Introduction
Cardiac hypertrophy is characterized by the
enlargement of cardiomyocytes and interstitial fibrisis (1–4).
Hypertension, myocardial infarction and cardiomyopathy are often
associated with interstitial fibrosis, contractile and diastolic
dysfunction, and the re-expression of fetal cardiac genes, such as
atrial natriuretic peptides (ANPs), brain natriuretic peptides or
B-type natriuretic peptides (BNPs) and the β-myosin heavy chain
(MHC), which can finally result in heart failure (5–8).
In the initial stage, myocardial hypertrophy is an adaptive and
compensatory process which maintains cardiac output; however,
sustained cardiac hypertrophy leads to detrimental cardiac
remodeling and heart failure, which is the main reason of morbidity
and mortality worldwide (1,2,5,6,8).
New strategies to prevent or attenuate the enlargement of
cardiomyocytes and cell apoptosis, and prevent the transition
between adaptive hypertrophy and heart failure are necessary
(1,9–11).
Over the years, there have many efforts made to elucidate the
molecular mechanisms of action of the intracellular signaling
pathways involved in the progression of cardiac hypertrophy
(1–8), in order to identify novel
pharmacological agents with which to prevent heart failure.
Syringin (C17H24O9,
molecular weight, 372.37), also known as eleutheroside B, is the
major biologically active component extracted from
Eleutherococcus senticosus (12–14). Various studies have certified the
multiple pharmacological properties of syringin. Cui et al
demonstrated that syringin decreased sleep latency and prolonged
sleep duration in mice; the molecular mechanisms involved may be
associated with the nitric oxide synthase (NOS)/nitric oxide (NO)
pathway (13). Furthermore, there
is evidence proving that syringin exerts remarkable
anti-inflammatory and antioxidant effects, and possesses
immunomodulatory properties (15–17). However, the effects of syringin on
cardiac hypertrophy and its potential mechanisms of action have not
yet been elucidated. The results of our study demonstrated that
syringin attenuates the progression of cardiac hypertrophy induced
by hypertrophic stimuli via the AMP-activated protein kinase α
(AMPKα) and autophagy-related signaling pathways.
The autophagy pathway is essential to myocardium
homeostasis, and regulates cell survival and cell death pathways
through the turnover of organelles and proteins (18–20). Nevertheless, under pathological
conditions, the specific role of autophagy in cardiac geometry and
function remains controversial. Zhu et al found that the
autophagic response was activated by pressure overload, and they
provided direct evidence that load triggered cardiac autophagy is a
maladaptive response that contributes to the progression of heart
failure (21). Another study
demonstrated that multiple forms of stress, including pressure
overload provoked an increase in autophagic activity in
cardiomyocytes (22). On the
contrary, other studies have suggested that autophagy activation
may exert protective effects in cardiac hypertrophy and heart
failure (23). Thus this study
was designed to examine the role of syringin in the development of
cardiac hypertrophy following pressure overload, and to elucidate
the underlying mechanisms with a particular focus on autophagy.
Materials and methods
Chemicals
Syringin was obtained from Shanghai Winherb Medical
S&T Development Co., Ltd. (Shanghai, China). High-performance
liquid chromatography analysis was performed to confirm the purity
of syringin (98%; data not shown).
Animals
All animal experiments were performed in accordance
with the institutional guidelines of the Animal Care and Use
Committee of Renmin Hospital of Wuhan University, Wuhan, China.
Male C57BL/6 mice weighing 23.5–27.5 g and aged 8–10 weeks were
obtained from the Institute of Laboratory Animal Science, Chinese
Academy of Medical Sciences and Peking Union Medical College
(Beijing, China). The mice were housed in the Cardiovascular
Research Institute of Wuhan University in an environment with
controlled temperature and humidity. The mice were randomly divided
into 5 groups of 20 mice each. Three of the groups were subjected
to aortic banding (AB) and the two other groups were subjected to
sham surgery (Sham group). The aortic arch branch was exposed with
a chest expander upon opening of the second and third intercostals
by an incision. The vessel was ligated using a 26G/27 G syringe
needle placed parallel above the vessel. Subsequent to the rapid
withdrawal of the needle to achieve aortic constriction, the chest
was closed in layers and a total of 0.1 ml 0.5% bupivacaine
(Sigma-Aldrich, St. Louis, MO, USA) was injected subcutaneously
close to the edges of the skin incision to alleviate post-operative
pain. The procedure for sham surgery was the same as that for AB
but without vessel ligation. The mice in three of the treatment
groups were gavaged with syringin at doses of 50 mg/kg body weight
(L-syringin) or 100 mg/kg body weight (syringin) for 7 weeks. The
mice in the remaining groups were gavaged with the same doses of
normal saline (vehicle) for 7 weeks as a control. The 5 groups were
designated as follows: sham + syringin, sham + vehicle, AB +
vehicle, AB + L-syringin and AB + syringin. The mice were subjected
to heart function determination by echocardiography and
catheter-based measurements of hemodynamic parameters [heart
weight/body weight (HW/BW; mg/g), lung weight/body weight (LW/BW;
mg/g) and heart weight/tibia length (HW/TL; mg/ml) ratios; and the
cross-sectional area of the cardiomyocytes (the areas of individual
myocytes were calculated using Image Pro-Plus 6.0 software)] before
being sacrificed (by cervical dislocation) at 8 weeks following
surgery.
The mice were also subjected to heart function
determination by echocardiography and catheter-based measurements
of hemodynamic parameters before being sacrificed at 8 weeks
following surgery. The mice were anesthetized with 1.5% isoflurane
before performing echocardiography using a MyLab™ 30CV (Esaote
S.p.A., Genoa, Switzerland) and a 10 MHz linear array ultrasound
transducer (10). End-systole and
end-diastole pressure were measured at the end of the left
ventricular systole and diastole, respectively. Left ventricular
end-diastolic diameter (LVEDd), interventricular septum depth
(IVSd), fractional shortening percentage (FS%) and ejection
fraction percentage (EF%) were also measured.
Echocardiography and hemodynamics
The mice were anesthetized with 1.5% isoflurane
before being subjected to echocardiography using a MyLab™ 30CV
cardiovascular ultrasound system (Esaote S.p.A., Genoa,
Switzerland) and a 10 MHz linear array ultrasound transducer.
End-systolic and end-diastolic volume were measured at the end of
the left ventricular systole and diastole, respectively. Left
ventricular end-diastolic diameter (LVEDd), interventricular septum
depth (IVSd) were measured from the left ventricle (LV) M-mode
tracing with a sweep speed of 50 mm/sec at the mid-papillary muscle
level. The mice were also anesthetized with 1.5% isoflurane before
being subjected to hemodynamic measurements. A microtip catheter
transducer (SPR-839; Millar Instruments, Houston, TX, USA) was
inserted into the right carotid artery and advanced into the left
ventricle. The dPdt max and dPdt min end systolic volume and
end-diastolic volume were determined. Data were continuously
recorded with a Millar Pressure-Volume System (MPVS-400; Millar
Instruments) and analyzed by PVAN analysis software.
