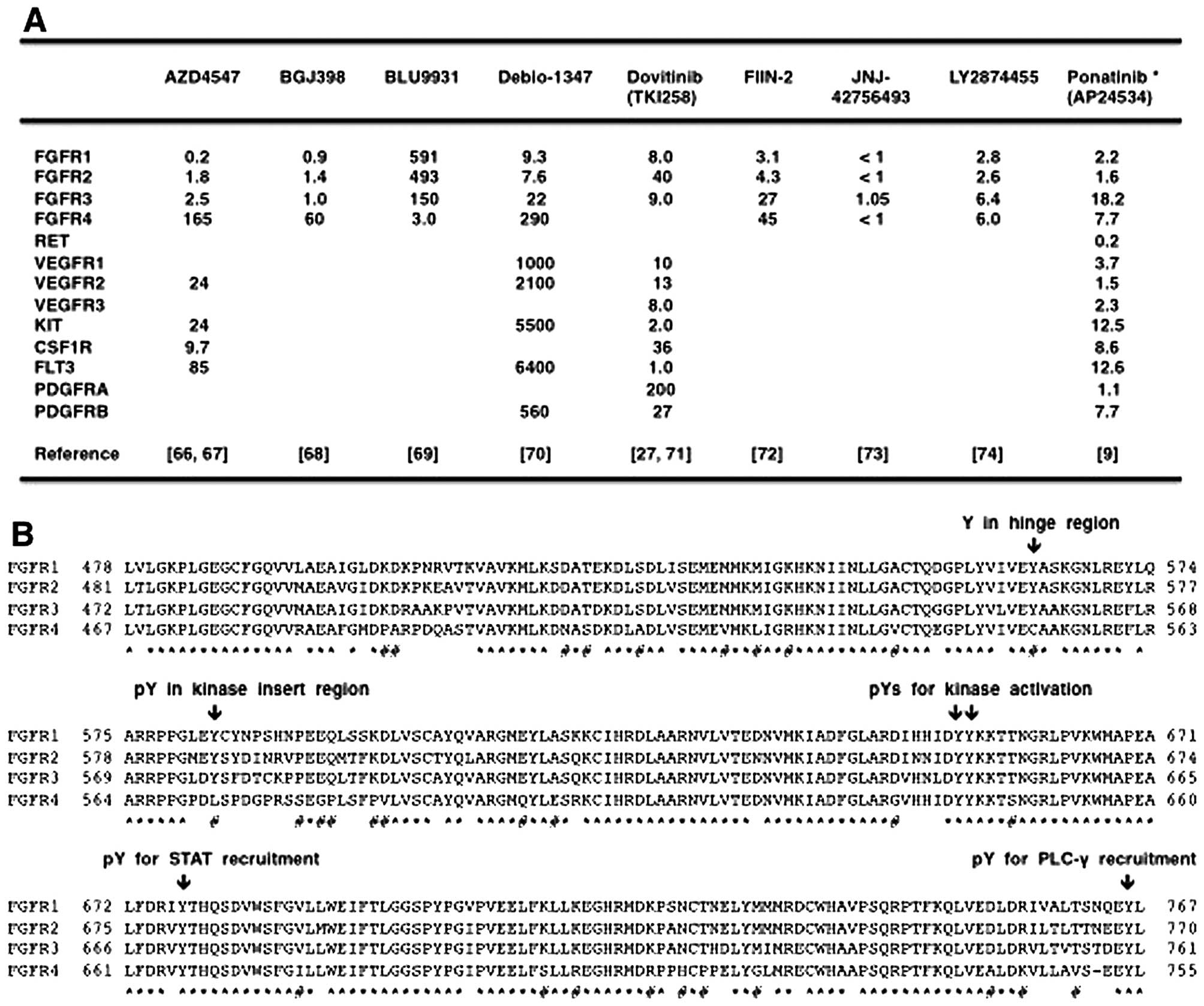
AZD4547, BGJ398, Debio-1347 and dovitinib are
FGFR1/2/3 inhibitors that are less effective on FGFR4; BLU9931 is a
selective FGFR4 inhibitor; FIIN-2, JNJ-42756493, LY2874455 and
ponatinib are pan-FGFR inhibitors (Fig. 5A). Phylogenetic analysis on 54
RTKs revealed diversification of FGFR4 from other FGFRs (Fig. 2), and amino-acid alignment of the
tyrosine kinase domains in the FGFR family members revealed
relatively frequent amino-acid substitutions specifically in FGFR4
(Fig. 4B). Phospho-tyrosine
residues involved in catalytic activation (Y653 and Y654 in FGFR1),
STAT recruitment (Y677 in FGFR1) and PLC-γ recruitment (Y766 in
FGFR1) are conserved in all members of the FGFR family. By
contrast, one tyrosine residue in the hinge region (Y563 in FGFR1,
Y566 in FGFR2 and Y557 in FGFR3) is changed to C552 in FGFR4, and
phospho-tyrosine residue in the kinase insert region (Y583 in
FGFR1, Y586 in FGFR2 and Y577 in FGFR3) is changed to L572 in FGFR4
(Fig. 4B). Y563 in FGFR1 is
necessary for the interaction with Debio-1347 (70), whereas C552 in FGFR4 is necessary
for the covalent binding with BLU9931 (69). As the diversification of FGFR4
significantly affects the biding affinities of TKIs, it is
reasonable to functionally classify FGFR-targeting TKIs into
FGFR1/2/3 inhibitors, FGFR4 inhibitor and pan-FGFR inhibitors.
AZD4547, BGJ398, Debio-1347, dovitinib, JNJ-42756493
and ponatinib are currently being investigated in clinical trials
(https://clinicaltrials.gov): phase II
studies of AZD4547 in patients with breast, gastric and
squamous-cell lung cancer (FGFR1 or FGFR2
amplification) and metastatic breast or non-small-cell lung cancer
(FGFR genetic alterations, umbrella trial); phase II studies
of BGJ398 in patients with solid tumors or hematological
malignancies (FGFR genetic alterations); phase II study of
dovitinib in patients with gastric cancer (FGFR2
amplification); phase II study of JNJ-42756493 in patients with
urothelial cancer (FGFR genetic alterations); phase II study
of ponatinib in patients with advanced biliary cancer (FGFR2
fusion) or refractory metastatic solid tumors (genetic alterations
in FGFRs and other targets).
TKIs have been approved for cancer therapy by
regulatory authorities in expectation of an improved risk/benefit
ratio; however, adverse effects on viral organs, such as the
cardiovascular system and liver, are serious issues that may occur
in the clinic (75).
Hypertension, bleeding and thrombosis are adverse effects of
anti-angiogenic therapy targeting the VEGF signaling pathway
(76), while cardiovascular
events are serious adverse effects of ponatinib for the treatment
of chronic myeloid leukemia (77). AZD4547, dovitinib and ponatinib
are representative multi-kinase inhibitors targeting FGFRs and
other tyrosine kinases (Fig. 5A).
Selective FGFR targeting is expected to reduce adverse effects,
whereas the dual targeting of FGFR and VEGFR/CSF1R is expected to
enhance the anti-tumor effects indirectly through the normalization
of tumor microenvironment.
