Introduction
Inflammatory lung diseases, such as acute lung
injury (ALI), acute respiratory distress syndrome and pneumonia are
life-threatening diseases which cause high morbidity and mortality
worldwide (1,2). ALI is most often seen as part of a
systemic inflammatory process, particularly systemic sepsis, where
the lung manifestations parallel those of other tissues, such as
the widespread destruction of the capillary endothelium,
extravasations of protein-rich fluid and interstitial edema. In
addition, when the alveolar basement membrane is damaged and fluid
seeps into the airspaces, it stiffens the lungs and causes
ventilation-perfusion mismatch (3). A major cause for the development of
ALI is sepsis, of which Gram-negative bacteria are a prominent
cause (4). In mice, the
inhalation of endotoxins, such as lipopolysaccharide (LPS) can be
used to mimic human Gram-negative bacteria-induced ALI, leading to
neutrophil recruitment, pulmonary edema and finally in the
impairment of gas exchange; this model has been used extensively
used for testing new anti-ALI drugs. Despite extensive
investigations revealing the pathogenetic factors of ALI (5,6),
current treatments do not significantly reduce lung injury and
mortality (5). Therefore, new and
effective treatment strategies are required for patients with
ALI.
The excessive production of pro-inflammatory
cytokines, including tumor necrosis factor-α (TNF-α) and
interleukin (IL)-1β, is a key event in the development of
LPS-induced ALI (7). Hence, these
pro-inflammatory mediators and the upstream nuclear factor-κB
(NF-κB) signaling pathway may play critical roles in the
pathogenesis of ALI (8). The
injury and inflammation of the lung tissues in ALI can be
attenuated by inhibiting the activation of NF-κB (9–11).
NF-κB has been considered as a promising pharmacological target in
inflammatory diseases, including sepsis, ALI and asthma (12).
Apart from NF-κB, the production of IL-1β is also
controlled by NLR family pyrin domain containing 3 (NLRP3). NLPR3,
which binds the adaptor apoptosis-associated speck-like protein
containing a CARD domain (ASC) to induce pro- caspase-1
recruitment, autoactivation and pro-IL-1β processing, responds to
highly diverse stimuli, including ATP, bacterial toxins,
microcrystalline substances, lipid particles, bacteria and viruses
(13). It has also been
identified as an important target for sepsis, asthma or chronic
obstructive pulmonary disease (COPD) (14–16).
Silybin is the major flavonolignan from the extracts
of milk thistle seed, Silybum marianum. The whole extract,
known as silymarin, as well as silybin, has been found to protect
the liver from both acute and chronic toxicity and injury (17–20). Previous studies have demonstrated
that silybin inhibits various inflammatory responses by suppressing
the NF-κB pathway. For example, silybin has been shown to
effectively suppress tumorigenesis by attenuating oxidative stress
and deregulating the activation of inflammatory mediators (21). Silybin also inhibits the
production of pro-inflammatory cytokines through the inhibition of
the NF-κB signaling pathway in HMC-1 human mast cells (22). In addition, silybin has been shown
to attenuate asthma by decreasing antigen-specific IgE production
through the modulation of the Th1/Th2 balance in ovalbumin
(OVA)-sensitized mice (23).
However, the effects of silybin in LPS-induced ALI remain unclear.
Thus, in this study, we aimed to assess the possible protective
effects of silybin against LPS-induced lung injury and to eludicate
the possible underlying mechanisms. The results from our in
vivo and in vitro experiments suggest that silybin
attenuates LPS-induced ALI through the inhibition of NF-κB
signaling and NLPR3 inflammasome activation.
Materials and methods
Animals
Female C57/BL6 mice, 6–8 weeks of age, were
purchased from Shanghai Laboratory Animal Centre at the Chinese
Academy of Sciences, Shanghai, China. The mice were maintained in a
temperature-controlled room (22±2°C) with a 12-h light/dark cycle
and a relative humidity of 40–60%. The mice were given free access
to food and water. All animal experiments were approved by the
Institutional Animal Care and Use Committee of Tianjin Key
Laboratory of Cerebral Vascular and Neurodegenerative Diseases (no.
SKL20160021) and the animal protocol was designed to minimize the
pain and discomfort of the animals.
Reagents and antibodies
Silybin and LPS (Escherichia coli: Serotype
O55:B5) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Enzyme-linked immunosorbent assay (ELISA) kits for TNF-α and IL-1β
were purchased from R&D Systems, Inc. (Minneapolis, MN, USA).
Anti-actin (4970), anti-phosphorylated (p-)p65 (3033) and anti-p65
(8242) antibodies were purchased from Cell Signaling Technology
(Beverly, MA, USA). Anti-NLRP3 (ab4270) and anti-caspase-1
(ab179515) antibodies were purchased from Abcam (Burlingame, CA,
USA). Anti-ASC (sc-514414) antibody was purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-mouse CD3-FITC
(11-0032), CD11b-PE (12-0112) and Gr1-APC (53-5931) antibodies were
obtained from eBioscience (San Diego, CA, USA). Alexa Fluor 488
anti-mouse IgG (A-11001) was from Invitrogen, Thermo Fisher
Scientific (Waltham, MA, USA). The DAB staining kit was from KeyGen
(Nanjing, China). All other chemicals were obtained from
Sigma-Aldrich.
Induction of ALI by LPS
The mice were randomly divided into 4 groups
(n=8/group): the control group receiving saline, the model group
receiving LPS only, and the 2 experimental groups receiving silybin
followed by LPS. The animals inhaled 0.9% NaCl, or 1,000
µg/ml LPS for 1 h at 3 h intervals over a period of 8 h. The
mice received silybin (50, 100 mg/kg), dissolved in the vehicle
(0.5% carboxy methyl cellulose) via intragastric (i.g.)
administration once per day for 3 consecutive days prior to LPS
sensitization. We selected the dose of silybin according to the
findings of previous studies in which the dose of silybin for
OVA-sensitized mice ranged from 25–200 mg/kg (19,23,24). The mice were sacrificed at 6 h
post-LPS administration under anaesthesia by an intraperitoneal
injection of 30 mg/kg pentobarbital to collect bronchoalveolar
lavage fluid (BALF), blood plasma and tissue samples. Serum was
isolated from whole blood to assess cytokine levels. After the
trachea was cannulated and the chest cavity was opened via a
midline incision, the lung was lavaged 3 times with 1 ml of
ice-cold sterile saline. The total BALF cell number was counted and
the BALF composition was evaluated by FACS analysis as follows:
BALF cells were resuspended with 100 µl PBS containing 1
µl CD3-FITC, 1 µl CD11b-PE and 1 µl Gr1-APC
antibodies for 30 min on ice. The cells were then washed twice and
subjected to FACS analysis. The remaining BALF was centrifuged at
1,000 × g for 5 min at 4°C, and the cell-free supernatant was
stored at −80°C for ELISA.