Cell culture
The H9c2 cell line was acquired from the Cell Bank
of Chinese Academy of Sciences (Shanghai, China). The cells were
cultured in standard DMEM-basic, supplemented with 10% calf serum,
1% penicillin (100 U/ml) and streptomycin (100 mg/ml) (all from
Gibco, Grand Island, NY, USA). The cells were incubated in a
CO2 incubator (18 M; Sanyo, Osaka, Japan) with 5% CO2 at
37°C. Syringin was dissolved in dimethyl sulfoxide. The
pharmaceuticals [angiotensin II (Ang II; Cat. no. A9525;
Sigma-Aldrich), 2 µM; syringin, 15 µM and rapamycin
(Cat. no. 53123-88-9; Sigma-Aldrich), 100 nM] were added to the
medium and the cells were incubated for 24 h. Total RNA was then
extracted from the H9c2 cells. The mRNA expression levels of ANP,
β-MHC, α-MHC and BNP were examined by reverse
transcription-quantitative PCR (RT-qPCR). We characterized the
cells by immunocytochemistry for cardiac α-actin using α-actin
antibody (Cat. no. 2207266; EMD Millipore, Billerica, MA, USA) to
assess cardiomyocyte hypertrophy. The cells also were detected by
primary antibody to light chain 3 (LC3) A/B (Cat. no. 12741, Cell
Signaling Technology Inc., Danvers, MA, USA) to assess
autophagy.
Histological analysis
After the 8-week treatment period described above,
the mouse hearts were rapidly excised, washed with
phosphate-buffered saline, weighed and then immersed by perfusion
with 10% neutral buffered formalin. To visualize the left and right
ventricles, the hearts were cut transversely. Slides were prepared
for hematoxylin and eosin (H&E) staining for morphological
evaluation. The slides were then visualized and photographed under
a light microscope (Olympus FSX100; Olympus Corp., Tokyo, Japan).
The cross-sections of myocytes stained with fluorescein
isothiocyanate-conjugated wheat germ agglutinin (WGA; W849;
Invitrogen, Carlsbad, CA, USA) to visualize membranes and DAPI to
visualize nuclei were assessed. The areas of individual myocytes
were calculated using Image Pro-Plus 6.0 software, a quantitative
digital image analysis system.
RT-qPCR
The mRNA levels of the hypertrophic and autophagy
markers, ANP, BNP, α-MHC, β-MHC, autophagy-related gene (ATG)5,
ATG7, beclin 1, LC3 A/B and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) were analyzed by RT-qPCR. Total RNA was
extracted from the cardiac tissues or H9c2 cells using TRIzol
reagent following the manufacturer's instructions (Invitrogen). The
RNA purities were evaluated based on the OD260/OD280 ratios
detected with the SmartSpec Plus Spectrophotometer (Bio-Rad,
Hercules, CA, USA). The mRNA was reverse transcribed into cDNA
using oligo(dT) primers and the Transcriptor First Strand cDNA
Synthesis kit (Roche, Basel, Switzerland). The primers used are
listed in Table I. PCR
amplifications were quantified using the LightCycler 480 SYBR-Green
I Master Mix (Roche) in the LightCycler® 480 Real-Time
Quantitative PCR System (Roche). Briefy, subsequent to a 5-min
initial denaturation at 95°C, a total of 42 primer-extension cycles
were conducted. Each cycle consisted of a 10-sec denaturation step
at 95°C, a 20-sec annealing step at 60°C and a 20-sec incubation at
72°C for extension. A final extension step was thene conducted at
72°C for 10 min. The double standard curve was used to quantify the
PCR results. The results were normalized to the mRNA expression of
glyceraldehydes-3-phosphate dehydrogenase (GAPDH).
 | Table ISequences of primers used for
PCR. |
Table I
Sequences of primers used for
PCR.
| mRNA | Forward primer | Reverse primer |
|---|
| ANP (rat) |
5′-AAAGCAAACTGAGGGCTCTGCTCG-3′ |
5′-TTCGGTACCGGAAGCTGTTG CA-3′ |
| β-MHC (rat) |
5′-TCTGGACAGCTCCCCATTCT-3′ |
5′-CAAGGCTAACCTGGAGAAGATG-3′ |
| α-MHC (rat) |
5′-CAGAAAATGCACCATGAGGA-3′ |
5′-TCAAGCATTCATATTTATTGTGGC-3′ |
| BNP (rat) |
5′-CAGCAGCTTCTGCATCGTGGAT-3′ |
5′-TTCCTTAATCTGTCGCCGCTGG-3′ |
| GAPDH (rat) |
5′-GACATGCCGCCTGGAGAAAC-3′ |
5′-AGCCCAGGATGCCCTTTAGT-3′ |
| ATG 5 (rat) |
5′-GGGACCTCCTGAATCTCACA-3′ |
5′-CCAACAGGACAGCAGAGACA-3′ |
| ATG 7 (rat) |
5′-GTGTACGATCCCTGTAACCTAACCC-3′ |
5′-CGAAAGCAGAGAACTTCAACAGACT-3′ |
| LC3 A/B (rat) |
5′-GACCCTCTACGATGCTGGTG-3′ |
5′-TGCTGTCCTCAATGTCCTTCT-3′ |
| Beclin 1(rat) |
5′-GTGCTCCTGTGGAATGGAAT-3′ |
5′-GCTGCACACAGTCCAGAAAA-3′ |
| ANP (mouse) |
5′-ACCTGCTAGACCACCTGGAG-3′ |
5′-CCTTGGCTGTTATCTTCGGTACCGG-3′ |
| BNP (mouse) |
5′-GAGGTCACTCCTATCCTCTGG-3′ |
5′-GCCATTTCCTCCGACTTTTCTC-3′ |
| α-MHC (mouse) |
5′-GTCCAAGTTCCGCAAGGT-3′ |
5′-AGGGTCTGCTGGAGAGGTTA-3′ |
| β-MHC (mouse) |
5′-CCGAGTCCCAGGTCAACAA-3′ |
5′-CTTCACGGGCACCCTTGGA-3′ |
| GAPDH (mouse) |
5′-ACTCCACTCACGGCAAATTC-3′ |
5′-TCTCCATGGTGGTGAAGACA-3′ |
| ATG 5 (mouse) |
5′-ATATCAGACCACGACGGAGCG-3′ |
5′-CAGCATTGGCTCTATCCCGTG-3′ |
| ATG 7 (mouse) |
5′-CAGTTGGCTAGCTGTGTAA-3′ |
5′-GGCTATCGACCCACTTGGA-3′ |
| LC3 A/B
(mouse) |
5′-GCCATGGATCAACTATGG-3′ |
5′-GGCACCTCACGGATCGGTA-3′ |
| Beclin 1
(mouse) |
5′-CTGGCCAGCAGCAGTTAGAC-3′ |
5′-TCACGCTCCTCCGACGTCAA-3′ |
Western blot analysis
Ice-cold radioimmunoprecipitation assay buffer was
used to extract protein from tissue homogenates or cell lysates.
Protein quantification was carried out using the Pierce BCA protein
assay kit (Thermo Fisher Scientific, Waltham, MA, USA). The
extracted protein was subjected to SDS-PAGE, after which the
proteins were transferred to Immobilon-FL Transfer Membranes (Merck
Millipore, Billerica, MA, USA) and incubated with various primary
antibodies for 24 h in 4°C. The primary antibodies were the
following: GAPDH (Cat. no. sc-25778; Santa Cruz Biotechnology, Inc.
Dallas, TX, USA), p-AMPKα (Cat. no. 2535; Cell Signaling
Technology, Inc.), AMPKα (Cat. no. 2603P; Cell Signaling
Technology, Inc. Danvers, MA, USA); ATG7 (Cat. no. 2631S; Cell
Signaling Technology, Inc.), beclin 1 (Cat. no. ab55878; Abcam,
Cambridge, UK), p62 (Cat. no. ab91526; Abcam), LC3 A/B (Cat. no.