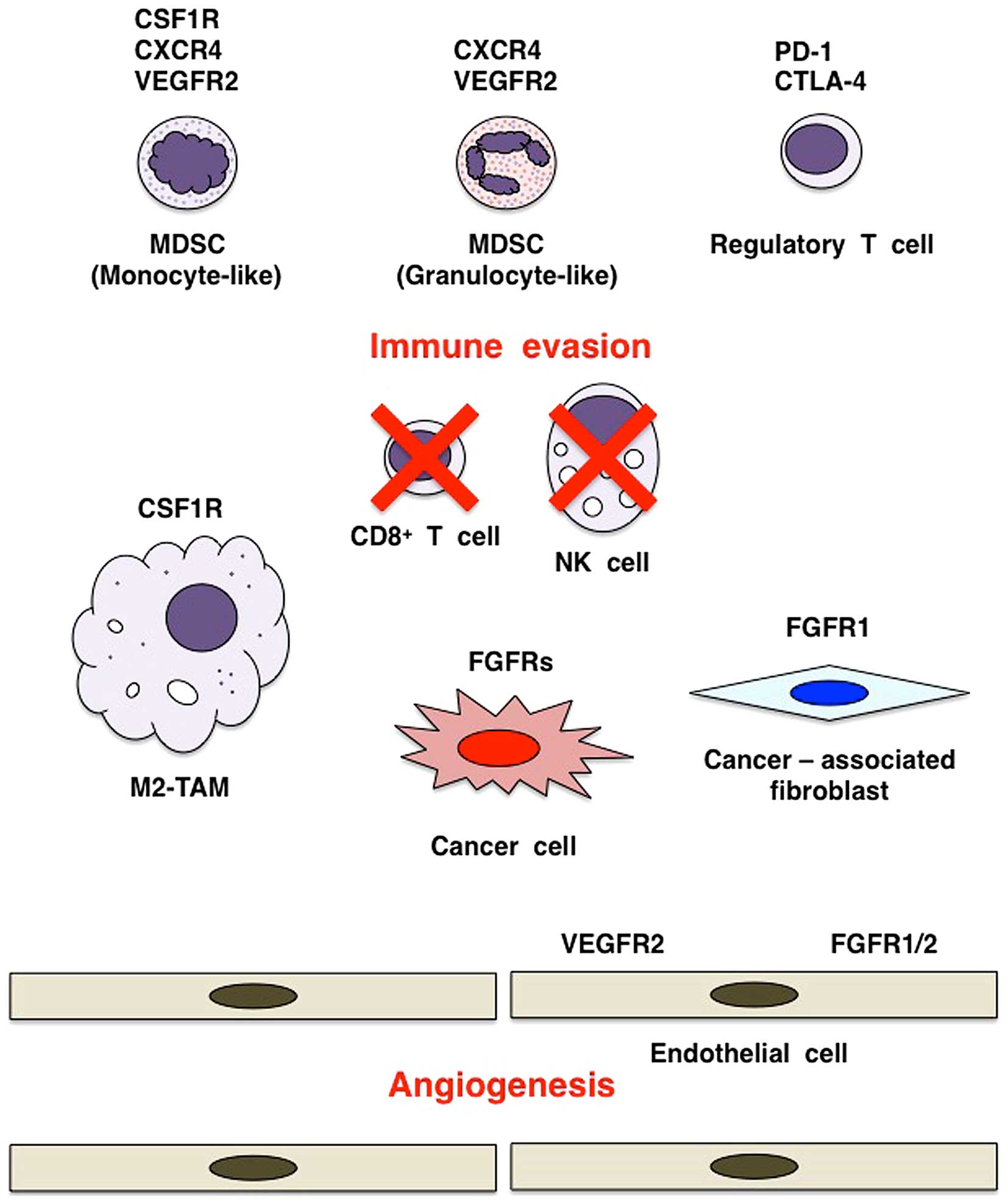
The tumor microenvironment consists of cancer cells
and stromal/immune cells, such as fibroblasts, endothelial cells,
lymphocytes, macrophages, monocytes and neutrophils (Fig. 6). The interactions of tumor cells
and stromal cells are involved in almost all stages of tumor
development, including neoplastic transformation, proliferation,
invasion and metastasis, through the regulation of various cellular
processes in a context-dependent manner (78–80). FGFs derived from cancer cells, as
well as stromal cells play a key role in the tumor
microenvironment.
Cancer-associated fibroblasts (CAFs) are activated
stromal fibroblasts that support tumorigenesis (Fig. 6). FGF2 activates human dermal
fibroblasts through transcriptional downregulation of the
TP53 gene, whereas BGJ398 or ponatinib treatment induces
their senescence through the upregulation and activation of TP53
(81). By contrast, FGF2
signaling through FGFR1 causes resistance to EGFR inhibitor in lung
cancer cells, and combination therapy using EGFR inhibitor and
AD4547 is effective to overcome drug resistance (82). Multiple myeloma cells induce FGF23
secretion from osteocytes, and then FGF23 signaling through FGFR3
to multiple myeloma cells promotes proliferation and induces
heparanase upregulation, which explains the pathogenesis of
osteolytic 'punched-out lesion' in patients with multiple myeloma.
BGJ398 treatment inhibits FGF23-dependent growth and heparanase
expression of multiple myeloma cells (83). These results indicate the rational
for the application of FGFR inhibitors to target paracrine FGF
signaling in the tumor microenvironment.
Tumor angiogenesis is largely classified into
sprouting angiogenesis and vasculogenesis (84). Sprouting angiogenesis is the
formation of new blood vessels as a result of endothelial sprouting
from preexisting blood vessels, whereas vasculogenesis is the de
novo formation of blood vessels owing to endothelial
differentiation of progenitor cells and endothelial-like
differentiation of cancer cells. Tumor angiogenesis is involved in
the supply of oxygen and nutrient (mature blood vessels), as well
as the formation of hypoxic environment (immature blood
vessels).
VEGF, FGF, angiopoietin (ANGPT) and Notch signaling
cascades are major players of tumor angiogenesis (85,86). VEGF signaling through VEGFR2
promotes endothelial cell proliferation via the DAG-PKC signaling
cascade, endothelial cell survival via the PI3K-AKT signaling
cascade, endothelial cell migration via the FAK-Paxillin signaling
cascade as well as vascular permeability and vasodilatation via the
IP3-eNOS (NOS3) signaling cascade (76,87,88). Pro-angiogenic FGF2 also promotes
the proliferation and migration of endothelial cells directly
through FGFR1 (or FGFR2) signaling activation (89,90) and indirectly through the
induction/secretion of VEGF and ANGPT2 from endothelial cells
(91,92). ANGPT1 is secreted from pericytes
and maintains endothelial quiescence or stabilization through TIE2
signaling activation. ANGPT2 is secreted from endothelial cells and
promotes the endothelial activation or sprouting through TIE2
signaling inhibition (92,93).
VEGF signaling in endothelial tip cells induces DLL4 expression,
which subsequently activates Notch signaling in endothelial stalk
cells for vascular quiescence through VEGFR downregulation
(94–96). VEGF, FGF2 and ANGPT2 are involved
in endothelial activation, whereas ANGPT1 and Notch are involved in
endothelial quiescence. VEGFR2 and FGFR1/2 on endothelial cells are
representative RTKs that promote tumor angiogenesis (Fig. 6).