Histological analysis
Lungs from 4 animals in each experimental group were
fixed by 10% buffered formalin, embedded in paraffin, and then
sectioned to reveal the maximum longitudinal view of the main
intrapulmonary bronchus of the left lung lobe. Histopathological
analysis was carried out using hematoxylin and eosin
(H&E)-stained lung sections.
Cell culture
RAW264.7 mouse macrophages and THP-1 human monocytes
were purchased from the Cell Bank of the Shanghai Institute of
Biochemistry and Cell Biology at the Chinese Academy of Sciences
and cultured in Dulbecco's modified Eagle's medium supplemented
with 10% heat-inactivated fetal bovine serum (both from Gibco,
Paisley, UK), 100 U/ml penicillin G, and 100 µg/ml
streptomycin at 37°C with 5% CO2. The RAW264.7 cells
were treated with silybin in the absence or presence of 500
µg/ml LPS for 24 h and RNA and cell culture supernatant was
collected for use in reverse transcription-quantitative PCR
(RT-qPCR) and ELISA for the analysis of cytokine levels. For the
determination of the phosphorylation levels of NF-κB and its
nuclear translocation by western blot analysis and
immnuofluorescence staining, the RAW264.7 cells were treated with
silybin (50 and 100 µM) for 3 h and stimulated with 500
µg/ml LPS for 30 min. For the analysis of NLPR3 inflammasome
activation, the THP-1 cells were first induced to differentiate by
500 nM PMA for 3 h and then stimulated with 100 ng/ml LPS for 3 h,
followed by treatment with various concentrations of silybin for 1
h, and subsequent incubation with 5 mM ATP for a further 1 h. The
cells and cell culture supernatant were then collected for use in
western blot analysis, immunoprecipitation and FACS analysis.
RT-qPCR
Total RNA from the lung tissue of each animal and
the cells using TRIzol reagent (Takara, Dalian, China). RNA samples
were reverse transcribed into cDNA and analyzed by qPCR, and the
relative expression of specific genes was determined using the
real-time PCR master mix (Roche, Bromma, Sweden) on an ABI 7500
Fast Real-Time PCR system. The program for amplification was 1
cycle of 95°C for 2 min followed by 40 cycles of 95°C for 10 sec,
60°C for 30 sec and 72°C for 30 sec. The glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) gene was used as an endogenous control to
normalize for differences in the amount of total RNA present in the
samples. The primer sequences used in this study were as follows:
IL-1β, 5′-CTTCAGGCAGGCAGTATCACTC-3′ (forward) and
5′-TGCAGTTGTCTAATGGGAACGT-3′ (reverse); IL-6,
5′-ACAACCACGGCCTTCCCTAC-3′ (forward) and
5′-TCTCATTTCCACGATTTCCCAG-3′ (reverse); TNF-α,
5′-CGAGTGACAAGCCTGTAGCCC-3′ (forward) and
5′-GTCTTTGAGATCCATGCCGTTG-3′ (reverse); IL-17,
5′-TCGAGAAGATGCTGGTGGGT-3′ (forward) and 5′-CTCTGTTTAGGCTGCCTGGC-3′
(reverse); GAPDH, 5′-AACGACCCCTTCATTGAC-3′ (forward) and
5′-CACGACTCATACAGCACCT-3′ (reverse).
Western blot analysis
Freshly isolated lung tissues were homogenized in
the presence of protease inhibitors and protein content of the
supernatant was determined by BCA protein assay kit (Pierce,
Rockford, IL, USA). The protein sample (20 µg) from the lung
homogenates was loaded per lane on a 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel.
Electrophoresis was then performed. The proteins were then
transferred onto polyvinylidene fluoride membranes. The membranes
were blocked with 5% non-fat milk for 1 h at room temperature. The
blocked membranes were then incubated with the indicated primary
antibodies overnight at 4°C, followed by incubation with
HRP-coupled secondary antibody (7074, Cell Signaling Technology).
The binding of all the antibodies was detected using an ECL
detection system. For the extraction of nucleoprotein, the cells
were collected and lysed in lysis buffer (10 mM HEPES pH 7.9, 1.5
mM MgCl2, 10 mM KCl, 0.5 mM DTT, 2% NP-40 and 1 mM PMSF)
for 30 min. The homogenate was centrifuged for 10 min at 3,000 rpm
at 4°C. The supernatant was collected as cytoplasmic protein. The
pellet was the nuclear fraction. The pellet was washed twice before
being resuspended in 50 µl lysis buffer containing Triton
X-100 for 30 min on ice with vortexing at 10 min intervals. This
was followed by centrifugation for 30 min at 14,000 × g at 4°C. The
supernatant was collected as the nuclear fraction. Actin was used
as the internal control for cytoplasmic protein and histone for the
nuclear fraction (Santa Cruz Biotechnology, Inc.).
Immunohistochemical analysis
Immunohistochemical analysis was performed on
paraffin-embedded colonic tissue sections (5-µm-thick).
Briefly, the sections were deparaffinised, rehydrated and washed in
1% phosphate-buffered saline (PBS)-Tween-20, and they were thyen
treated with 2% hydrogen peroxide, blocked with 3% goat serum and
incubated overnight at 4°C with monoclonal rabbit anti-p-NF-κB
antibody (1:200). The slides were then processed using the DAB
staining kit from KeyGen according to the manufacturer's
instructions. Images were obtained using an Olympus IX51 light
microscope (Olympus, Tokyo, Japan). The settings for image
acquisition were identical for the control and experimental
tissues.
Immunofluorescence staining
The cells grown on cover glasses were fixed with 4%
paraformaldehyde (20 min, room temperature), stained with the
anti-NF-κB antibody (1:100), and detected with secondary antibody
(Alexa Fluor 488 anti-mouse IgG; Invitrogen). The coverslips were
counterstained with DAPI and imaged using a confocal laser scanning
microscope (Olympus).
