Stroke is the third‑leading cause of death,
following coronary heart disease and cancer, and the main cause of
permanent adult disablement in most Western countries (1). In China, it has become the first
leading cause of death in recent years (2). Generally, between two main types of
stroke, ischemic and hemorrhagic, ischemic stroke accounts for the
majority of cases (~80-90%) (3-5).
Ischemic injury often leads to irreversible cerebral infarction
depending on the location, severity, and duration of cerebral blood
flow (CBF) reduction, causing cognitive and motor impairment. Based
on the pathophysiological characteristics of ischemic stroke, there
are currently two major therapeutic strategies: Reperfusion and
neuroprotection. Using thrombolytic, antithrombotic and
anti‑aggregation agents, the blood flow of the compromised region
can be restored. However, only one drug, IV Alteplase, is
recommended to treat ischemic stroke, and the drug has a very short
therapeutic time window and a high risk of hemorrhagic
transformation, which severely limits its clinical use (6). The second therapeutic strategy,
neuroprotection, aims at preventing neuronal death by regulating
multiple intra- or extracellular signals that lead to cell injury.
The concept that ischemic penumbra is potentially salvageable is
the basis of numerous searches for neuroprotective medications that
can save the penumbra tissue and limit the negative consequences of
stroke (7). However, no drug with
proven neuroprotective efficiency and without harmful adverse side
effects has been discovered yet. Therefore, it is urgent to develop
novel drugs with potential and effective targets.
Inflammation is a defense response aiming at
eliminating the primary causes of cell damage. Although
inflammation aids in clearing infections and other toxic stimuli,
inflammatory responses in the cerebral tissue during an ischemic
injury increase the damage for a few hours following the onset of
cerebral ischemia (8). During
transient or permanent vascular obstruction, all the functions and
molecules in the neurons, glial cells (oligodendrocytes, microglia
and astrocytes) and vascular cells (endothelial cells and
pericytes), identified as components of the neurovascular unit, are
affected (9). Dangerous molecular
signals are released from the damaged cells following multiple
types of cellular stress (10).
These signals, including danger signals denominated
damage-associated molecular patterns (DAMPs) and
pathogen-associated molecular patterns (PAMPs), activate the innate
immune system via intracellular and extracellular pattern
recognition receptors (PRRs), which have been highlighted recently
as an important inflammatory mechanism that may contribute to
cerebral ischemic injury (11).
Inflammasomes are intracellular protein complexes associated with
innate immunity. Activated inflammasomes are able to cleave the
precursor of interleukin (IL)-1β and IL‑18 to mature forms and
initiate a newly discovered programmed inflammatory cell death,
pyroptosis, via cleaved caspase-1 (12). In specific, the nucleotide-binding
oligomerization domain (NOD)-like receptor (NLR) family pyrin
domain containing 3 (NLRP3), which is plentifully expressed in the
brain, is regarded as one of the predominant inflammasomes, due to
its critical role in recognizing cellular damage and modulating
inflammatory responses to ischemia reperfusion injury during
ischemic stroke (13). In the
current review, we survey the existing evidence for the structure
of NLRP3 inflammasome, its activation process in the ischemic brain
and the therapeutic potential of blocking or inhibiting NLRP3
inflammasome signaling.
The inflammasome was first proposed in 2002 as an
activator complex of caspase-1 that generates IL-1β via cleavage of
its preform (14). Inflammation
is now considered as an important innate immune reaction to
multiple types of infection and tissue injury (15). Two kinds of inflammasomes have
been established to date: Pyrin and HIN domain-containing protein
(PYHIN) inflammasomes and NLR inflammasomes (12). NLRP3 inflammasome is one of the
best characterized inflammasomes to date and the most relevant to
aseptic inflammation (16).