VEGF signaling is targeted using anti-VEGF
monoclonal antibody (mAb) or small-molecule VEGFR inhibitors in
cancer patients; however, some tumors do not respond to the VEGF
blockade therapy and other tumors recur after transient response
[(Gacche and Meshram (76); Jain
(78)]. As FGF signaling
activation in endothelial cells is one of the mechanisms
responsible for intrinsic and acquired resistance to the VEGF
blockade therapy (97), FGFR
inhibitors may be applicable to overcome the resistance to the VEGF
blockade therapy. There are two options for the dual blockade of
FGF and VEGF signaling cascades. Combination therapy using FGFR
inhibitor and anti-VEGF mAb is a preferable choice to reduce
adverse effects, whereas monotherapy using small-molecule
FGFR/VEGFR2 dual inhibitors, such as AZD4547 and dovitinob, may be
a preferable choice to reduce medical cost. FGF/VEGF dual blockade
therapy should be optimized in consideration of safety issues and
medical costs.
Cancer immunity and immune tolerance in the tumor
microenvironment are regulated by the interaction between cancer
cells and immune cells (98).
CD8+ T cells, NK cells and NKT cells are immune effector
cells involved in tumor elimination (98), whereas myeloid-derived suppressor
cells (MDSCs) (99),
tumor-associating macrophages of M2 type (M2-TAMs) (100) and regulatory T (Treg) cells
(101) are immune modifier cells
involved in immune evasion and tumor growth (Fig. 6).
MDSCs are heterogeneous populations of immature
myeloid cells, including monocyte-like MDSCs (CD14+,
CXCR4+, CSF1R+ and VEGFR2+),
granulocyte-like MDSCs (CD15+, CXCR4+,
KIT+ and VEGFR2+) and endothelial progenitor
cells (CD31+, CXCR4+, KIT+ and
VEGFR2+) (99). CSF1
(M-CSF), CSF2 (GM-CSF) and CSF3 (G-CSF) are secreted from the tumor
microenvironment and stimulate the growth and survival of MDSCs and
other myeloid-lineage cells, while CXCL12 (SFD-1α) and VEGF promote
the recruitment of MDSCs to the tumor microenvironment (102–106). MDSCs activate M2-TAMs and Treg
cells, but inhibit CD8+ T cells and NK cells, leading to
immune evasion in the tumor microenvironment. In addition,
endothelial progenitor cell-like MDSCs are involved in tumor
angiogenesis (104). MDSC
infiltration and tumor angiogenesis during mammary tumorigenesis in
MMTV-Wnt1/iFGFR1 bi-genic mice are significantly enhanced in
comparison with MMTV-Wnt1 transgenic mice, and BGJ398 treatment
results in tumor regression and disappearance of MDSCs from the
residual mammary gland (107).
By contrast, AZD4547 treatment inhibits the proliferation and lung
metastasis of 4T1 mouse mammary tumor cells, and reduces MDSCs in
the tumor microenvironment and systemic circulation (108). FGFR inhibitors induce the
reduction or disappearance of MDSCs from the tumor
microenvironment, partly by targeting cytokine-producing CAFs.
CSF1 signaling through CSF1R on
monocyte/macrophage-lineage cells are involved in their
proliferation, survival and differentiation (109–111). CSF1R inhibitors (GW2580 and
PLX3397) and anti-CSF1R mAb (RG7155) have been developed as
therapeutics for CSF1 signaling blockade in monocyte-like MDSCs and
M2-TAM (112–115). Combination therapy of CSF1R
inhibitor PLX3397 and paclitaxel inhibits tumor-infiltration of
MDCSs and M2-TAM and suppresses mammary tumorigenesis (113,114). IC50 value of PLX3397
to CSF1R is 20 nM (113),
whereas IC50 values of AZD4547 (67), ponatinib (9) and dovitinib (71) to CSF1R are 9.7, 8.7 and 36 nM,
respectively. By contrast, PI3K is one of common signaling
effectors CSF1R and FGFRs (Fig.
1A), and PI3K activation enhances immune suppressor and
pro-angiogenic potentials of M2-TAMs (116). Therefore, as CSF1 and FGF
signals are both involved in the accumulation of
tumor-infiltrating/promoting MDSCs and M2-TAMs, the dual inhibition
of CSF1R and FGFRs may be more effective for cancer therapy than
selective CSF1R inhibition.
FGF19, FGF21 and FGF23 are endocrine FGFs that
transduce signals to target organs through FGFRs and the Klotho
family of co-receptors, α-Klotho (KL) and β-Klotho (KLB) (117). FGF19 is upregulated by bile acid
in the intestine to transduce endocrine signaling through FGFR4 and
β-Klotho in the liver. FGF21 is upregulated by fasting in the liver
and adipose tissue to transduce paracrine signaling through FGFR1
and β-Klotho locally and endocrine signaling to the pancreas and
brain. FGF23 is upregulated by serum phosphate, vitamin D and
parathyroid hormone in bone to transduce endocrine signaling
through FGFR1 and α-Klotho in the kidneys and negative feedback
signaling through FGFR3 and α-Klotho in the parathyroid gland
(118). As endocrine FGFs are
involved in the maintenance of whole-body homeostasis, FGFR
inhibitors elicit endocrine or metabolic abnormalities. This
section will be focused on adverse effects of FGFR inhibitors on
endocrine FGF signaling in cancer patients.
FGF19 signaling through FGFR4 in the liver
stimulates hepatocyte proliferation and glycogen synthesis but
reduces bile acid synthesis and triglyceride synthesis (69,119). FGF19-FGFR4 signaling blockade in
cynomolgus monkeys using anti-FGF19 monoclonal antibody causes
hepatotoxicity, increased bile acid secretion and severe diarrhea
(120). Fgfr4 knockout in
mice also causes increased bile acid secretion in the liver, which
leads to induction of Fgf15 (mouse ortholog of human FGF19) in the
intestine and subsequent improvement of insulin resistance and
glucose metabolism (121). The
selective FGFR4 inhibitor, BLU9931, may be applied for the
treatment of patients with hepatocellular carcinoma depending on
FGF19-FGFR4 signaling. By contrast, as FGFR4 blockade is associated
with a risk of liver toxicity, FGFR1/2/3 inhibitors rather than
pan-FGFR inhibitors are preferable for the treatment of cancer
patients with genetic alterations in FGFR1, FGFR2 or
FGFR3, especially those with liver dysfunction (Fig. 5B).
Serum FGF23 elevation is a biomarker indicating
on-target effects of FGFR1/2/3 and pan-FGFR inhibitors in cancer
patients, whereas serum FGF23 levels are also elevated in patients
with non-cancerous diseases, such as hypophosphatemic rickets and
chronic kidney diseases (118).
Physiological FGF23 signaling through FGFR1 and α-Klotho in the
kidneys decreases the serum phosphate level through the
downregulation of phosphate reabsorption. FGFR inhibitors,
hindering FGF23 signaling in the kidneys, promote hyperphosphatemia
and subsequent FGF23 secretion from bone and soft-tissue
mineralization. Pathological FGF23 upregulation is associated with
endothelial dysfunction and arterial stiffness (122). Pathological FGF23 signaling
through FGFR4 in cardiac myocytes then induces phosphorylation of
PLC-γ and activation of the IP3-Ca2+
signaling cascade, which results in cardiac remodeling, such as
cardiac hypertrophy and cardiac fibrosis (123). As FGF23-FGFR4 signaling
activation is associated with a risk of cardiac toxicity, pan-FGFR
inhibitors rather than FGFR1/2/3 inhibitors may be selected for the
treatment of cancer patients with FGFR genetic alterations,
particularly those with heart dysfunction (Fig. 5B).