Co-immunoprecipitation assay
For co-immunoprecipitation, the cells were lysed in
lysis buffer containing Triton X-100 and cell lysates were
immunoprecipitated with antibody to ASC or control IgG with protein
A/G-Sepharose (sc-2003; Santa Cruz Biotechnology, Inc.). The beads
were washed, separated by SDS-PAGE and analyzed by western blot
analysis with antibodies to caspase-1 and NLRP3. Protein bands were
visualized using the western blotting detection system (ChemiDoc™
MP Imaging System 17001402; Bio-Rad, Hercules, CA, USA).
Measurement of intracellular ROS
levels
LPS-primed THP-1 cells were treated with various
concentrations of silybin for 1 h, and then incubated with 5 mM ATP
for 1 h. The cells were then harvested and incubated with
2′,7′-dichlorofluorescein diacetate (DCFH-DA, Life Technologies,
Carlsbad, CA, USA) at 37°C for 20 min and washed twice with cold
PBS. DCF fluorescence level was detected by FACS.
Statistical analysis
The data are presented as the means ± SD. The
significance of differences between experimental groups was
assessed using the two-tailed Student's t-test. The value of
statistical significance was set at p<0.05.
Results
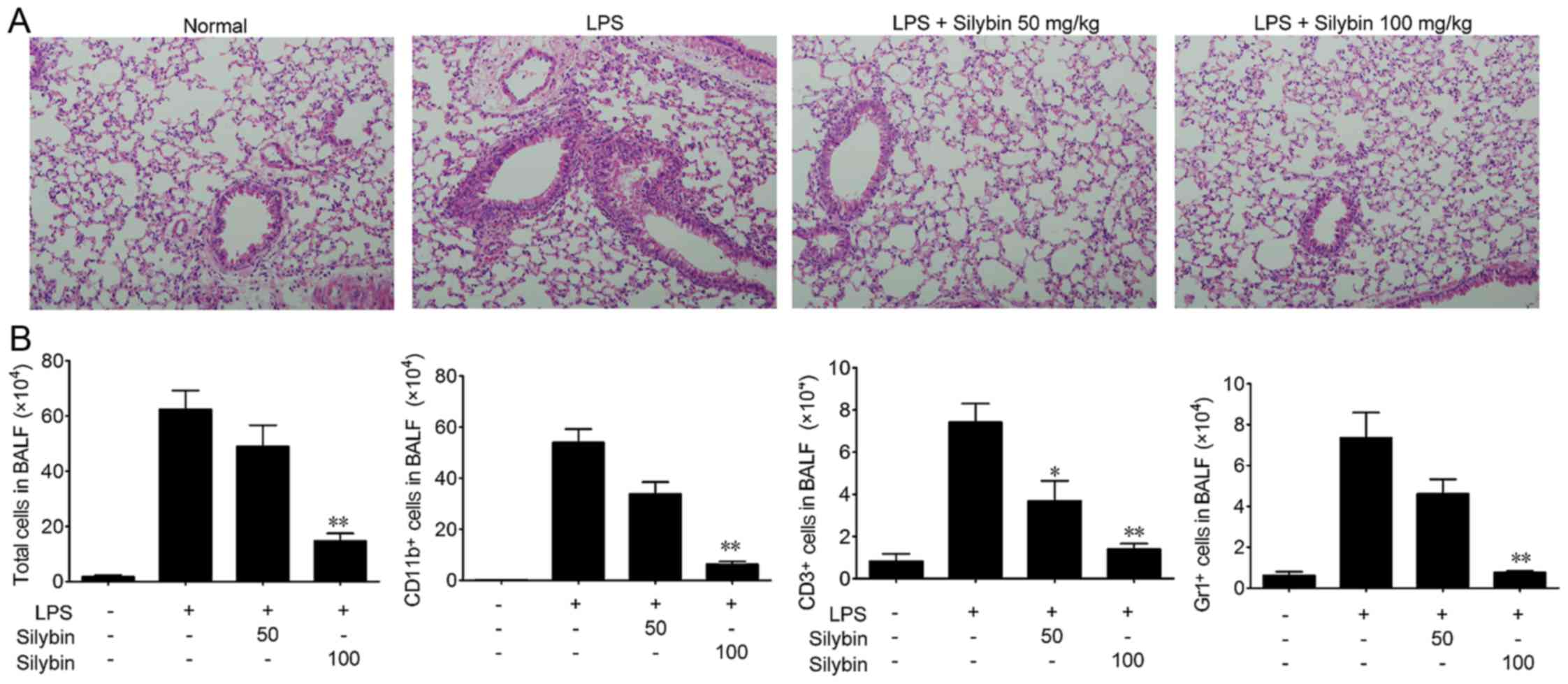
Silybin attenuates disease progression
and pathological changes in mice with LPS-induced lung injury
In the present study, we used a mouse model of
LPS-induced ALI to evaluate the therapeutic effects of silybin.
Mice were challenged with LPS and lung tissue samples were
collected, and the sections stained with H&E as shown in
Fig. 1A. The lung tissues from
the LPS group exhibited significant pathological alterations,
including notable inflammatory cell infiltration, interstitial and
intra-alveolar edema, and patchy hemorrhage, inter-alveolar septal
thickening, hyaline membrane formation and some collapsed alveoli.
By contrast, a marked attenuation of these responses was observed
in the lungs of mice treated with either 50 or 100 mg/kg silybin
(Fig. 1A). Next, the infiltration
of inflammatory cells in BALF was examined. As shown in Fig. 1B, the number of total cells,
macrophages (CD11b+), T cells (CD3+) and
neutrophils (Gr1+) in BALF was significantly increased
following LPS stimulation, while treatment with silybin
dose-dependently reduced the infiltration of inflammatory
cells.
Silybin suppresses inflammatory cytokine
levels in mice with LPS-induced lung injury
Next, the anti-inflammatory properties of silybin
were evaluated by determining the mRNA and protein levels of
inflammatory cytokines, including TNF-α and IL-1β. LPS stimulation
significantly increased the levels of TNF-α and IL-1β in BALF, as
well as the serum levels, whereas treatment with silybin
significantly decreased these cytokine levels (Fig. 2A and B). In addition, LPS
stimulation significantly elevated the mRNA levels of TNF-α IL-1β,
IL-17 and IL-6 in lung tissue, whereas treatment with silybin
dose-dependently decreased the mRNA levels of these cytokines
(Fig. 2C).