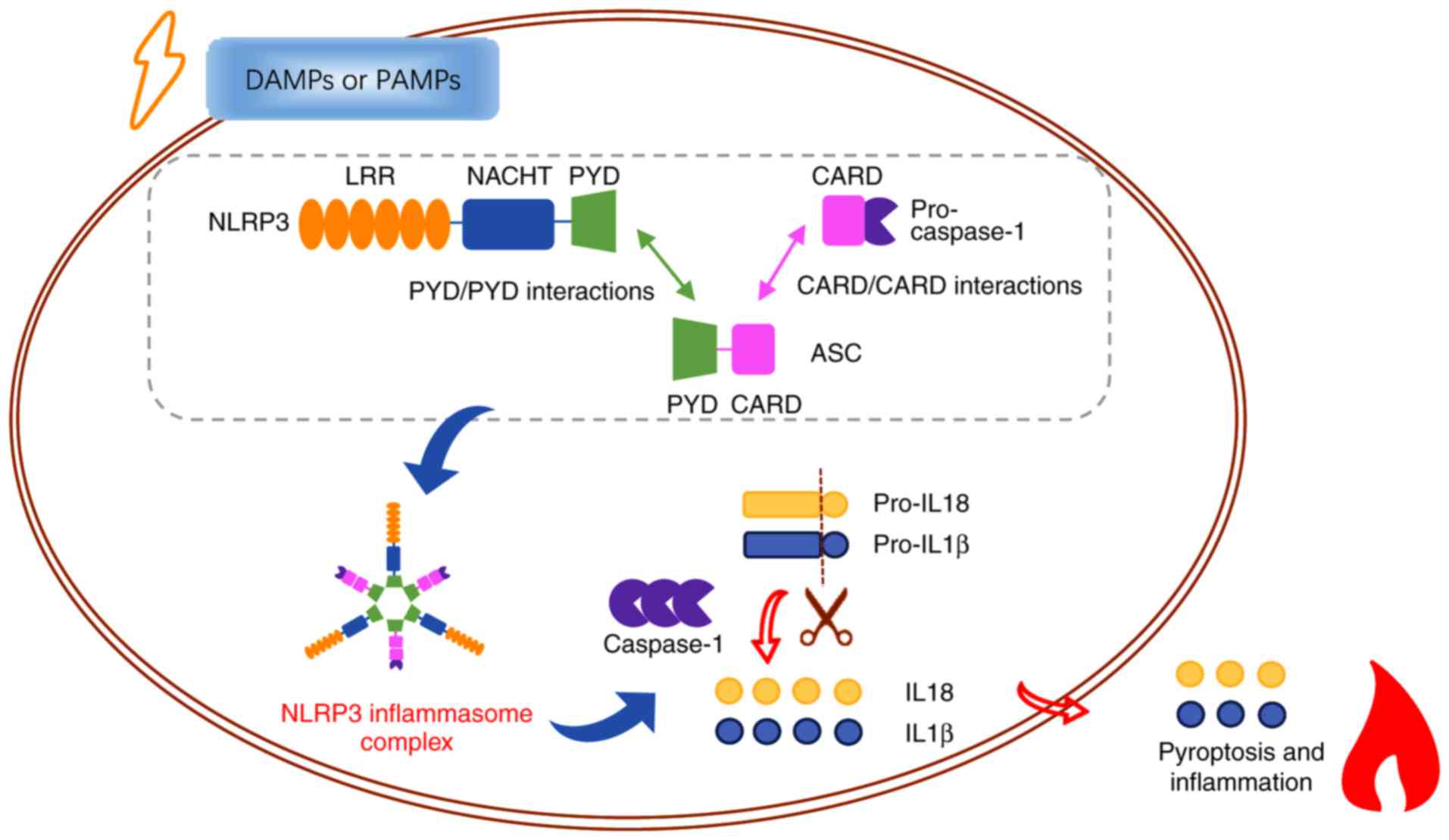
NLRP3, also known as NALP3, is encoded by the cold‑induced
auto‑inflammatory syndrome‑1 (CIAS‑1) gene and is
plentifully expressed in cells of the nervous and immune systems
(17). It is commonly comprised
of three domains: A C-terminal domain containing leucine-rich
repeats (LRRs), a central NACHT domain, and an N-terminal PYD. The
LRR domain is implicated in the progress of ligand sensing
(18,19). The NACHT domain is responsible for
oligomerization and assembly of the central core of the NLRP3
inflammasome, which depends on ATP (20). The N-terminal PYD domain
encourages homotypic PYD/PYD interactions with apoptosis‑associated
speck‑like protein containing CARD (ASC) (21,22), which comprises both PYD and CARD
domains and regulates inflammatory response and cell death
(23,24). Following danger signals,
activation of upstream signals and oligomerization of NLRP3 result
in the formation of NLRP3 inflammasome. The NLRP3 inflammasome
consists of three cytoplasmic proteins: NLRP3, ASC and the
precursor of caspase-1 (25,26). Once binding to NLRP3 via PYD
domain, ASC recruits the precursor of caspase-1 via homotypic
CARD/CARD interactions (22).