Massively parallel sequencing technology for the
whole-exome or whole-genome sequencing has been used to clarify
genomic landscapes in various types of human cancer (124). The over-expression of FGFR
occurs in human cancers through gene amplification, as well as
other types of aberrations. Rearrangement in the distal enhancer
region and point mutation in the proximal promoter region are both
able to induce FGFR overexpression. Repression of FGFR-targeting
microRNA (miRNA) precursor gene or upregulation of long-non-coding
RNA (lncRNA) sequestering FGFR-targeting miRNA leads to FGFR
overexpression. SWI/SNF mutation dysregulating chromatin
remodeling, as well as cancer-associated fusion transcription
factor also cause FGFR overexpression. For example, lung cancer
cells with FGFR1 upregulation rather than FGFR1 copy number
gain are sensitive to ponatinib (125). Rhabdoid tumor cells with FGFR
overexpression as a result of SMARCB1 (SNF5) deletion
are sensitive to BGJ398 (126).
Myxoid liposarcoma cells with FGFR2 upregulation owing to FUS-DDIT3
or EWS1R-DDIT3 fusion are sensitive to BGJ398 and dovitinib
(127). Taken together, these
facts clearly indicate that FGFR inhibitors are applicable for the
treatment of cancers with FGFR overexpression in the absence of
gene amplification, particularly rare cancers with their specific
alterations inducing FGFR overexpression. However, development of
biomarkers for FGF dependence is necessary before clinical
application of FGFR inhibitors for the treatment of cancers with
FGFR overexpression.
Genomic heterogeneity is the major mechanism of
tumor evolution for recurrence after cancer therapy (128). FGFR2-BICC1 and
FGFR2-PPHLN1 fusions in mostly distinct, but some
overlapping cases of cholangiocarcinomas (65) suggest convergent evolution and
intra-tumor heterogeneity, respectively. Resistance to
EGFR-targeted therapy occurs based on paracrine FGF signaling from
tumor-stromal cells (82) or
activating FGFR alterations, such as FGFR1
amplification and FGFR3 mutation, in cancer cells (129,130). On the other hand, resistance to
FGFR-targeted therapy occurs based on paracrine signaling through
EGFR/HER2/MET (29,107) or secondary FGFR
alterations (131). FGFR1 V561M
(34), FGFR2 V564M (72), FGFR3 V555M (131) and FGFR4 V550M (132) are gatekeeper mutations that
cause resistance to ATP-competitive FGFR inhibitors, such as
AZD4547 and BGJ398, whereas FGFR2 V564M is sensitive to a covalent
pan-FGFR inhibitor, FIIN-2 (72).
Because antitumor effects of FIIN-2 are limited to cell-based
assays, orally bioavailable derivatives of FIIN-2 should be
developed for the treatment of cancer resistant to ATP-competitive
FGFR inhibitors.
Immune-checkpoint blockade therapy is a frontier in
the field of clinical oncology. PD-1 ligand (PD-L1) is expressed on
cancer cells and stromal/immune cells, whereas PD-1 and CTLA-4 are
expressed on CD8+ T cells and Treg cells (133–136). As PD-1 signaling and CTLA-4
signaling are both involved in functional suppression of cytotoxic
T cells directly or indirectly through Treg cells, anti-PD-1 mAb,
anti-PD-L1 mAb and anti-CTLA-4 mAb are clinically applied for
cancer immunotherapy, which leads to sustainable remission in a
fraction of patients. By contrast, FGFR and CSF1R inhibitors are
shown to target immune cells, such as MDSCs and M2-TAMs, in the
tumor microenvironment (Fig. 6)
and are expected to indirectly repress PD-L1 expression on tumor
cells and stromal/immune cells through normalization of tumor
microenvironment. Therefore, combination therapy using TKI (FGFR or
CSF1R inhibitor) and immune checkpoint blocker (anti-PD-1 or
anti-CTLA-4 mAb) may be a promising choice for cancer patients.
Cancer patients are prescribed appropriate drug
based on their genetic alterations to reduce the costs of diagnosis
and to increase the amounts of knowledge (Fig. 7). Partial-exome sequencing of a
panel of cancer-associated genes are utilized for therapeutic
optimization of cancer patients in the field of clinical oncology,
which is relatively inexpensive but unable to detect cis-acting
enhancer/promoter alterations and transacting rare coding
alterations. By contrast, integrative genomic analyses based on
whole-genome sequencing are utilized for precise characterization
of human cancers in the field of basic oncology, which is expensive
but comprehensive and informative. As a benefit-cost ratio is a
critical issue to sustain health care system of aging society, it
is necessary to discuss the benefit-cost issue with a focus on
disease-free survival and total medical cost before implementation
of genome-based precision medicine for cancer patients.
This study was financially supported in part by a
Grant-in-Aid for the Knowledgebase Project from the M. Katoh's
Fund.
|
1
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Roskoski R Jr: The ErbB/HER family of
protein-tyrosine kinases and cancer. Pharmacol Res. 79:34–74. 2014.
View Article : Google Scholar
|
|
4
|
Rugo HS, Herbst RS, Liu G, Park JW, Kies
MS, Steinfeldt HM, Pithavala YK, Reich SD, Freddo JL and Wilding G:
Phase I trial of the oral antiangiogenesis agent AG-013736 in
patients with advanced solid tumors: Pharmacokinetic and clinical
results. J Clin Oncol. 23:5474–5483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi
Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al: Cabozantinib
(XL184), a novel MET and VEGFR2 inhibitor, simultaneously
suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer
Ther. 10:2298–2308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marsilje TH, Pei W, Chen B, Lu W, Uno T,
Jin Y, Jiang T, Kim S, Li N, Warmuth M, et al: Synthesis,
structure-activity relationships, and in vivo efficacy of the novel
potent and selective anaplastic lymphoma kinase (ALK) inhibitor
5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)
pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2
clinical trials. J Med Chem. 56:5675–5690. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung
PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, et al: Structure
based drug design of crizotinib (PF-02341066), a potent and
selective dual inhibitor of mesenchymal-epithelial transition
factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med
Chem. 54:6342–6363. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tohyama O, Matsui J, Kodama K, Hata-Sugi
N, Kimura T, Okamoto K, Minoshima Y, Iwata M and Funahashi Y:
Antitumor activity of lenvatinib (e7080): An angiogenesis inhibitor
that targets multiple receptor tyrosine kinases in preclinical
human thyroid cancer models. J Thyroid Res. 2014:6387472014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
O'Hare T, Shakespeare WC, Zhu X, Eide CA,
Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al:
AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia,
potently inhibits the T315I mutant and overcomes mutation-based
resistance. Cancer Cell. 16:401–412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wilhelm SM, Dumas J, Adnane L, Lynch M,
Carter CA, Schütz G, Thierauch KH and Zopf D: Regorafenib (BAY
73-4506): A new oral multikinase inhibitor of angiogenic, stromal
and oncogenic receptor tyrosine kinases with potent preclinical
antitumor activity. Int J Cancer. 129:245–255. 2011. View Article : Google Scholar
|
|
11
|
Chow LQ and Eckhardt SG: Sunitinib: From
rational design to clinical efficacy. J Clin Oncol. 25:884–896.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wells SA Jr, Gosnell JE, Gagel RF, Moley
J, Pfister D, Sosa JA, Skinner M, Krebs A, Vasselli J and
Schlumberger M: Vandetanib for the treatment of patients with
locally advanced or metastatic hereditary medullary thyroid cancer.
J Clin Oncol. 28:767–772. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eswarakumar VP, Lax I and Schlessinger J:
Cellular signaling by fibroblast growth factor receptors. Cytokine
Growth Factor Rev. 16:139–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Katoh M and Nakagama H: FGF receptors:
Cancer biology and therapeutics. Med Res Rev. 34:280–300. 2014.