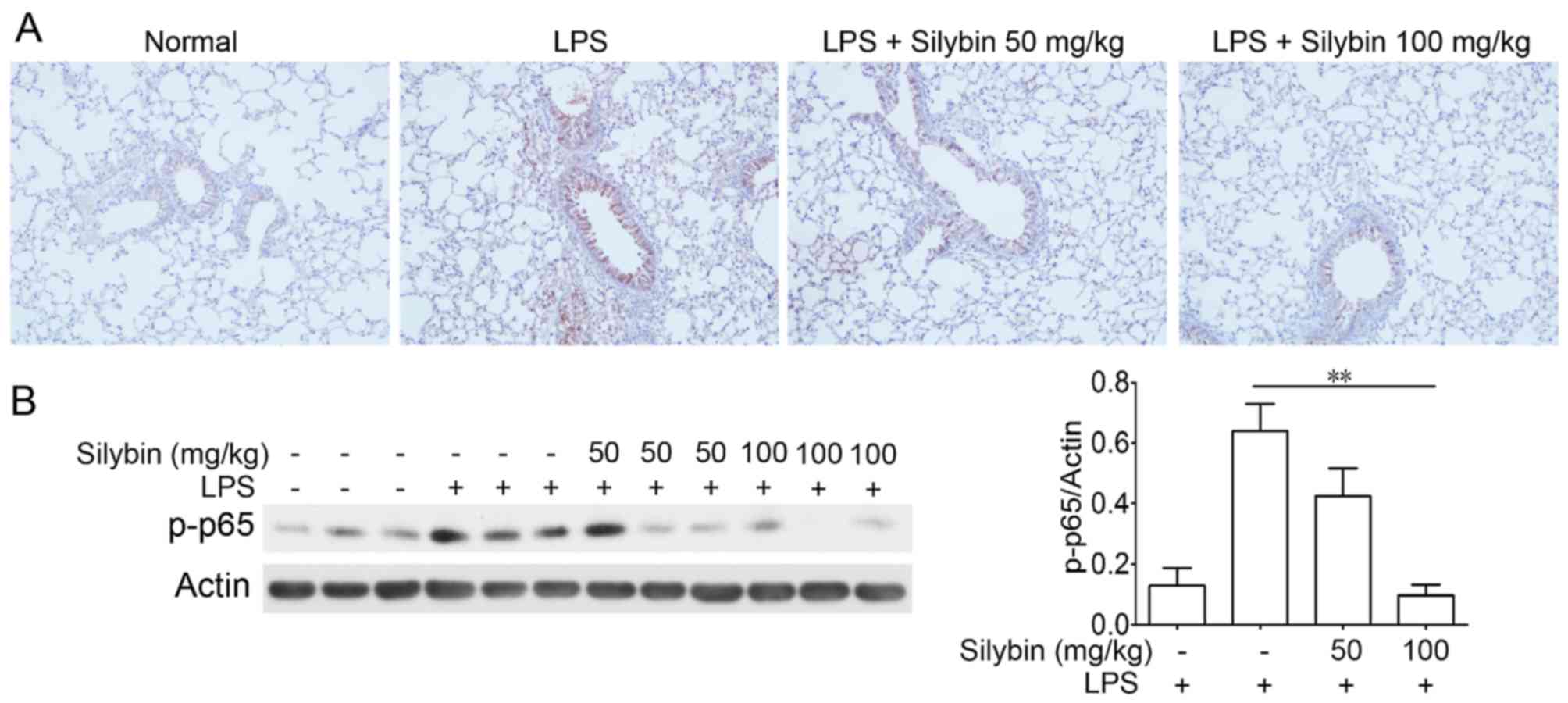
Silybin suppresses NF-κB phosphorylatin
in vivo
NF-κB is one of the major components mediating the
inflammatory process. Thus, we examined NF-κB activation in lung
samples from mice with LPS-induced lung injury. Immunohistochemical
analysis revealed an increased level of p-NF-κB (p-p65) following
LPS stimulation; however, treatment with silybin markedly inhibited
the phosphorylation of NF-κB (Fig.
3).
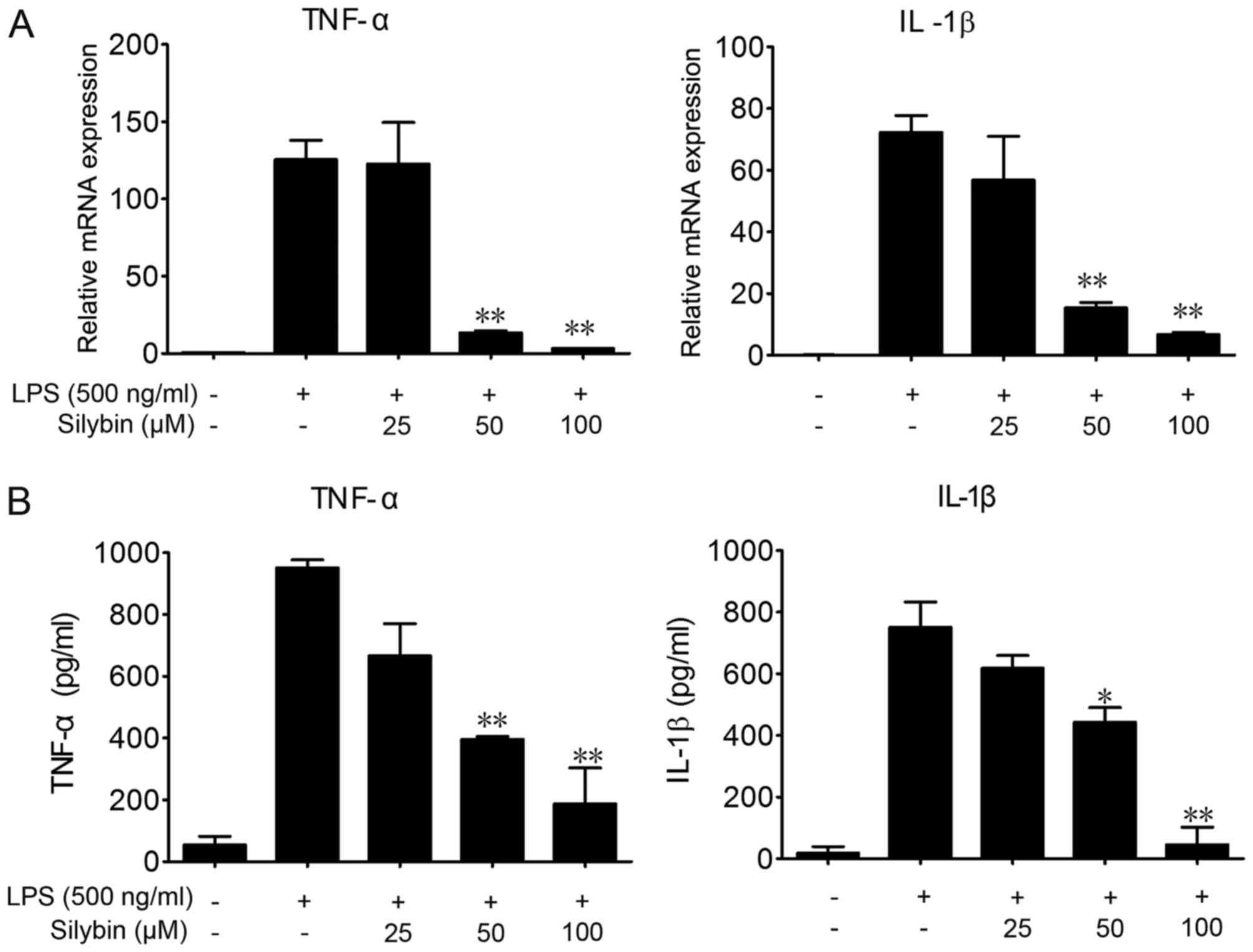
Silybin inhibits the secretion of
inflammatory cytokines in vitro
To further investigate the protective effects of
silybin against LPS-induced lung injury and the underlying
mechanisms, we used RAW264.7 cells. LPS stimulation significantly
elevated the mRNA and protein levels of TNF-α and IL-1β in the
RAW264.7 cells (Fig. 4).