12741; Cell Signaling Technology, Inc.). Following the removal of
the primary antibody, the blots were incubated with the
corresponding peroxidase-conjugated secondary antibodies [IRdye
800CW-conjugated goat anti-rabbit immunoglobulin (Ig)G (Cat. no.
926-32211; LI-COR Biosciences, Lincoln, NE, USA) and IRdye
800CW-conjugated goat anti-mouse IgG (Cat. no. 926-32210; LI-COR
Biosciences)] for 2 h. The blots were then scanned using a
two-color infrared imaging system (Odyssey, Danvers, MA, USA).
Target protein expression levels were normalized to GAPDH
protein.
Statistical analysis
Data were analyzed using SPSS 16.0 (SPSS, Inc.,
Chicago, IL, USA). Differences among the groups were determined by
one-way ANOVA, and a value of p<0.05 was considered to indicate
a statistically significant difference.
Results
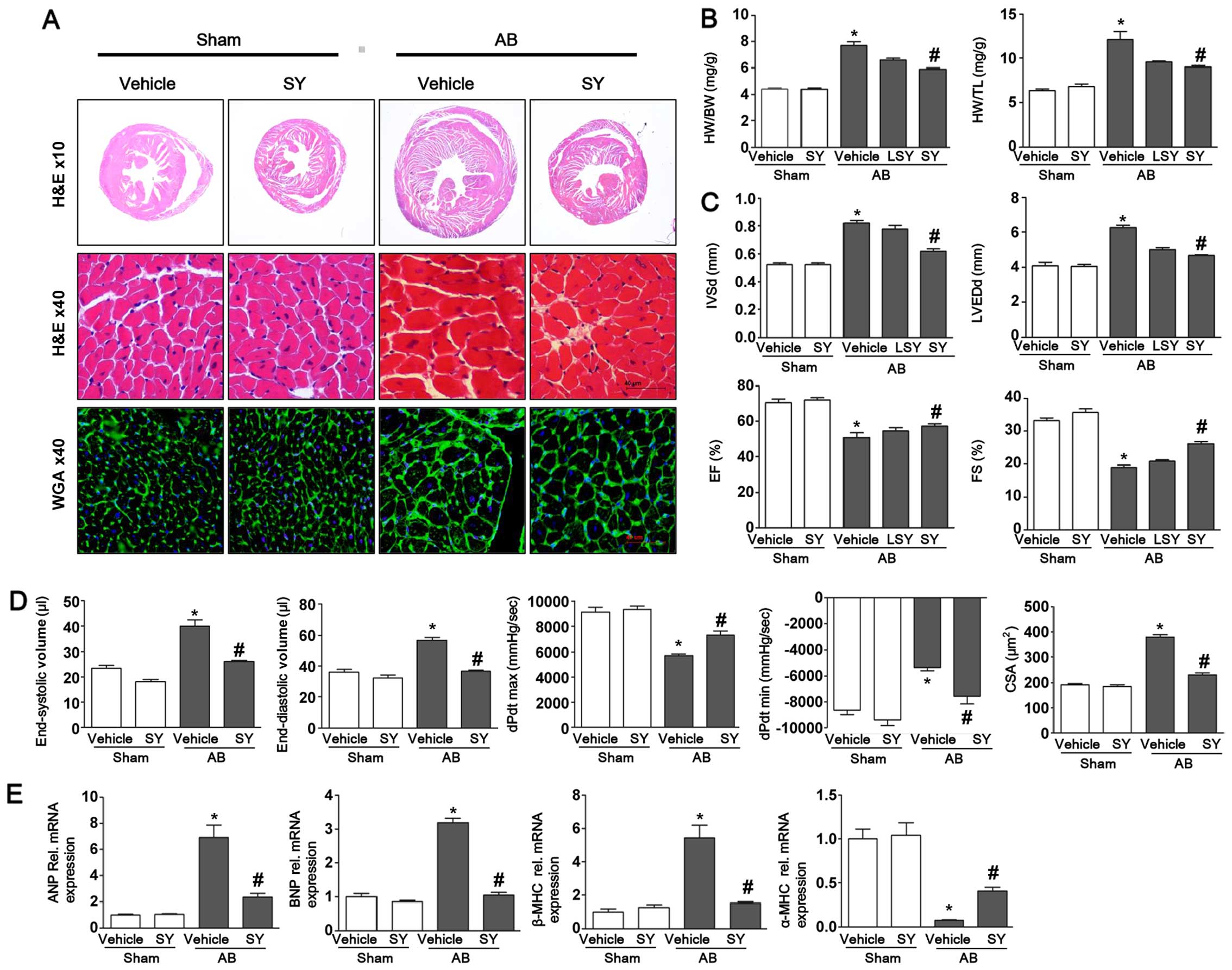
Syringin attenuates cardiac hypertrophy
induced by pressure overload in mice
To determine whether syringin attenuates the
hypertrophic response to pressure overload, mice were administered
syringin after 7 days of AB or sham surgery. The administration of
syringin markedly decreased the heart weight/body weight ratio
(HW/BW), heart weight/tibial length ratio (HW/TL) and the
cross-sectional area (CSA) of the cardiomyocytes. As shown in
Fig. 1, the protective effects of
syringin on cardiac hypertrophy was confirmed by morphological
analysis, H&E staining and WGA staining. The results of H&E
and WGA staining revealed that syringin decreased cardiac mass and
the myocyte cross-sectional area induced by pressure overload. The
results of echocardiographic and pressure-volume loop analyses
revealed that the LVEDd and IVSd increased, and the FS% and EF%
decreased in the vehicle-treated mice following AB. Pressure
overload significantly increased the LV mass and exacerbated
cardiac function in the vehicle-treated group; however, treatment
with syringin attenuated cardiac function and chamber dilation. The
vehicle-treated mice exhibited ventricular dysfunction 8 weeks
after AB, as well as an increase in diastolic blood pressure and a
decrease in systolic and diastolic functions. Syringin attenuated
the dysfunction of the hemodynamic parameters, dP/dt max and dP/dt
min. In addition, the expression levels of hypertrophic markers,
such as ANP, BNP and β-MHC significantly increased following AB.
However, the administration of syringin decreased the expression
levels of hypertrophic markers. In addition, the expression of
α-MHC decreased following AB and was elevated by the administration
of syringin.