View Article : Google Scholar
|
|
15
|
Coleman SJ, Bruce C, Chioni AM, Kocher HM
and Grose RP: The ins and outs of fibroblast growth factor receptor
signalling. Clin Sci (Lond). 127:217–231. 2014. View Article : Google Scholar
|
|
16
|
Ornitz DM and Itoh N: The fibroblast
growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol.
4:215–266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Katoh M: FGFR2 abnormalities underlie a
spectrum of bone, skin, and cancer pathologies. J Invest Dermatol.
129:1861–1867. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kelleher FC, O'Sullivan H, Smyth E,
McDermott R and Viterbo A: Fibroblast growth factor receptors,
developmental corruption and malignant disease. Carcinogenesis.
34:2198–2205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Helsten T, Schwaederle M and Kurzrock R:
Fibroblast growth factor receptor signaling in hereditary and
neoplastic disease: Biologic and clinical implications. Cancer
Metastasis Rev. 34:479–496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brooks AN, Kilgour E and Smith PD:
Molecular pathways: fibroblast growth factor signaling: a new
therapeutic opportunity in cancer. Clin Cancer Res. 18:1855–1862.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang J, Liu X, Wang S, Zhang Z, Wu Z,
Zhang X and Li J: Prognostic value of FGFR gene amplification in
patients with different types of cancer: A systematic review and
meta-analysis. PLoS One. 9:e1055242014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Parker BC and Zhang W: Fusion genes in
solid tumors: An emerging target for cancer diagnosis and
treatment. Chin J Cancer. 32:594–603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Feng S, Zhou L, Nice EC and Huang C:
Fibroblast growth factor receptors: Multifactorial-contributors to
tumor initiation and progression. Histol Histopathol. 30:13–31.
2015.
|
|
25
|
Liang G, Chen G, Wei X, Zhao Y and Li X:
Small molecule inhibition of fibroblast growth factor receptors in
cancer. Cytokine Growth Factor Rev. 24:467–475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
André F and Cortés J: Rationale for
targeting fibroblast growth factor receptor signaling in breast
cancer. Breast Cancer Res Treat. 150:1–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Porta C, Giglione P, Liguigli W and
Paglino C: Dovitinib (CHIR258, TKI258): Structure, development and
preclinical and clinical activity. Future Oncol. 11:39–50. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carter EP, Fearon AE and Grose RP:
Careless talk costs lives: Fibroblast growth factor receptor
signalling and the consequences of pathway malfunction. Trends Cell
Biol. 25:221–233. 2015. View Article : Google Scholar
|
|
29
|
Chang J, Wang S, Zhang Z, Liu X, Wu Z,
Geng R, Ge X, Dai C, Liu R, Zhang Q, et al: Multiple receptor
tyrosine kinase activation attenuates therapeutic efficacy of the
fibroblast growth factor receptor 2 inhibitor AZD4547 in FGFR2
amplified gastric cancer. Oncotarget. 6:2009–2022. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang J, Mikse O, Liao RG, Li Y, Tan L,
Janne PA, Gray NS, Wong KK and Hammerman PS: Ligand-associated
ERBB2/3 activation confers acquired resistance to FGFR inhibition
in FGFR3-dependent cancer cells. Oncogene. 34:2167–2177. 2015.
View Article : Google Scholar
|
|
31
|
Ronca R, Giacomini A, Rusnati M and Presta
M: The potential of fibroblast growth factor/fibroblast growth
factor receptor signaling as a therapeutic target in tumor
angiogenesis. Expert. Opin Ther Targets. 19:1361–1377. 2015.
View Article : Google Scholar
|
|
32
|
Salazar L, Kashiwada T, Krejci P, Meyer
AN, Casale M, Hallowell M, Wilcox WR, Donoghue DJ and Thompson LM:
Fibroblast growth factor receptor 3 interacts with and activates
TGFβ-activated kinase 1 tyrosine phosphorylation and NFκB signaling
in multiple myeloma and bladder cancer. PLoS One. 9:e864702014.
View Article : Google Scholar
|
|
33
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weiss J, Sos ML, Seidel D, Peifer M,
Zander T, Heuckmann JM, Ullrich RT, Menon R, Maier S, Soltermann A,
et al: Frequent and focal FGFR1 amplification associates with
therapeutically tractable FGFR1 dependency in squamous cell lung
cancer. Sci Transl Med. 2:62ra932010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cihoric N, Savic S, Schneider S, Ackermann
I, Bichsel-Naef M, Schmid RA, Lardinois D, Gugger M, Bubendorf L,
Zlobec I, et al: Prognostic role of FGFR1 amplification in
early-stage non-small cell lung cancer. Br J Cancer. 110:2914–2922.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Preusser M, Berghoff AS, Berger W,
Ilhan-Mutlu A, Dinhof C, Widhalm G, Dieckmann K, Wöhrer A, Hackl M,
von Deimling A, et al: High rate of FGFR1 amplifications in brain
metastases of squamous and non-squamous lung cancer. Lung Cancer.
83:83–89. 2014. View Article : Google Scholar
|
|
37
|
Seo JS, Ju YS, Lee WC, Shin JY, Lee JK,
Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, et al: The
transcriptional landscape and mutational profile of lung
adenocarcinoma. Genome Res. 22:2109–2119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu YM, Su F, Kalyana-Sundaram S, Khazanov
N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin SF, et al:
Identification of targetable FGFR gene fusions in diverse cancers.
Cancer Discov. 3:636–647. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tanizaki J, Ercan D, Capelletti M, Dodge
M, Xu C, Bahcall M, Tricker EM, Butaney M, Calles A, Sholl LM, et
al: Identification of oncogenic and drug-sensitizing mutations in
the extracellular domain of FGFR2. Cancer Res. 75:3139–3146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Maxmen A: The hard facts. Nature.
485:S50–S51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ellis MJ and Perou CM: The genomic
landscape of breast cancer as a therapeutic roadmap. Cancer Discov.
3:27–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Khoo BL, Lee SC, Kumar P, Tan TZ, Warkiani
ME, Ow SG, Nandi S, Lim CT and Thiery JP: Short-term expansion of
breast circulating cancer cells predicts response to anti-cancer
therapy. Oncotarget. 6:15578–15593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yu M, Bardia A, Aceto N, Bersani F, Madden
MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, et al:
Cancer therapy. Ex vivo culture of circulating breast tumor cells
for individualized testing of drug susceptibility. Science.
345:216–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
André F, Bachelot T, Commo F, Campone M,
Arnedos M, Dieras V, Lacroix-Triki M, Lacroix L, Cohen P, Gentien
D, et al: Comparative genomic hybridisation array and DNA
sequencing to direct treatment of metastatic breast cancer: A
multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol.
15:267–274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Turner N, Pearson A, Sharpe R, Lambros M,
Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A,
et al: FGFR1 amplification drives endocrine therapy resistance and
is a therapeutic target in breast cancer. Cancer Res. 70:2085–2094.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ferro A, Peleteiro B, Malvezzi M, Bosetti
C, Bertuccio P, Levi F, Negri E, La Vecchia C and Lunet N:
Worldwide trends in gastric cancer mortality 1980–2011 with
predictions to 2015, and incidence by subtype. Eur J Cancer.