Co-incubation with silybin and LPS dose-dependently decreased the
mRNA expression levels of TNF-α and IL-1β (Fig. 4A), as well as the protein levels
in the cell culture medium (Fig.
4B).
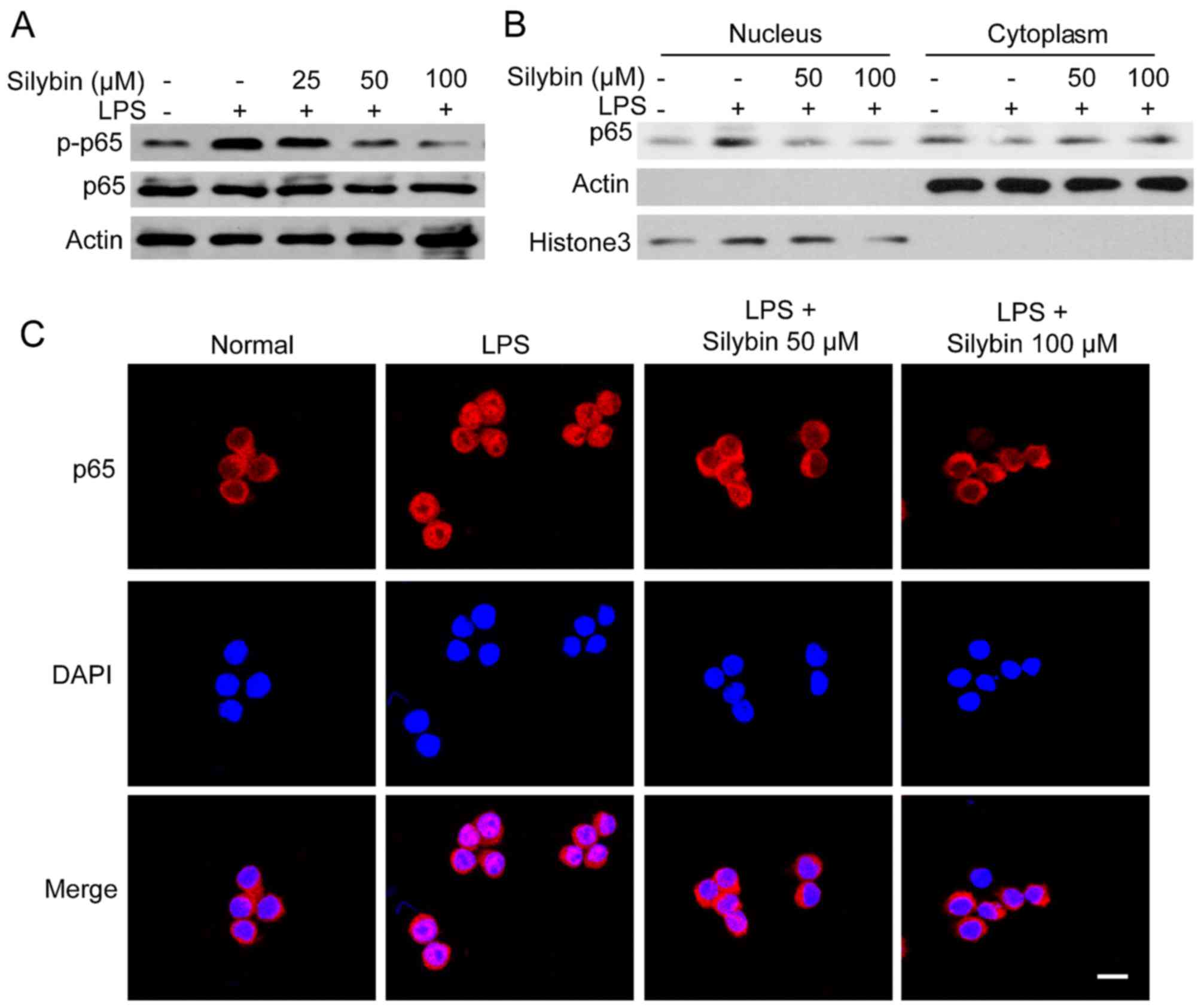
Silybin inhibits NF-κB activation in
vitro
As observed in our mouse model, treatment with
silybin inhibited the phosphorylation of NF-κB. Thus, we wished to
determine whether it can also inhibit NF-κB signaling in
vitro. Upon exposure to LPS, the levels of phosphorylated NF-κB
(p65) were significantly increased. However, pre-treatment with
silybin for 3 h dose-dependently suppressed the phosphorylation of
NF-κB (Fig. 5A). Moreover,
silybin decreased the nuclear trans-location of NF-κB (p65)
(Fig. 5B). The results of western
blot analysis of the cytoplasmic and nuclear content revealed that
the level of NF-κB (p65) in the nucleus was reduced by silybin
treatment (Fig. 5B). Using
immunofluorescence staining, strong NF-κB (p65) staining in the
nucleus was observed following stimulation with LPS, whereas in the
silybin-treated cells, NF-κB (p65) was mainly located in the
cytosol (Fig. 5C). These results
suggest that the anti-inflammatory effects of silybin may result
from the inhibition of NF-κB.