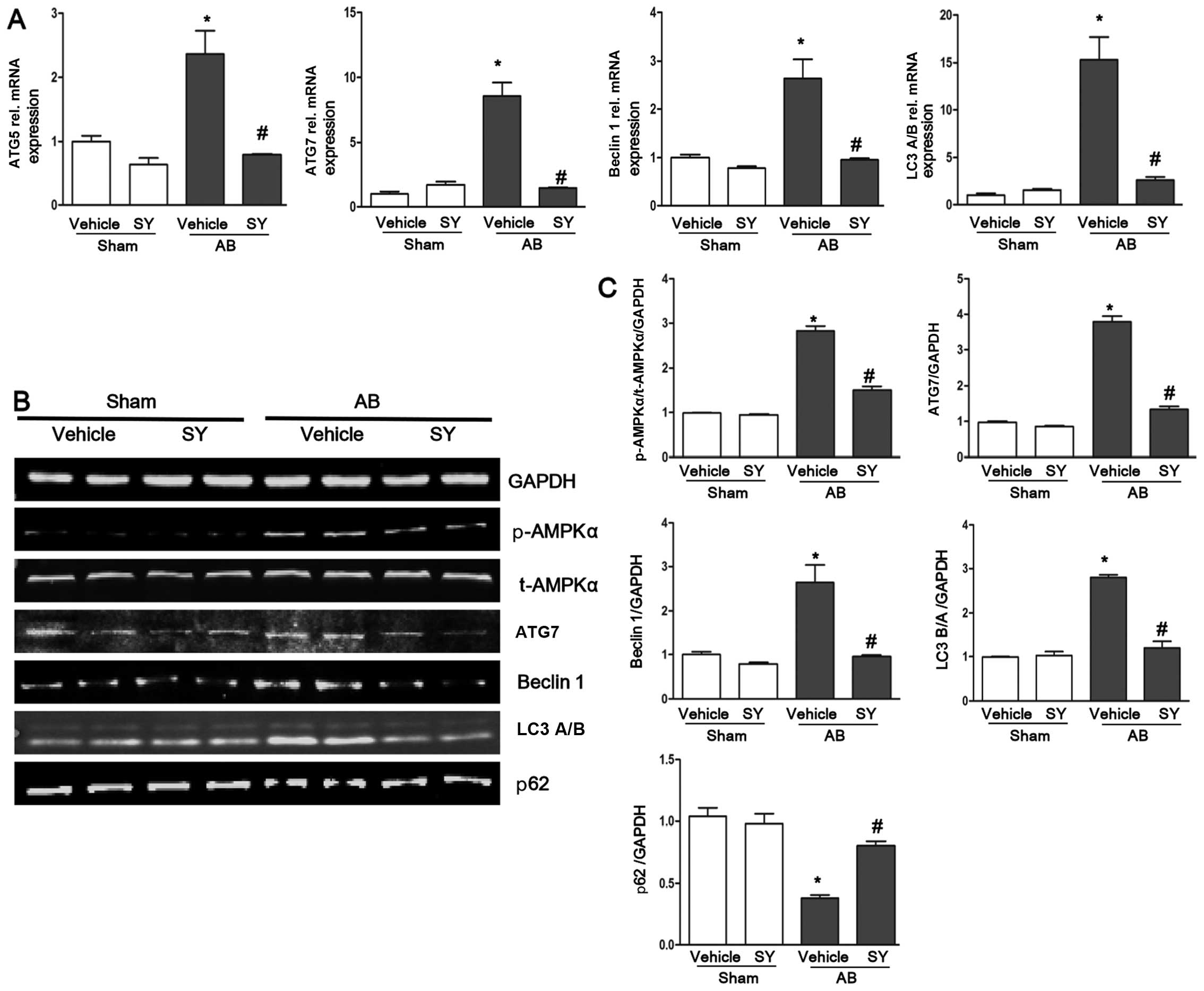
Syringin alleviates cardiac autophagy
induced by pressure overload
The expression levels of the autophagy markers,
ATG5, ATG7, beclin 1 and LC3 A/B, were detected by RT-qPCR in the
present study. The results revealed that the expression levels of
ATG5, ATG7, beclin 1 and LC3 A/B significantly increased following
AB, but were attenuated by the administration of syringin as shown
in Fig. 2. We examined the
protein expression levels of ATG7, beclin 1, p62 and LC3 A/B by
western blot analysis. We found that the protein expression levels
of ATG7 and beclin 1 were higher in the vehicle-treated mice than
in the syringin-treated mice in response to AB. The ratio of LC3
B/A was elevated following AB and was attenuated by the
administration of syringin. In addition, p62 was found to be in
inverse proportion to the ATG7 and beclin 1 (Fig. 2). These findings illustrated that
syringin alleviated cardiac autophagy induced by pressure overload.
To examine the molecular mechanisms responsible for the protective
effects of syringin against the autophagy response, we examined the
activation of AMPKα. We found that AMPKα was significantly
activated in the mice subjected to AB. The administration of
syringin decreasesd the phosphorylation levels of AMPKα (Fig. 2).
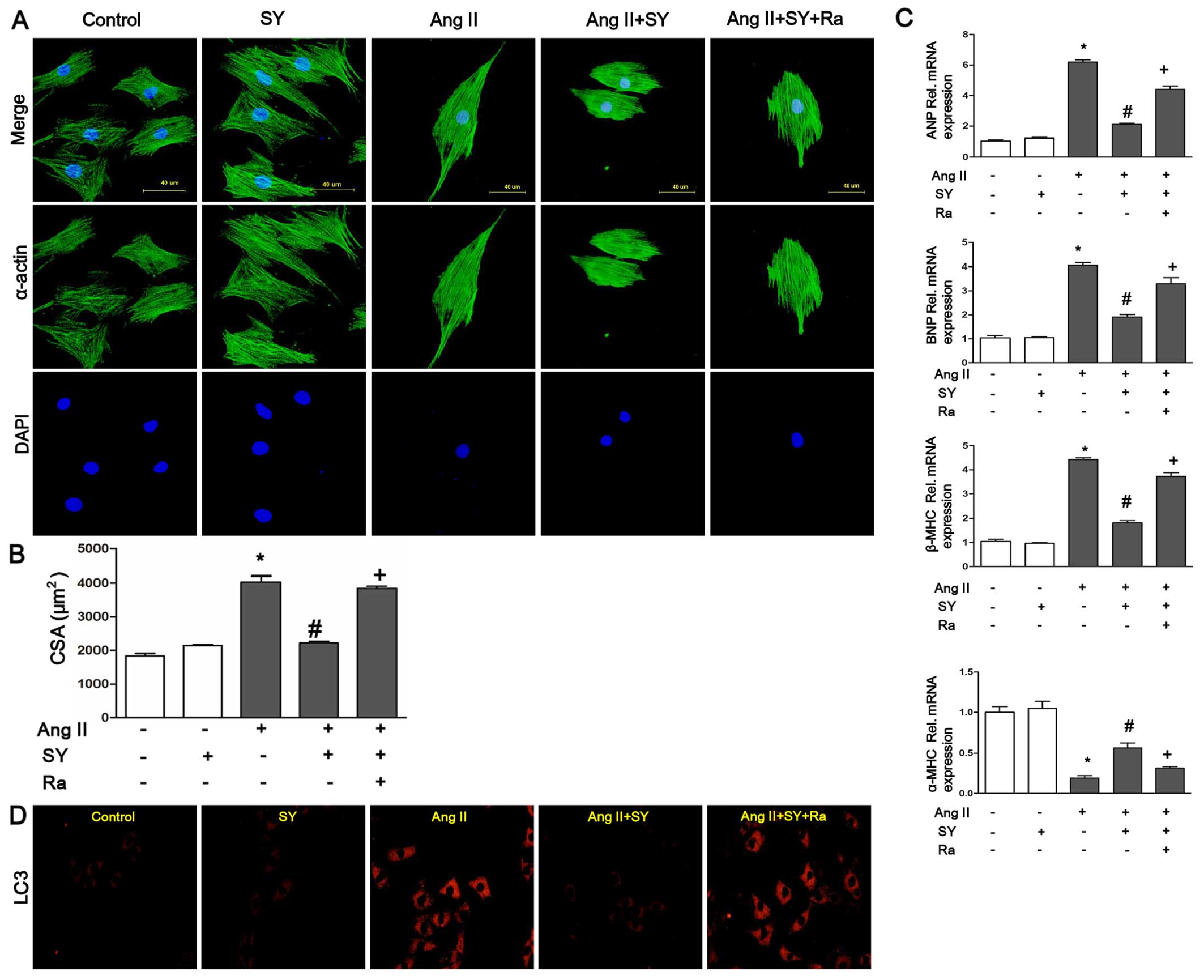
Protective effects of syringin on
cardiomyocyte hypertrophy in H9C2 cells
In in vitro experiments, the mRNA expression
levels of hypertrophic markers, including ANP, BNP and β-MHC, were
increased following stimulation with Ang II (2 µM, 24 h).