50:1330–1344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Holbrook JD, Parker JS, Gallagher KT,
Halsey WS, Hughes AM, Weigman VJ, Lebowitz PF and Kumar R: Deep
sequencing of gastric carcinoma reveals somatic mutations relevant
to personalized medicine. J Transl Med. 9:1192011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Deng N, Goh LK, Wang H, Das K, Tao J, Tan
IB, Zhang S, Lee M, Wu J, Lim KH, et al: A comprehensive survey of
genomic alterations in gastric cancer reveals systematic patterns
of molecular exclusivity and co-occurrence among distinct
therapeutic targets. Gut. 61:673–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jung EJ, Jung EJ, Min SY, Kim MA and Kim
WH: Fibroblast growth factor receptor 2 gene amplification status
and its clinicopathologic significance in gastric carcinoma. Hum
Pathol. 43:1559–1566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Su X, Zhan P, Gavine PR, Morgan S, Womack
C, Ni X, Shen D, Bang YJ, Im SA, Ho Kim W, et al: FGFR2
amplification has prognostic significance in gastric cancer:
Results from a large international multicentre study. Br J Cancer.
110:967–975. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xie L, Su X, Zhang L, Yin X, Tang L, Zhang
X, Xu Y, Gao Z, Liu K, Zhou M, et al: FGFR2 gene amplification in
gastric cancer predicts sensitivity to the selective FGFR inhibitor
AZD4547. Clin Cancer Res. 19:2572–2583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang T, Zhang L, Fan S, Zhang M, Fu H,
Liu Y, Yin X, Chen H, Xie L, Zhang J, et al: Patient-derived
gastric carcinoma xenograft mouse models faithfully represent human
tumor molecular diversity. PLoS One. 10:e01344932015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Agelopoulos K, Richter GH, Schmidt E,
Dirksen U, von Heyking K, Moser B, Klein HU, Kontny U, Dugas M,
Poos K, et al: Deep sequencing in conjunction with expression and
functional analyses reveals activation of FGFR1 in Ewing sarcoma.
Clin Cancer Res. 21:4935–4946. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Reintjes N, Li Y, Becker A, Rohmann E,
Schmutzler R and Wollnik B: Activating somatic FGFR2 mutations in
breast cancer. PLoS One. 8:e602642013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Byron SA, Gartside M, Powell MA, Wellens
CL, Gao F, Mutch DG, Goodfellow PJ and Pollock PM: FGFR2 point
mutations in 466 endometrioid endometrial tumors: Relationship with
MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological
features. PLoS One. 7:e308012012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ross JS, Wang K, Al-Rohil RN, Nazeer T,
Sheehan CE, Otto GA, He J, Palmer G, Yelensky R, Lipson D, et al:
Advanced urothelial carcinoma: Next-generation sequencing reveals
diverse genomic alterations and targets of therapy. Mod Pathol.
27:271–280. 2014. View Article : Google Scholar
|
|
57
|
Gartside MG, Chen H, Ibrahimi OA, Byron
SA, Curtis AV, Wellens CL, Bengston A, Yudt LM, Eliseenkova AV, Ma
J, et al: Loss-of-function fibroblast growth factor receptor-2
mutations in melanoma. Mol Cancer Res. 7:41–54. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Koika V, Varnavas P, Valavani H, Sidis Y,
Plummer L, Dwyer A, Quinton R, Kanaka-Gantenbein C, Pitteloud N,
Sertedaki A, et al: Comparative functional analysis of two
fibroblast growth factor receptor 1 (FGFR1) mutations affecting the
same residue (R254W and R254Q) in isolated hypogonadotropic
hypogonadism (IHH). Gene. 516:146–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Villanueva C, Jacobson-Dickman E, Xu C,
Manouvrier S, Dwyer AA, Sykiotis GP, Beenken A, Liu Y, Tommiska J,
Hu Y, et al: Congenital hypogonadotropic hypogonadism with split
hand/foot malformation: A clinical entity with a high frequency of
FGFR1 mutations. Genet Med. 17:651–659. 2015. View Article : Google Scholar :
|
|
60
|
Jackson CC, Medeiros LJ and Miranda RN:
8p11 myeloproliferative syndrome: A review. Hum Pathol. 41:461–476.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kumar KR, Chen W, Koduru PR and Luu HS:
Myeloid and lymphoid neoplasm with abnormalities of FGFR1
presenting with trilineage blasts and RUNX1 rearrangement: A case
report and review of literature. Am J Clin Pathol. 143:738–748.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yagasaki F, Wakao D, Yokoyama Y, Uchida Y,
Murohashi I, Kayano H, Taniwaki M, Matsuda A and Bessho M: Fusion
of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell
lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer
Res. 61:8371–8374. 2001.PubMed/NCBI
|
|
63
|
Ren M, Qin H, Kitamura E and Cowell JK:
Dysregulated signaling pathways in the development of
CNTRL-FGFR1-induced myeloid and lymphoid malignancies associated
with FGFR1 in human and mouse models. Blood. 122:1007–1016. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang J, Wu G, Miller CP, Tatevossian RG,
Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, et
al: St Jude Children's Research Hospital-Washington University
Pediatric Cancer Genome Project: Whole-genome sequencing identifies
genetic alterations in pediatric low-grade gliomas. Nat Genet.
45:602–612. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sia D, Losic B, Moeini A, Cabellos L, Hao
K, Revill K, Bonal D, Miltiadous O, Zhang Z, Hoshida Y, et al:
Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion
and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun.
6:60872015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gavine PR, Mooney L, Kilgour E, Thomas AP,
Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, et
al: AZD4547: An orally bioavailable, potent, and selective
inhibitor of the fibroblast growth factor receptor tyrosine kinase
family. Cancer Res. 72:2045–2056. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kwak Y, Cho H, Hur W and Sim T: Antitumor
effects and mechanisms of AZD4547 on FGFR2-deregulated endometrial
cancer cells. Mol Cancer Ther. 14:2292–2302. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Guagnano V, Kauffmann A, Wöhrle S, Stamm
C, Ito M, Barys L, Pornon A, Yao Y, Li F, Zhang Y, et al: FGFR
genetic alterations predict for sensitivity to NVP-BGJ398, a
selective pan-FGFR inhibitor. Cancer Discov. 2:1118–1133. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hagel M, Miduturu C, Sheets M, Rubin N,
Weng W, Stransky N, Bifulco N, Kim JL, Hodous B, Brooijmans N, et
al: First selective small molecule inhibitor of FGFR4 for the
treatment of hepatocellular carcinomas with an activated FGFR4
signaling pathway. Cancer Discov. 5:424–437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nakanishi Y, Akiyama N, Tsukaguchi T,
Fujii T, Sakata K, Sase H, Isobe T, Morikami K, Shindoh H, Mio T,
et al: The fibroblast growth factor receptor genetic status as a
potential predictor of the sensitivity to CH5183284/Debio 1347, a
novel selective FGFR inhibitor. Mol Cancer Ther. 13:2547–2558.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lee SH, Lopes de Menezes D, Vora J, Harris
A, Ye H, Nordahl L, Garrett E, Samara E, Aukerman SL, Gelb AB, et
al: In vivo target modulation and biological activity of CHIR-258,
a multitargeted growth factor receptor kinase inhibitor, in colon
cancer models. Clin Cancer Res. 11:3633–3641. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tan L, Wang J, Tanizaki J, Huang Z, Aref
AR, Rusan M, Zhu SJ, Zhang Y, Ercan D, Liao RG, et al: Development
of covalent inhibitors that can overcome resistance to
first-generation FGFR kinase inhibitors. Proc Natl Acad Sci USA.