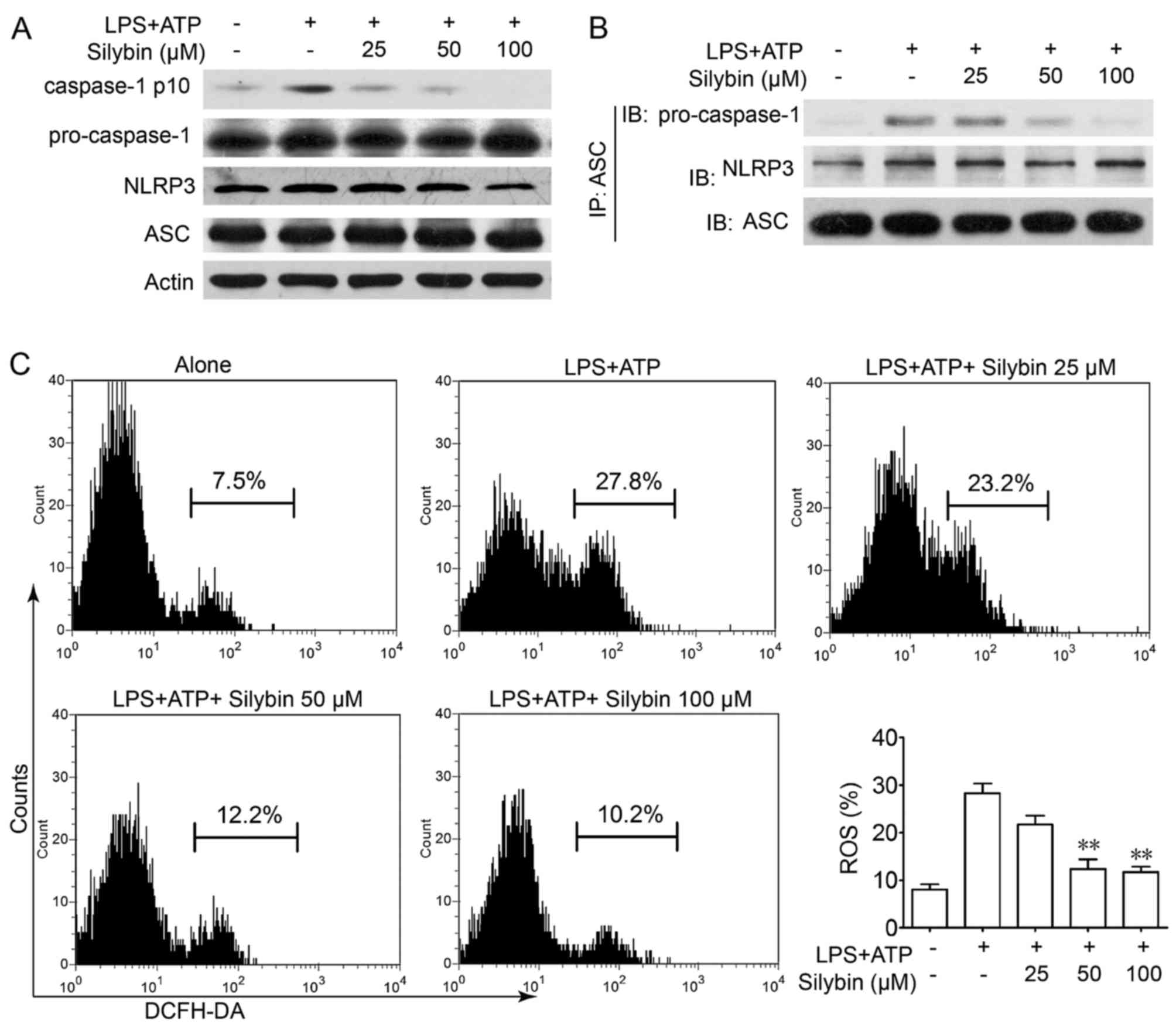
Silybin inhibits the activation of the
NLRP3 inflammasome in THP-1 cells by reducing the production of
intracellular ROS
It is worth noting that there is evidence to
indicate that IL-1β is processed as an inactive cytoplasmic
precursor (pro-IL-1β and pro-IL-18) that requires cleavage by
caspase-1 to produce the mature active forms (25,26). As caspase-1 needs to be activated
by the NLRP3 inflammasome, we then evaluated the effects of silybin
on caspase-1 activation in THP-1 cells. Treatment with silybin
inhibited caspase-1 activation in a concentration-dependent manner
(Fig. 6A). Furthermore,
immunoprecipitation analysis revealed that the process of NLRP3
inflammasome formation was also interrupted by silybin (Fig. 6B). It has been reported that in
the presence of signal I (NF-κB signaling), the NLRP3 inflammasome
is activated by ROS signaling which leads to the production of
caspase-1 (27). Treatment with
LPS and ATP led to ROS generation, whereas treatment with silybin
significantly scavenged ROS (Fig.
6C). These results indicate that silybin may reduce ROS
generation and interrupt the activation of the inflammasome in
macrophages, thereby inhibiting the cleavage of caspase-1 and the
production of cytokines.
Discussion
Despite the development of innovative therapy and
intensive care, ALI induced by bacteria or microbe infection
remains an unsresolved issue. Silybin, the major active molecule of
silymarin, is a very potent antioxidant compound capable of
scavenging free radicals and ROS, and has been shown to exert
neuroprotective (28–30), hepatoprotective (31,32) and anti-carcinogenic effects
(33–35). It is now being used as a
traditional medicine for the treatment of various liver disorders
in China. In the current study, our data revealed that silybin
inhibited the LPS-induced production of TNF-α and IL-1β, and the
activation of NF-κB and the NLRP3 inflammasome, thus protecting
mice against LPS-induced ALI.
ALI is a disorder of acute inflammation that causes
the disruption of the lung endothelial and epithelial barriers.
Following infection or trauma, the increased permeability of the
alveolar-capillary barrier, and the excessive production of
inflammatory mediators from inflammatory cells, particularly
cytokines, chemokines and adhesion molecules occurs as a direct
response and/or as a marker of ongoing cellular injury. These
symptoms can be mimicked in experimental animals by LPS inhalation
(36,37).
The treatment of ALI is based in both ventilatory
and non-ventilatory strategies. To date, the most significant
advances in the supportive care of patients with lung injury have
been associated with improved ventilator management (38). The results of this study
demonstrated that silybin induced a significant decrease in
thickened intra-alveolar septa, and a decrease in the infiltration
of prominent inflammatory cells (including alveolar macrophages,
neutrophils and T cells) (Fig.