The levels of α-MHC were decreased following stimulation with Ang
II. Treatment with syringin (15 µM) resulted in a marked
decrease in the expression levels of hypertrophic markers and an
inrease in the levels of α-MHC. The cardiomyocytes were examined by
immunocytochemistry using primary antibodies to cardiac α-actin to
assess cardiomyocyte hypertrophy. Thye H9c2 cells exposed to Ang II
exhibited an enlarged cell surface area as compared with those
treated with syringin (15 µM). As shown in Fig. 3, syringin alleviated cardiomyocyte
hypertrophy induced by Ang II. The quantification of the cell CSA
(Fig. 3B), indicated that the
enlarged cell surface area induced by Ang II was significantly
reduced by treatment with syringin; however, when rapamycin was
added to the medium, the effect of syringin was reversed.
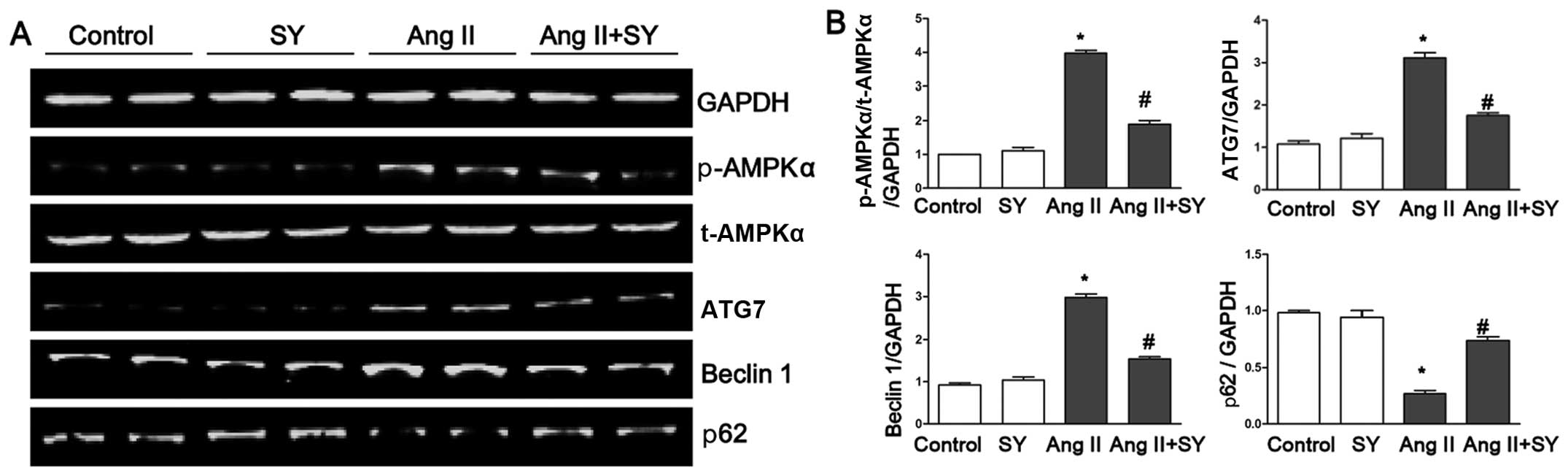
Furthermore, syringin reduced the protein expression levels of ATG7
and beclin 1 induced by Ang II and elevated the levels of p62
(Fig. 4). To explore the
molecular mechanisms through which syringin exerts its
anti-hypertrophy and anti-autophagy effects, rapamycin, an
autophagy agonist, was used in further experiments. Treatment of
the cells with rapamycin attenauted the protective effects of
syringin on cardiomyocytes. Treatment with rapamycin increased the
levels of ANP, BNP and β-MHC, and decreased the levels of α-MHC. We
found that treatment of the H9c2 cells with syringin decreased the
single cardiomyocyte area and the levels of hypertrophic markers.
In order to examine the role of autophagy, the cells were examined
by immunofluorescence using primary antibodies to LC3. As shown in
Fig. 3D, syringin decreased the
expresstion of LC3. However, rapamycin attenuated this effect.
Thus, our data prove that the protective effects of syringin
against cardiac hypertrophy are closely related to autophagy.
Discussion
Syringin is a major biologically active component
extracted from Eleutherococcus senticosus in Chinese herbs
and possessed several biological functions, including
anti-inflammatory, immunomodulatory actions and exerts antioxidant
effects (13,15–17,24). However, whether syringin has an
effect on cardiac hypertrophy remains unclear. In this study, we
found that syringin attenuated cardiac remodeling induced by
pressure overload. The beneficial effects of syringin may be
mediated by inhibition of the AMPKα and autophagy-related signaling
pathways.
Syringin is widely regarded as the major constituent
of eleutherosides (14,25). Over the years, this plant has been
used extensively as a pharmacological agent to help the body to
adapt to stress by supporting healthy adrenal gland function in
many Asian countries (13,16).
In recent years, a number of studies have demonstrated that
syringin promotes a wide range of biological activities. A previous
study indicated that syringin injection at the desired doses (100
g/kg) decreased plasma glucose and increased plasma insulin levels
in fasted Wistar rats. This effect may be associated with the
release of ACh from the nerve terminals, which in turn enhances
insulin secretion (15). Another
study by Niu et al indicated that in conscious rats with a
regular sympathetic tone, the effect of syringin on plasma glucose
regulation was impaired. Whereas in anesthetized animals, the
decreased sympathetic tone may be helpful to the therapeutic
benefits of syringin (16). Cui
et al demonstrated that the administration of syringin
decreased sleep latency and prolonged sleep duration, and that
these effects may be mediated by the NOS/NO pathway (13). Another study indicated that
syringin may alleviate the fulminant hepatic failure induced by
lipopolysaccharide/D-galactosamine by attenuating NF-κB activation
to reduce inflammatory factor production, such as TNF-α (12). In conclusion, syringin promotes a
wide range of biological activities. In this study, syringin
attenuated the cardiac hypertrophyhic response induced by pressure
overload and Ang II, as evidenced by changes in HW/BW, HW/TL and
CSA in cardiomyocytes. Furthermore, our data indicated that
syringin ameliorated LVEDd, IVSd, FS%, EF%, dP/dt max and dP/dt min
which had been affected by AB. Thus, syringin attenuated cardiac
dilation and improved LV function. Syringin also attenuated the
increase in diastolic blood pressure and the decrease in systolic
and diastolic functions. ANP and BNP, belong to the natriuretic
peptides family, and are synthetized mainly in the heart (26). The expression levels of ANP and
BNP, as in parallel with the degree of LV dysfunction and
hemodynamic stress, have become the main index for the measurement
of conventional cardiovascular disease risk factors and in parallel
with the degree of LV dysfunction and hemodynamic stress (26–28). The present study demonstrated that
syringin decreased the expression levels of ANP and BNP which were
increased by AB, suggesting that syringin attenuated cardiac
hypertrophy induced by pressure overload.