111:E4869–E4877. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tabernero J, Bahleda R, Dienstmann R,
Infante JR, Mita A, Italiano A, Calvo E, Moreno V, Adamo B, Gazzah
A, et al: Phase I dose-escalation study of JNJ-42756493, an oral
pan-fibroblast growth factor receptor inhibitor, in patients with
advanced solid tumors. J Clin Oncol. 33:3401–3408. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhao G, Li WY, Chen D, Henry JR, Li HY,
Chen Z, Zia-Ebrahimi M, Bloem L, Zhai Y, Huss K, et al: A novel,
selective inhibitor of fibroblast growth factor receptors that
shows a potent broad spectrum of antitumor activity in several
tumor xenograft models. Mol Cancer Ther. 10:2200–2210. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Shah RR and Morganroth J: Update on
cardiovascular safety of tyrosine kinase inhibitors: With a special
focus on QT interval, left ventricular dysfunction and overall
risk/benefit. Drug Saf. 38:693–710. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gacche RN and Meshram RJ: Angiogenic
factors as potential drug target: Efficacy and limitations of
anti-angiogenic therapy. Biochim Biophys Acta. 1846:161–179.
2014.PubMed/NCBI
|
|
77
|
Douxfils J, Haguet H, Mullier F, Chatelain
C, Graux C and Dogné JM: Association between BCR-ABL tyrosine
kinase inhibitors for chronic myeloid leukemia and cardiovascular
events, major molecular response, and overall survival: A
systematic review and meta-analysis. JAMA Oncol. Feb 4–2016.Epub
ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Jain RK: Normalizing tumor
microenvironment to treat cancer: Bench to bedside to biomarkers. J
Clin Oncol. 31:2205–2218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Junttila MR and de Sauvage FJ: Influence
of tumour micro-environment heterogeneity on therapeutic response.
Nature. 501:346–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Procopio MG, Laszlo C, Al Labban D, Kim
DE, Bordignon P, Jo SH, Goruppi S, Menietti E, Ostano P, Ala U, et
al: Combined CSL and p53 downregulation promotes cancer-associated
fibroblast activation. Nat Cell Biol. 17:1193–1204. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ware KE, Hinz TK, Kleczko E, Singleton KR,
Marek LA, Helfrich BA, Cummings CT, Graham DK, Astling D, Tan AC,
et al: A mechanism of resistance to gefitinib mediated by cellular
reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth
loop. Oncogenesis. 2:e392013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Suvannasankha A, Tompkins DR, Edwards DF,
Petyaykina KV, Crean CD, Fournier PG, Parker JM, Sandusky GE,
Ichikawa S, Imel EA, et al: FGF23 is elevated in multiple myeloma
and increases heparanase expression by tumor cells. Oncotarget.
6:19647–19660. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Weis SM and Cheresh DA: Tumor
angiogenesis: Molecular pathways and therapeutic targets. Nat Med.
17:1359–1370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Bridges E, Oon CE and Harris A: Notch
regulation of tumor angiogenesis. Future Oncol. 7:569–588. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Katoh M: Therapeutics targeting
angiogenesis: Genetics and epigenetics, extracellular miRNAs and
signaling networks (Review). Int J Mol Med. 32:763–767.
2013.PubMed/NCBI
|
|
87
|
Schmitt J and Matei D: Targeting
angiogenesis in ovarian cancer. Cancer Treat Rev. 38:272–283. 2012.
View Article : Google Scholar
|
|
88
|
Goel HL and Mercurio AM: VEGF targets the
tumour cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Oladipupo SS, Smith C, Santeford A, Park
C, Sene A, Wiley LA, Osei-Owusu P, Hsu J, Zapata N, Liu F, et al:
Endothelial cell FGF signaling is required for injury response but
not for vascular homeostasis. Proc Natl Acad Sci USA.
111:13379–13384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Xiao L, Yang S, Hao J, Yuan X, Luo W,
Jiang L, Hu Y, Fu Z, Zhang Y and Zou C: Endostar attenuates
melanoma tumor growth via its interruption of b-FGF mediated
angiogenesis. Cancer Lett. 359:148–154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Choi HJ, Armaiz Pena GN, Pradeep S, Cho
MS, Coleman RL and Sood AK: Anti-vascular therapies in ovarian
cancer: Moving beyond anti-VEGF approaches. Cancer Metastasis Rev.
34:19–40. 2015. View Article : Google Scholar :
|
|
92
|
Hilbert T and Klaschik S: The
angiopoietin/TIE receptor system: Focusing its role for
ischemia-reperfusion injury. Cytokine Growth Factor Rev.
26:281–291. 2015. View Article : Google Scholar
|
|
93
|
Fagiani E and Christofori G: Angiopoietins
in angiogenesis. Cancer Lett. 328:18–26. 2013. View Article : Google Scholar
|
|
94
|
Zhou W, Wang G and Guo S: Regulation of
angiogenesis via Notch signaling in breast cancer and cancer stem
cells. Biochim Biophys Acta. 1836:304–320. 2013.PubMed/NCBI
|
|
95
|
Rostama B, Peterson SM, Vary CP and Liaw
L: Notch signal integration in the vasculature during remodeling.
Vascul Pharmacol. 63:97–104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang P, Yan X, Chen Y, Yang Z and Han H:
Notch signaling in blood vessels: From morphogenesis to
homeostasis. Sci China Life Sci. 57:774–780. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Bertolini F, Marighetti P, Martin-Padura
I, Mancuso P, Hu-Lowe DD, Shaked Y and D'Onofrio A: Anti-VEGF and
beyond: Shaping a new generation of anti-angiogenic therapies for
cancer. Drug Discov Today. 16:1052–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Schreiber RD, Old LJ and Smyth MJ: Cancer
immunoediting: Integrating immunity's roles in cancer suppression
and promotion. Science. 331:1565–1570. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Talmadge JE and Gabrilovich DI: History of
myeloid-derived suppressor cells. Nat Rev Cancer. 13:739–752. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Mantovani A and Sica A: Macrophages,
innate immunity and cancer: Balance, tolerance, and diversity. Curr
Opin Immunol. 22:231–237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Sakaguchi S, Miyara M, Costantino CM and
Hafler DA: FOXP3+ regulatory T cells in the human immune
system. Nat Rev Immunol. 10:490–500. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Balkwill FR: The chemokine system and
cancer. J Pathol. 226:148–157. 2012. View Article : Google Scholar
|
|
103
|
Lippitz BE: Cytokine patterns in patients
with cancer: A systematic review. Lancet Oncol. e218–e228. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Condamine T, Ramachandran I, Youn JI and
Gabrilovich DI: Regulation of tumor metastasis by myeloid-derived
suppressor cells. Annu Rev Med. 66:97–110. 2015. View Article : Google Scholar :
|
|
105
|
Rivera LB and Bergers G: Intertwined
regulation of angiogenesis and immunity by myeloid cells. Trends
Immunol. 36:240–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chen Y, Ramjiawan RR, Reiberger T, Ng MR,
Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, et al:
CXCR4 inhibition in tumor microenvironment facilitates
anti-programmed death receptor-1 immunotherapy in sorafenib-treated
hepatocellular carcinoma in mice. Hepatology. 61:1591–1602. 2015.
View Article : Google Scholar :
|
|
107
|
Holdman XB, Welte T, Rajapakshe K, Pond A,
Coarfa C, Mo Q, Huang S, Hilsenbeck SG, Edwards DP, Zhang X, et al:
Upregulation of EGFR signaling is correlated with tumor stroma
remodeling and tumor recurrence in FGFR1-driven breast cancer.