1). Overall, these results suggest that silybin has potential
clinical applications for use in the treatment of ALI.
It is well known that NF-κB is important in terms of
directing the transcription of many inflammatory genes following
exposure to LPS, playing a crucial role in a number of inflammatory
disease processes (12,39,40). In unstimulated cells, NF-κB is
sequestered into the cytoplasm by IκBα. Upon stimulation, such as
with LPS or Toll like receptor (TLR), signal transduction events
rapidly lead to the degradation of IκBα, resulting in the nuclear
translocation of NF-κB (41).
NF-κB binds to specific DNA to initiate the transcription of
inflammatory genes. Our results revealed that silybin suppressed
the nuclear translocation of NF-κB in vivo and in
vitro, which then inhibited the expression of pro-inflammatory
cytokines, such as TNF-α and IL-1β. In this study, silybin was
concluded to successfully attenuate acute inflammation by
inhibiting the release of cytokines (such as TNF-α and IL-1β) in
BALF and serum (Fig. 2).
Additionally, the in vitro experimental results revealed
that silybin inhibited the transcription and protein levels of
TNF-α and IL-1β.
The production and secretion of pro-inflammatory
cytokines is governed not only by TLR-NF-κB signaling, but also by
the protein family containing a nucleotide-binding domain and a
leucine-rich repeat motif (NLR) (42). The NLRP3 inflammasome is a
cytosolic multiprotein complex and is increasingly being recognized
due to its clinical importance in autoimmune, infectious and
metabolic diseases (43,44). Previous studies have suggested
that ROS derived from damaged mitochondria are the major factor
that activate the NLRP3 inflammasome (45). A critical role for the NLRP3
inflammasome in the development of allergic airway inflammation in
mice has also been described. NLRP3−/−,
ASC−/− and caspase-1−/− mice have been found
to have significantly attenuated airway inflammation and cytokine
release in airway inflammation (14). Our in vitro experiments
suggested that sylibin inhibited NLRP3/ASC/caspase-1 complex
formation, thus suppressing the activation of caspase-1. Treatment
with sylibin also effectively prevented the LPS- and ATP-induced
ROS generation. We hypothesized that the beneficial effects of
sylibin against LPS-induced lung inflammation may be attributed to
its inhibition of the inflammasome activation.
Taken together, our data suggest that the
anti-inflammatory effects of silybin attenuate LPS-induced cytokine
production via the negative regulation NF-κB and NLRP3 inflammasome
activation in mice with ALI.
References
|
1
|
Global Burden of Disease Study 2013
Collaborators: Global, regional, and national incidence,
prevalence, and years lived with disability for 301 acute and
chronic diseases and injuries in 188 countries, 1990–2013: A
systematic analysis for the Global Burden of Disease Study 2013.
Lancet. 386:743–800. 2015. View Article : Google Scholar
|
|
2
|
Han S and Mallampalli RK: The acute
respiratory distress syndrome: From mechanism to translation. J
Immunol. 194:855–860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sadowitz B, Roy S, Gatto LA, Habashi N and
Nieman G: Lung injury induced by sepsis: lessons learned from large
animal models and future directions for treatment. Expert Rev Anti
Infect Ther. 9:1169–1178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johnson ER and Matthay MA: Acute lung
injury: Epidemiology, pathogenesis, and treatment. J Aerosol Med
Pulm Drug Deliv. 23:243–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sweeney RM, Griffiths M and McAuley D:
Treatment of acute lung injury: current and emerging
pharmacological therapies. Semin Respir Crit Care Med. 34:487–498.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gekara NO, Dietrich N, Lyszkiewicz M,
Lienenklaus S and Weiss S: Signals triggered by a bacterial
pore-forming toxin contribute to toll-like receptor redundancy in
gram-positive bacterial recognition. J Infect Dis. 199:124–133.
2009. View
Article : Google Scholar
|
|
8
|
Jin LY, Li CF, Zhu GF, Wu CT, Wang J and
Yan SF: Effect of siRNA against NF-kappaB on sepsisinduced acute
lung injury in a mouse model. Mol Med Rep. 10:631–637.
2014.PubMed/NCBI
|
|
9
|
Zhu T, Wang DX, Zhang W, Liao X, Guan X,
Bo H, Sun J, Huang N, He J, Zhang Y, et al: Andrographolide
protects against LPS-induced acute lung injury by inactivation of
NF-kappaB. PLoS One. 8:e564072013. View Article : Google Scholar
|
|
10
|
Xiao M, Zhu T, Zhang W, Wang T, Shen YC,
Wan QF and Wen FQ: Emodin ameliorates LPS-induced acute lung
injury, involving the inactivation of NF-kappaB in mice. Int J Mol
Sci. 15:19355–19368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen X, Yang X, Liu T, Guan M, Feng X,
Dong W, Chu X, Liu J, Tian X, Ci X, et al: Kaempferol regulates
MAPKs and NF-kappaB signaling pathways to attenuate LPS-induced
acute lung injury in mice. Int Immunopharmacol. 14:209–216. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rahman A and Fazal F: Blocking NF-kappaB:
An inflammatory issue. Proc Am Thorac Soc. 8:497–503. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Elliott EI and Sutterwala FS: Initiation
and perpetuation of NLRP3 inflammasome activation and assembly.
Immunol Rev. 265:35–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Besnard AG, Guillou N, Tschopp J, Erard F,
Couillin I, Iwakura Y, Quesniaux V, Ryffel B and Togbe D: NLRP3
inflammasome is required in murine asthma in the absence of
aluminum adjuvant. Allergy. 66:1047–1057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sohn SH, Lee JM, Park S, Yoo H, Kang JW,
Shin D, Jung KH, Lee YS, Cho J and Bae H: The inflammasome
accelerates radiation-induced lung inflammation and fibrosis in
mice. Environ Toxicol Pharmacol. 39:917–926. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Birrell MA and Eltom S: The role of the
NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacol
Ther. 130:364–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jia R, Cao L, Du J, Xu P, Jeney G and Yin
G: The protective effect of silymarin on the carbon tetrachloride
(CCl4)-induced liver injury in common carp (Cyprinus carpio). In
Vitro Cell Dev Biol Anim. 49:155–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sozmen M, Devrim AK, Tunca R, Bayezit M,
Dag S and Essiz D: Protective effects of silymarin on fumonisin B1
induced hepatotoxicity in mice. J Vet Sci. 15:51–60. 2014.