To further investigate the molecular mechanisms
through which syringin attenuates cardiac hypertrophy, we examined
activation of the AMPKα signaling pathway in response to stress
stimuli. Our data demonstrated that syringin attenuated the
phosphorylation of AMPKα. Thus, we made the conjecture that
syringing may play an important role in regulating the activation
of AMPKα. AMPK mediates energy metabolism by modulating the
activities of key enzymes or their transcription factors in
metabolic pathways (29–32) and also plays an important role in
protein synthesis, myocyte apoptosis and myocardial angiogenesis.
AMPK is composed of α, β and γ subunits; each α subunit contains a
phosphorylation site that plays a critical role in regulating AMPK
function (30,31). The specific role that AMPKα plays
in myocardial metabolism remains incompletely understood. The study
by Dyck and Lopaschuk demonstrated that AMPKα activation can
influence cardiac metabolism by regulating oxidative
phosphorylation and the uptake of fatty acids (33). Another study indicated that AMPKα
was highly expressed in the embryonic stages and reduced to the
adult level after birth; interestingly, in heart failure, the
expression of AMPKα is increased (30,34). Over the years, studies have
demonstrated important roles of AMPKα in protecting the heart
during ischemia/reperfusion injury, pathological hypertrophy and
heart failure using both genetic and pharmacological approaches
(35,36). In a study on a model of
ischemia/reperfusion, it was shown that AMPK clearly plays a
cardioprotective role during ischemic episodes, by increasing
glycolytic ATP production. However, AMPK can also play a
deleterious role in the reperfused heart (37). Indeed, during early reperfusion,
the still existing AMPK activation helps fatty acid oxidation to
predominate over glucose oxidation by phosphorylating and
inactivating acetyl-CoA carboxylase. Fatty acid oxidation,
occurring in parallel with the still present glycolytic
stimulation, can promote a deleterious uncoupling of glucose
oxidation and glycolysis (38).
This activation of AMPK results in the stimulation of glucose
uptake, glycolysis and fatty acid oxidation (32,34). These metabolic effects can be both
beneficial and harmful during ischemia and during reperfusion
following ischemia. In cardiac hypertrophy, AMPK activation can
inhibit cardiac protein synthesis and the cardiac hypertrophic
process (33,39,40); however, on the other hand, both
activating and inactivating AMPK mutations have been shown to
contribute to cardiac hypertrophy (41–44). As our data demonstrated, it is
possible that increased AMPK activity in the hypertrophic heart may
actually be detrimental to cardiac function by accelerating fatty
acid oxidation rates, such that they become the major oxidative
substrate in the heart at the expense of glucose oxidation;
syringin ameliorated the process. Furthermore, it has been proven
that the anti-apoptotic and pro-apoptotic effects of AMPK,
myocardial hypertrophy and ischemia can also induce apoptosis in
the heart (33,45,46). The role of AMPK in apoptosis or
the role of apoptosis in myocardial hypertrophy is still not clear.
It is possible that AMPK activation may play dual roles in the
development of cardiac hypertrophy (33). That is, the pharmacological
activation of AMPK during the early phase of cardiac hypertrophy
may be able to prevent hypertrophic growth, while AMPK activation
during pathological hypertrophy may be an adaptive response to the
metabolic stress (33,47,48). In addition, studies have
elaborated that AMPKα plays an improtant role in cardiac
hypertrophy by adjusting the process of autophagy (49–51). A previous study demonstrated that
the elevation of autophagy during myocardial ischemia and glucose
deprivation was accompanied by the activation of AMPKα, and the
inhibition of AMPKα significantly attenuated the induction of
autophagy (52). In this study,
syringin decreased hypertrophy induced by pressure overload by
attenuating the activation of AMPKα and autophagy. The role of
autophagy is complex, but is indispensable to maintain cardiac
homeostasis (22,49,53); it is difficult to confirm whether
autophagy is protective or deleterious in the setting of
cardiomyopathies (53,54). The variationd in the expression of
LC3II reveal that the level of autophagy may change in the
different periods of left ventricular hypertrophy induced by
transverse aortic constriction. Autophagy has been shown to be
reduced in hypertrophied hearts following this type of surgery for
1 week, and elevated for 4 weeks (55,56). Furthermore, our data indicated
that cardiac hypertrophy induced by aortic banding involved
AMPK-dependent autophagy. Another study indicated that autophagy
was elevated in patient cardiac tissues affected by hypertrophic
cardiomyopathy by detecting the expression of LC3II and beclin 1,
which proved that miR-451 regulates cardiac hypertrophy and
autophagy by targeting TSC1 (56). However, our data demonstrated that
syringin reduced the autophagy level to alleviate cardiac
hypertrophy. Our findings broaden our understanding of the
protective role of syringing in cardiac hypertrophy, and provide a
novel and potential pharmacotherapeutic strategy with which to
mitigate cardiac hypertrophy induced by pressure overload and
attenuate the progression of heart failure.
In conclusion, our data indicate that the long-term
oral administration of syringin attenuates the development of
cardiac hypertrophy induced by pressure overload and improves
cardiac functions. The protective effects of syringin may
potentially be attributed to the inhibition of the AMPKα and
autophagy-related signaling pathways. Our results indicated the use
of syringin may provide a potentially effective strategy with which
to attenuate the progression of cardiac hypertrophy.
Acknowledgments
This study was supported by the Hubei Province
Outstanding Medical Academic Leader program.
Abbreviations:
|
AMPKα
|
AMP-activated protein kinase α
|
|
AB
|
aortic banding
|
|
LVEDd
|
left ventricular end-diastolic
diameter
|
|
IVSd
|
interventricular septum depth
|
|
H&E
|
hematoxylin and eosin
|
|
WGA
|
wheat germ agglutinin
|
|
ANP
|
atrial natriuretic peptide
|
|
BNP
|
B-type natriuretic peptide
|
|
α-MHC
|
α-myosin heavy chain
|
|
β-MHC
|
β-myosin heavy chain
|
|
ATG
|
autophagy-related gene
|
|
LC3
|
light chain 3
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
CSA
|
cross-sectional area
|
|
FS%
|
fractional shortening percentage
|
|
EF%
|
ejection fraction percentage
|
|
Ang II
|
angiotensin II
|
References
|
1
|
Wu QQ, Xu M, Yuan Y, Li FF, Yang Z, Liu Y,
Zhou MQ, Bian ZY, Deng W, Gao L, et al: Cathepsin B deficiency
attenuates cardiac remodeling in response to pressure overload via
TNF-α/ASK1/JNK pathway. Am J Physiol Heart Circ Physiol.
308:H1143–H1154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou H, Guo H, Zong J, Dai J, Yuan Y, Bian
ZY and Tang QZ: ATF3 regulates multiple targets and may play a dual
role in cardiac hypertrophy and injury. Int J Cardiol. 174:838–839.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deng W, Zong J, Wei L, Guo H, Cheng Z,
Zhang R, Lin Y and Tang Q: 3,3′-Diindolylmethane improves
myocardial energy metabolism imbalance induced by pressure overload
via AMPKα in mice. Int J Cardiol. 177:235–237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bian Z, Dai J, Hiroyasu N, Guan H, Yuan Y,
Gan L, Zhou H, Zong J, Zhang Y, Li F, et al: Disruption of tumor
necrosis factor receptor associated factor 5 exacerbates pressure
overload cardiac hypertrophy and fibrosis. J Cell Biochem.