Subsequently, activated caspase‑1 is generated, which then cleaves
the precursor of proinflammatory cytokines IL‑1β and IL-18, and
results in the maturation and secretion of these cytokines,
inducing cellular pyroptosis (Fig.
1) (27). Recently, multiple
studies have reported that excessive activation of NLRP3
inflammasome is closely associated with pathophysiological changes
in a majority of inflammatory and non‑inflammatory illnesses,
including ischemic stroke (28-31).
An increasing number of studies have indicated a key
role of NLRP3 inflammasome in recognizing cellular damage and
modulating inflammatory responses that eventually result in cell
death (19,32). The NLRP3 inflammasome was first
associated with ischemic injury in an animal model of renal
ischemic injury, which occurs as blockage of blood flow (33,34). Following renal ischemic injury,
plenty of evidence proves that activation of NLRP3 is critical in
mediating myocardial and liver ischemic damage (35). In the central nervous system
(CNS), NLRP3 inflammasome was first reported to be activated in
cortical neurons under ischemic conditions and the expression of
NLRP3, ASC, caspase-1, IL-1β and IL-18 was upregulated in
vitro and in vivo (36). The latter study also demonstrated
that suppression of NLRP3 inflammasome activity and neuroprotection
resulted from intravenous immunoglobulin (IVIg) and anti‑caspase‑1
treatment, respectively (36).
Another study indicated that deficiency of the NLRP3 gene protected
mice from ischemic damage with improved functional outcomes,
decreased infarction volume and edema formation, preserved blood
brain barrier (BBB) permeability, and reduced inflammatory
pathology in a transient middle cerebral artery occlusion (tMCAO)
mouse model (37,38), which was first developed in 1986
to mimic ischemic stroke in patients by Koizumi (39). However, several researchers have
questioned the role of NLRP3 in the progress of cerebral ischemic
injury (40). The conflict
between these results could be attributed to differences in
ischemic time that may modify the inflammatory response.
After the NLRP3 inflammasome is activated,
caspase‑1, an evolutionarily conserved enzyme that proteolytically
cleaves other proteins, becomes mature. Following ischemic injury
in a permanent animal model of stroke, the levels of activated
caspase‑1 increase at 30 min following surgery, and a second wave
of activation comes 12 h later (41). The upregulated levels of cleaved
caspase-1 have been observed in neurons and astrocytes following
cerebral ischemia, and in microglia 24 h after a stroke (42). Using transgenic mice, the role of
caspase-1 has been highlighted in the progress of ischemic stroke.
Knockout or dominant-negative mutants of caspase-1 inhibited brain
damage in contrast to wild type, following experimental stroke
(43,44). The therapeutic effects of
caspase-1 molecule inhibitors have also been observed in an
oxygen/glucose deprivation model in rat hippocampal slices
(45). Collectively, these
studies demonstrate a critical role of caspase-1 during ischemic
stroke.
Several studies have demonstrated upregulated
concentrations of pro‑inflammatory factors in the blood,
cerebrospinal fluid, and location of blockages of the brain in both
clinical patients and experimental animals (46-49), suggesting a localized CNS
inflammatory response to ischemic injury. In pathological
conditions, premature IL-1β and IL-18 proteins without biological
function need to be processed and secreted to exert their
pro-inflammatory effects (50).