View Article : Google Scholar :
|
|
19
|
Schumann J, Prockl J, Kiemer AK, Vollmar
AM, Bang R and Tiegs G: Silibinin protects mice from T
cell-dependent liver injury. J Hepatol. 39:333–340. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tzeng JI, Chen MF, Chung HH and Cheng JT:
Silymarin decreases connective tissue growth factor to improve
liver fibrosis in rats treated with carbon tetrachloride. Phytother
Res. 27:1023–1028. 2013. View
Article : Google Scholar
|
|
21
|
Khan AQ, Khan R, Tahir M, Rehman MU,
Lateef A, Ali F, Hamiza OO, Hasan SK and Sultana S: Silibinin
inhibits tumor promotional triggers and tumorigenesis against
chemically induced two-stage skin carcinogenesis in Swiss albino
mice: Possible role of oxidative stress and inflammation. Nutr
Cancer. 66:249–258. 2014. View Article : Google Scholar
|
|
22
|
Kim BR, Seo HS, Ku JM, Kim G-J, Jeon CY,
Park JH, Jang BH, Park SJ, Shin YC and Ko SG: Silibinin inhibits
the production of pro-inflammatory cytokines through inhibition of
NF-kappaB signaling pathway in HMC-1 human mast cells. Inflamm Res.
62:941–950. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi YH, Jin GY, Guo HS, Piao HM, Li L, Li
GZ, Lin ZH and Yan GH: Silibinin attenuates allergic airway
inflammation in mice. Biochem Biophys Res Commun. 427:450–455.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuo FH and Jan TR: Silibinin attenuates
antigen-specific IgE production through the modulation of Th1/Th2
balance in ovalbumin-sensitized BALB/c mice. Phytomedicine.
16:271–276. 2009. View Article : Google Scholar
|
|
25
|
Schroder K and Tschopp J: The
Inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sutterwala FS, Ogura Y, Szczepanik M,
Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ,
Galán JE, Askenase PW, et al: Critical role for
NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its
regulation of caspase-1. Immunity. 24:317–327. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sorbara MT and Girardin SE: Mitochondrial
ROS fuel the inflammasome. Cell Res. 21:558–560. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jangra A, Kasbe P, Pandey SN, Dwivedi S,
Gurjar SS, Kwatra M, Mishra M, Venu AK, Sulakhiya K, Gogoi R, et
al: Hesperidin and silibinin ameliorate aluminum-induced
neurotoxicity: modulation of antioxidants and inflammatory
cytokines level in mice hippocampus. Biol Trace Elem Res.
168:462–471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan WJ, Tan YC, Xu JC, Tang XP, Zhang C,
Zhang PB and Ren ZQ: Protective effects of silibinin and its
possible mechanism of action in mice exposed to chronic
unpredictable mild stress. Biomol Ther (Seoul). 23:245–250. 2015.
View Article : Google Scholar
|
|
30
|
Duan S, Guan X, Lin R, Liu X, Yan Y, Lin
R, Zhang T, Chen X, Huang J, Sun X, et al: Silibinin inhibits
acetylcholinesterase activity and amyloid beta peptide aggregation:
A dual-target drug for the treatment of Alzheimer's disease.
Neurobiol Aging. 36:1792–1807. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Raghu R, Jesudas B, Bhavani G, Ezhilarasan
D and Karthikeyan S: Silibinin mitigates zidovudine-induced
hepatocellular degenerative changes, oxidative stress and
hyperlipidaemia in rats. Hum Exp Toxicol. 34:1031–1042. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Braun DL, Rauch A, Aouri M, Durisch N,
Eberhard N, Anagnostopoulos A, Ledergerber B, Müllhaupt B, Metzner
KJ, Decosterd L, et al: A lead-in with silibinin prior to
triple-therapy translates into favorable treatment outcomes in
difficult-to-treat HIV/hepatitis C coinfected patients. PLoS One.
10:e01330282015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheung CWY, Gibbons N, Johnson DW and
Nicol DL: Silibinin - A promising new treatment for cancer.
Anticancer Agents Med Chem. 10:186–195. 2010. View Article : Google Scholar
|
|
34
|
Momeny M, Malehmir M, Zakidizaji M,
Ghasemi R, Ghadimi H, Shokrgozar MA, Emami AH, Nafissi S,
Ghavamzadeh A and Ghaffari SH: Silibinin inhibits invasive
properties of human glioblastoma U87MG cells through suppression of
cathepsin B and nuclear factor kappa B-mediated induction of matrix
metalloproteinase 9. Anticancer Drugs. 21:252–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li L, Zeng J, Gao Y and He DL: Targeting
silibinin in the antiproliferative pathway. Expert Opin Investig
Drugs. 19:243–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hakansson HF, Smailagic A, Brunmark C,
Miller-Larsson A and Lal H: Altered lung function relates to
inflammation in an acute LPS mouse model. Pulm Pharmacol Ther.
25:399–406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mouratis MA, Magkrioti C, Oikonomou N,
Katsifa A, Prestwich GD, Kaffe E and Aidinis V: Autotaxin and
endotoxin-induced acute lung injury. PLoS One. 10:e01336192015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
No authors listed. Ventilation with lower
tidal volumes as compared with traditional tidal volumes for acute
lung injury and the acute respiratory distress syndrome. The Acute
Respiratory Distress Syndrome Network. N Engl J Med. 342:1301–1308.
2000. View Article : Google Scholar
|
|
39
|
McKenna S and Wright CJ: Inhibiting
IkappaBbeta-NFkappaB signaling attenuates the expression of select
pro-inflammatory genes. J Cell Sci. 128:2143–2155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee IT and Yang CM: Inflammatory
signalings involved in airway and pulmonary diseases. Mediators
Inflamm. 2013:7912312013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Alvira CM: Nuclear factor-kappa-B
signaling in lung development and disease: One pathway, numerous
functions. Birth Defects Res A Clin Mol Teratol. 100:202–216. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Petrilli V, Papin S and Tschopp J: The
inflammasome. Curr Biol. 15:R5812005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wen H, Ting JP and O'Neill LA: A role for
the NLRP3 inflammasome in metabolic diseases - did Warburg miss
inflammation? Nat Immunol. 13:352–357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shaw PJ, McDermott MF and Kanneganti TD:
Inflammasomes and autoimmunity. Trends Mol Med. 17:57–64. 2011.
View Article : Google Scholar :
|
|
45
|
Martinon F: Signaling by ROS drives
inflammasome activation. Eur J Immunol. 40:616–619. 2010.
View Article : Google Scholar : PubMed/NCBI
|