115:349–358. 2014. View Article : Google Scholar
|
|
5
|
Zong J, Wu QQ, Zhou H, Zhang JY, Yuan Y,
Bian ZY, Deng W, Dai J, Li FF, Xu M, et al: 3,3′-Diindolylmethane
attenuates cardiac H9c2 cell hypertrophy through 5′-adenosine
monophosphate-activated protein kinase-α. Mol Med Rep.
12:1247–1252. 2015.PubMed/NCBI
|
|
6
|
Zhou H, Yuan Y, Liu Y, Ni J, Deng W, Bian
ZY, Dai J and Tang QZ: Icariin protects H9c2 cardiomyocytes from
lipopoly-saccharide induced injury via inhibition of the reactive
oxygen species dependent c Jun N terminal kinases/nuclear factor-κB
pathway. Mol Med Rep. 11:4327–4332. 2015.PubMed/NCBI
|
|
7
|
Deng W, Wang X, Xiao J, Chen K, Zhou H,
Shen D, Li H and Tang Q: Loss of regulator of G protein signaling 5
exacerbates obesity, hepatic steatosis, inflammation and insulin
resistance. PLoS One. 7:e302562012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zong J, Salim M, Zhou H, Bian ZY, Dai J,
Yuan Y, Deng W, Zhang JY, Zhang R, Wu QQ and Tang QZ: NOD2 deletion
promotes cardiac hypertrophy and fibrosis induced by pressure
overload. Lab Invest. 93:1128–1136. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yussman MG, Toyokawa T, Odley A, Lynch RA,
Wu G, Colbert MC, Aronow BJ, Lorenz JN and Dorn GW II:
Mitochondrial death protein Nix is induced in cardiac hypertrophy
and triggers apoptotic cardiomyopathy. Nat Med. 8:725–730.
2002.PubMed/NCBI
|
|
10
|
Fernandes RO, Dreher GJ, Schenkel PC,
Fernandes TR, Ribeiro MF, Araujo AS and Belló-Klein A: Redox status
and pro-survival/pro-apoptotic protein expression in the early
cardiac hypertrophy induced by experimental hyperthyroidism. Cell
Biochem Funct. 29:617–623. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adams JW, Sakata Y, Davis MG, Sah VP, Wang
Y, Liggett SB, Chien KR, Brown JH and Dorn GW II: Enhanced Galphaq
signaling: A common pathway mediates cardiac hypertrophy and
apoptotic heart failure. Proc Natl Acad Sci USA. 95:10140–10145.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gong X, Zhang L, Jiang R, Wang CD, Yin XR
and Wan JY: Hepatoprotective effects of syringin on fulminant
hepatic failure induced by D-galactosamine and lipopolysaccharide
in mice. J Appl Toxicol. 34:265–271. 2014. View Article : Google Scholar
|
|
13
|
Cui Y, Zhang Y and Liu G: Syringin may
exert sleep-potentiating effects through the NOS/NO pathway. Fundam
Clin Pharmacol. 29:178–184. 2015. View Article : Google Scholar
|
|
14
|
Li Q, Sun LX, Xu L, Jia Y, Wang ZW, Shen
ZD and Bi KS: Determination and pharmacokinetic study of syringin
and chlorogenic acid in rat plasma after administration of Aidi
lyophilizer. Biomed Chromatogr. 20:1315–1320. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu KY, Wu YC, Liu IM, Yu WC and Cheng JT:
Release of acetylcholine by syringin, an active principle of
Eleutherococcus senticosus, to raise insulin secretion in Wistar
rats. Neurosci Lett. 434:195–199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Niu HS, Hsu FL and Liu IM: Role of
sympathetic tone in the loss of syringin-induced plasma glucose
lowering action in conscious Wistar rats. Neurosci Lett.
445:113–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho JY, Nam KH, Kim AR, Park J, Yoo ES,
Baik KU, Yu YH and Park MH: In-vitro and in-vivo immunomodulatory
effects of syringin. J Pharm Pharmacol. 53:1287–1294. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yan L, Vatner DE, Kim SJ, Ge H, Masurekar
M, Massover WH, Yang G, Matsui Y, Sadoshima J and Vatner SF:
Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci
USA. 102:13807–13812. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Godar RJ, Ma X, Liu H, Murphy JT,
Weinheimer CJ, Kovacs A, Crosby SD, Saftig P and Diwan A:
Repetitive stimulation of autophagy-lysosome machinery by
intermittent fasting preconditions the myocardium to
ischemia-reperfusion injury. Autophagy. 11:1537–1560. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Higashi K, Yamada Y, Minatoguchi S, Baba
S, Iwasa M, Kanamori H, Kawasaki M, Nishigaki K, Takemura G,
Kumazaki M, et al: MicroRNA-145 repairs infarcted myocardium by
accelerating cardiomyocyte autophagy. Am J Physiol Heart Circ
Physiol. 309:H1813–H1826. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu H, Tannous P, Johnstone JL, Kong Y,
Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill
JA: Cardiac autophagy is a maladaptive response to hemodynamic
stress. J Clin Invest. 117:1782–1793. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Z, Wang J and Yang X: Functions of
autophagy in pathological cardiac hypertrophy. Int J Biol Sci.
11:672–678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu X, Hua Y, Nair S, Bucala R and Ren J:
Macrophage migration inhibitory factor deletion exacerbates
pressure overload-induced cardiac hypertrophy through mitigating
autophagy. Hypertension. 63:490–499. 2014. View Article : Google Scholar :
|
|
24
|
Tohda C, Ichimura M, Bai Y, Tanaka K, Zhu
S and Komatsu K: Inhibitory effects of Eleutherococcus senticosus
extracts on amyloid β(25–35)-induced neuritic atrophy and synaptic
loss. J Pharmacol Sci. 107:329–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma B, Zhang Q, Liu Y, Li J, Xu Q, Li X,
Yang X, Yao D, Sun J, Cui G, et al: Simultaneous determination of
Eleutheroside B and Eleutheroside E in rat plasma by high
performance liquid chromatography-electrospray ionization mass
spectrometry and its application in a pharmacokinetic study. J
Chromatogr B Analyt Technol Biomed Life Sci. 917–918:84–92. 2013.
View Article : Google Scholar
|
|
26
|
Sergeeva IA and Christoffels VM:
Regulation of expression of atrial and brain natriuretic peptide,
biomarkers for heart development and disease. Biochim Biophys Acta.
1832:2403–2413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu Q, Xu-Cai YO, Chen S and Wang W: Corin:
New insights into the natriuretic peptide system. Kidney Int.
75:142–146. 2009. View Article : Google Scholar :
|
|
28
|
Volpe M, Rubattu S and Burnett J Jr:
Natriuretic peptides in cardiovascular diseases: Current use and
perspectives. Eur Heart J. 35:419–425. 2014. View Article : Google Scholar :
|
|
29
|
Zhang Y, Mi SL, Hu N, Doser TA, Sun A, Ge
J and Ren J: Mitochondrial aldehyde dehydrogenase 2 accentuates
aging-induced cardiac remodeling and contractile dysfunction: Role
of AMPK, Sirt1, and mitochondrial function. Free Radic Biol Med.