Breast Cancer Res. 17:1412015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu L, Ye TH, Han YP, Song H, Zhang YK,
Xia Y, Wang NY, Xiong Y, Song XJ, Zhu YX, et al: Reductions in
myeloid-derived suppressor cells and lung metastases using AZD4547
treatment of a metastatic murine breast tumor model. Cell Physiol
Biochem. 33:633–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hume DA and MacDonald KP: Therapeutic
applications of macrophage colony-stimulating factor-1 (CSF-1) and
antagonists of CSF-1 receptor (CSF-1R) signaling. Blood.
119:1810–1820. 2012. View Article : Google Scholar
|
|
110
|
Sieweke MH and Allen JE: Beyond stem
cells: Self-renewal of differentiated macrophages. Science.
342:12429742013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Hamilton JA and Achuthan A: Colony
stimulating factors and myeloid cell biology in health and disease.
Trends Immunol. 34:81–89. 2013. View Article : Google Scholar
|
|
112
|
Moughon DL, He H, Schokrpur S, Jiang ZK,
Yaqoob M, David J, Lin C, Iruela-Arispe ML, Dorigo O and Wu L:
Macrophage blockade using CSF1R inhibitors reverses the vascular
leakage underlying malignant ascites in late-stage epithelial
ovarian cancer. Cancer Res. 75:4742–4752. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
DeNardo DG, Brennan DJ, Rexhepaj E,
Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD,
Junaid SA, et al: Leukocyte complexity predicts breast cancer
survival and functionally regulates response to chemotherapy.
Cancer Discov. 1:54–67. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Xu J, Escamilla J, Mok S, David J,
Priceman S, West B, Bollag G, McBride W and Wu L: CSF1R signaling
blockade stanches tumor-infiltrating myeloid cells and improves the
efficacy of radiotherapy in prostate cancer. Cancer Res.
73:2782–2794. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ries CH, Cannarile MA, Hoves S, Benz J,
Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I,
et al: Targeting tumor-associated macrophages with anti-CSF-1R
antibody reveals a strategy for cancer therapy. Cancer Cell.
25:846–859. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Rivera LB, Meyronet D, Hervieu V,
Frederick MJ, Bergsland E and Bergers G: Intratumoral myeloid cells
regulate responsiveness and resistance to antiangiogenic therapy.
Cell Rep. 11:577–591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Beenken A and Mohammadi M: The FGF family:
Biology, pathophysiology and therapy. Nat Rev Drug Discov.
8:235–253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Degirolamo C, Sabbà C and Moschetta A:
Therapeutic potential of the endocrine fibroblast growth factors
FGF19, FGF21 and FGF23. Nat Rev Drug Discov. 15:51–69. 2016.
View Article : Google Scholar
|
|
119
|
Liu WY, Xie DM, Zhu GQ, Huang GQ, Lin YQ,
Wang LR, Shi KQ, Hu B, Braddock M, Chen YP, et al: Targeting
fibroblast growth factor 19 in liver disease: A potential biomarker
and therapeutic target. Expert. Opin Ther Targets. 19:675–685.
2015. View Article : Google Scholar
|
|
120
|
Pai R, French D, Ma N, Hotzel K, Plise E,
Salphati L, Setchell KD, Ware J, Lauriault V, Schutt L, et al:
Antibody-mediated inhibition of fibroblast growth factor 19 results
in increased bile acids synthesis and ileal malabsorption of bile
acids in cynomolgus monkeys. Toxicol Sci. 126:446–456. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ge H, Zhang J, Gong Y, Gupte J, Ye J,
Weiszmann J, Samayoa K, Coberly S, Gardner J, Wang H, et al:
Fibroblast growth factor receptor 4 (FGFR4) deficiency improves
insulin resistance and glucose metabolism under diet-induced
obesity conditions. J Biol Chem. 289:30470–30480. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Silswal N, Touchberry CD, Daniel DR,
McCarthy DL, Zhang S, Andresen J, Stubbs JR and Wacker MJ: FGF23
directly impairs endothelium-dependent vasorelaxation by increasing
superoxide levels and reducing nitric oxide bioavailability. Am J
Physiol Endocrinol Metab. 307:E426–E436. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Grabner A, Amaral AP, Schramm K, Singh S,
Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, et al:
Activation of cardiac fibroblast growth factor receptor 4 causes
left ventricular hypertrophy. Cell Metab. 22:1020–1032. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wynes MW, Hinz TK, Gao D, Martini M, Marek
LA, Ware KE, Edwards MG, Böhm D, Perner S, Helfrich BA, et al:
FGFR1 mRNA and protein expression, not gene copy number, predict
FGFR TKI sensitivity across all lung cancer histologies. Clin
Cancer Res. 20:3299–3309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Wöhrle S, Weiss A, Ito M, Kauffmann A,
Murakami M, Jagani Z, Thuery A, Bauer-Probst B, Reimann F, Stamm C,
et al: Fibroblast growth factor receptors as novel therapeutic
targets in SNF5-deleted malignant rhabdoid tumors. PLoS One.
8:e776522013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Künstlinger H, Fassunke J, Schildhaus HU,
Brors B, Heydt C, Ihle MA, Mechtersheimer G, Wardelmann E, Büttner
R and Merkelbach-Bruse S: FGFR2 is overexpressed in myxoid
liposarcoma and inhibition of FGFR signaling impairs tumor growth
in vitro. Oncotarget. 6:20215–20230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zhang J, Fujimoto J, Zhang J, Wedge DC,
Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, et al: Intratumor
heterogeneity in localized lung adenocarcinomas delineated by
multiregion sequencing. Science. 346:256–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Bertotti A, Papp E, Jones S, Adleff V,
Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, et
al: The genomic landscape of response to EGFR blockade in
colorectal cancer. Nature. 526:263–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Crystal AS, Shaw AT, Sequist LV, Friboulet
L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A,
Greninger P, et al: Patient-derived models of acquired resistance
can identify effective drug combinations for cancer. Science.
346:1480–1486. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Chell V, Balmanno K, Little AS, Wilson M,
Andrews S, Blockley L, Hampson M, Gavine PR and Cook SJ: Tumour
cell responses to new fibroblast growth factor receptor tyrosine
kinase inhibitors and identification of a gatekeeper mutation in
FGFR3 as a mechanism of acquired resistance. Oncogene.
32:3059–3070. 2013. View Article : Google Scholar
|
|
132
|
Ang D, Ballard M, Beadling C, Warrick A,
Schilling A, O'Gara R, Pukay M, Neff TL, West RB, Corless CL, et
al: Novel mutations in neuroendocrine carcinoma of the breast:
Possible therapeutic targets. Appl Immunohistochem Mol Morphol.
23:97–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Okazaki T and Honjo T: PD-1 and PD-1
ligands: From discovery to clinical application. Int Immunol.
19:813–824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Sharma P and Allison JP: The future of
immune checkpoint therapy. Science. 348:56–61. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Topalian SL, Drake CG and Pardoll DM:
Immune checkpoint blockade: A common denominator approach to cancer
therapy. Cancer Cell. 27:450–461. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Mahoney KM, Rennert PD and Freeman GJ:
Combination cancer immunotherapy and new immunomodulatory targets.
Nat Rev Drug Discov. 14:561–584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Hovelson DH, McDaniel AS, Cani AK, Johnson
B, Rhodes K, Williams PD, Bandla S, Bien G, Choppa P, Hyland F, et
al: Development and validation of a scalable next-generation
sequencing.
|