71:208–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim M, Shen M, Ngoy S, Karamanlidis G,
Liao R and Tian R: AMPK isoform expression in the normal and
failing hearts. J Mol Cell Cardiol. 52:1066–1073. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Turdi S, Kandadi MR, Zhao J, Huff AF, Du M
and Ren J: Deficiency in AMP-activated protein kinase exaggerates
high fat diet-induced cardiac hypertrophy and contractile
dysfunction. J Mol Cell Cardiol. 50:712–722. 2011. View Article : Google Scholar :
|
|
32
|
Beauloye C, Bertrand L, Horman S and Hue
L: AMPK activation, a preventive therapeutic target in the
transition from cardiac injury to heart failure. Cardiovasc Res.
90:224–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dyck JR and Lopaschuk GD: AMPK alterations
in cardiac physiology and pathology: Enemy or ally? J Physiol.
574:95–112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meng R, Pei Z, Zhang A, Zhou Y, Cai X,
Chen B, Liu G, Mai W, Wei J and Dong Y: AMPK activation enhances
PPARα activity to inhibit cardiac hypertrophy via ERK1/2 MAPK
signaling pathway. Arch Biochem Biophys. 511:1–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li Y, Chen C, Yao F, Su Q, Liu D, Xue R,
Dai G, Fang R, Zeng J, Chen Y, et al: AMPK inhibits cardiac
hypertrophy by promoting autophagy via mTORC1. Arch Biochem
Biophys. 558:79–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kudo N, Barr AJ, Barr RL, Desai S and
Lopaschuk GD: High rates of fatty acid oxidation during reperfusion
of ischemic hearts are associated with a decrease in malonyl-CoA
levels due to an increase in 5′-AMP-activated protein kinase
inhibition of acetyl-CoA carboxylase. J Biol Chem. 270:17513–17520.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kudo N, Gillespie JG, Kung L, Witters LA,
Schulz R, Clanachan AS and Lopaschuk GD: Characterization of
5′AMP-activated protein kinase activity in the heart and its role
in inhibiting acetyl-CoA carboxylase during reperfusion following
ischemia. Biochim Biophys Acta. 1301:67–75. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fassett JT, Hu X, Xu X, Lu Z, Zhang P,
Chen Y and Bache RJ: AMPK attenuates microtubule proliferation in
cardiac hypertrophy. Am J Physiol Heart Circ Physiol.
304:H749–H758. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhu J, Ning RB, Lin XY, Chai DJ, Xu CS,
Xie H, Zeng JZ and Lin JX: Retinoid X receptor agonists inhibit
hypertension-induced myocardial hypertrophy by modulating
LKB1/AMPK/p70S6K signaling pathway. Am J Hypertens. 27:1112–1124.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Davies JK, Wells DJ, Liu K, Whitrow HR,
Daniel TD, Grignani R, Lygate CA, Schneider JE, Noël G, Watkins H
and Carling D: Characterization of the role of gamma2 R531G
mutation in AMP-activated protein kinase in cardiac hypertrophy and
Wolff-Parkinson-White syndrome. Am J Physiol Heart Circ Physiol.
290:H1942–H1951. 2006. View Article : Google Scholar
|
|
41
|
Patel VV, Arad M, Moskowitz IP, Maguire
CT, Branco D, Seidman JG, Seidman CE and Berul CI:
Electrophysiologic characterization and postnatal development of
ventricular pre-excitation in a mouse model of cardiac hypertrophy
and Wolff-Parkinson-White syndrome. J Am Coll Cardiol. 42:942–951.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arad M, Moskowitz IP, Patel VV, Ahmad F,
Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT,
et al: Transgenic mice overexpressing mutant PRKAG2 define the
cause of Wolff-Parkinson-White syndrome in glycogen storage
cardiomyopathy. Circulation. 107:2850–2856. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gollob MH, Green MS, Tang AS, Gollob T,
Karibe A, Ali Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L,
et al: Identification of a gene responsible for familial
Wolff-Parkinson-White syndrome. N Engl J Med. 344:1823–1831. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Capano M and Crompton M: Bax translocates
to mitochondria of heart cells during simulated ischaemia:
Involvement of AMP-activated and p38 mitogen-activated protein
kinases. Biochem J. 395:57–64. 2006. View Article : Google Scholar :
|
|
45
|
Russell RR III, Li J, Coven DL, Pypaert M,
Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ and Young LH:
AMP-activated protein kinase mediates ischemic glucose uptake and
prevents postischemic cardiac dysfunction, apoptosis, and injury. J
Clin Invest. 114:495–503. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nagata D and Hirata Y: The role of
AMP-activated protein kinase in the cardiovascular system.
Hypertens Res. 33:22–28. 2010. View Article : Google Scholar
|
|
47
|
Horman S, Beauloye C, Vanoverschelde JL
and Bertrand L: AMP-activated protein kinase in the control of
cardiac metabolism and remodeling. Curr Heart Fail Rep. 9:164–173.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xie S, Deng Y, Pan YY, Wang ZH, Ren J, Guo
XL, Yuan X, Shang J and Liu HG: Melatonin protects against chronic
intermittent hypoxia-induced cardiac hypertrophy by modulating
autophagy through the 5′ adenosine monophosphate-activated protein
kinase pathway. Biochem Biophys Res Commun. 464:975–981. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu L, Wang C, Sun D, Jiang S, Li H, Zhang
W, Zhao Y, Xi Y, Shi S, Lu F, et al: Calhex231
ameliorates cardiac hypertrophy by inhibiting cellular autophagy in
vivo and in vitro. Cell Physiol Biochem. 36:1597–1612. 2015.
View Article : Google Scholar
|
|
50
|
Liu B, Wu Z, Li Y, Ou C, Huang Z, Zhang J,
Liu P, Luo C and Chen M: Puerarin prevents cardiac hypertrophy
induced by pressure overload through activation of autophagy.
Biochem Biophys Res Commun. 464:908–915. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mellor KM, Reichelt ME and Delbridge LM:
Autophagy anomalies in the diabetic myocardium. Autophagy.
7:1263–1267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang X, Gibson ME, Li ZL, Zhu XY, Jordan
KL, Lerman A and Lerman LO: Autophagy portends the level of cardiac
hypertrophy in experimental hypertensive swine model. Am J
Hypertens. 29:81–89. 2016. View Article : Google Scholar
|
|
53
|
Yu P, Zhang Y, Li C, Li Y, Jiang S, Zhang
X, Ding Z, Tu F, Wu J, Gao X and Li L: Class III PI3K-mediated
prolonged activation of autophagy plays a critical role in the
transition of cardiac hypertrophy to heart failure. J Cell Mol Med.
19:1710–1719. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Oyabu J, Yamaguchi O, Hikoso S, Takeda T,
Oka T, Murakawa T, Yasui H, Ueda H, Nakayama H, Taneike M, et al:
Autophagy-mediated degradation is necessary for regression of
cardiac hypertrophy during ventricular unloading. Biochem Biophys
Res Commun. 441:787–792. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hariharan N, Ikeda Y, Hong C, Alcendor RR,
Usui S, Gao S, Maejima Y and Sadoshima J: Autophagy plays an
essential role in mediating regression of hypertrophy during
unloading of the heart. PLoS One. 8:e516322013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Song L, Su M, Wang S, Zou Y, Wang X, Wang
Y, Cui H, Zhao P, Hui R and Wang J: MiR-451 is decreased in
hypertrophic cardiomyopathy and regulates autophagy by targeting
TSC1. J Cell Mol Med. 18:2266–2274. 2014. View Article : Google Scholar : PubMed/NCBI
|