Secreted IL-1 in extracellular space has a direct impact on nearby
neurons via IL-1 receptors. High concentrations of IL-1β stimulates
phosphorylation and activation of the N‑methyl‑D‑aspartate (NMDA)
receptor, which induces excessive calcium flux and excitatory
toxicity (51).
The NLRP3 inflammasome complex, except for immune
organs, has also been recently found to be expressed in the brain
and spinal cord (47,50). Based on expression data from 11
types of tissues, it was demonstrated that the brain does not
express IL-1β and IL-18 constitutively, but expresses NLRP3
inflammasome in a constitutive state, indicating that the NLRP3
inflammasome can be assembled without upregulation of one or two
components (52). Expression of
the member proteins of the NLRP3 inflammasome complex and IL-1β and
IL-18 has been demonstrated to be upregulated in post-mortal brain
tissue from stroke patients. A higher level of activated caspase‑1
in ischemic brain tissues compared with control brain tissues from
patients was further confirmed by immunohistochemical analysis
(36,53). At the cellular level, similar to
bone marrow‑derived macrophages, microglia is the main cell type
that expresses NLRP3, ASC and caspase-1 in the brain. However,
neither NLRP3 nor ASC can be detected in astrocytes, which
highlights the important role of microglia‑dependent NLRP3
inflammasome activation under neuroinflammatory conditions
(54). In 2014, for the first
time, animals subjected to MCAO and cell cultures subjected to
oxygen-glucose deprivation (OGD) modeling ischemia/reper-fusion
injury in vivo and in vitro were used to confirm
NLRP3 expression in ischemic stroke. Another study has demonstrated
that NLRP3-related proteins are expressed in endotheliocytes and
microglia instead of neurons, suggesting that they are the main
sources of NLRP3 in the location of the ischemic incident (38). Others found that the levels of
NLRP3 inflam-masome proteins were also upregulated in primary
cortical neurons under ischemic injury (36). Although the specific expression
pattern of NLRP3-related proteins in different types of cells in
ischemic brain remains unclear, it is certain that NLRP3
inflammasome signaling has an important role in the pathogenesis of
ischemic stroke, at the neurovascular unit level. The underlying
causes for the differences in the distribution may due to diverse
models, ischemic duration and different interventions.
The specific intracellular and extracellular signals
leading to NLRP3 inflammasome activation are not yet fully
understood. Evidence has revealed that assembly of NLRP3
inflammasome and activation of downstream signals depend on two
complementary signals associated with cell injury: A priming
signal, required for upregulated expression of the NLRP3
inflammasome complex proteins and the precursors of IL-1β and IL-18
through nuclear factor (NF)-κB and mitogen‑activated protein kinase
(MAPK) signaling pathways (55);
and a second signal that results in NLRP3 activation and ASC
phosphorylation, thus triggering their assembly into the NLRP3
inflammasome complex (Fig. 2). In
addition, multiple activation mechanisms have been described for
the inflammasome, including K+ efflux, reactive oxygen
species (ROS) overproduction, mitochondrial dysfunction,
Ca2+ overload, and lysosome rupture. Notably, these
mechanisms also overlap with the two‑step activation of the NLRP3
inflammasome. It is well‑established that these mechanisms exist in
the progress of ischemic stroke. Several experts have confirmed
that reducing the ATP/ADP ratio following ischemic stroke opens the
channel and allows K+ ions to exit the cells via
potassium channels (56).
Mitochondrial dysfunction (57),
ROS overproduction (58) and
Ca2+ overload (59)
are also known to have important roles in the amplification and
transmission of ischemic injury.
NLRP3, pro-IL-1β and pro-IL-18 do not abound in
physiological conditions, and therefore a priming signal,
specifically the upregulation of NLRP3-related proteins, is
necessary for the activation of NLRP3 inflammasome. In addition,
several plasma membrane pattern recognition receptors (PRRs), such
as toll-like receptors (TLRs) and receptor for advanced glycation
end‑products (RAGE), and downstream adaptor proteins, such as
myeloid differentiation primary response gene 88 (MyD88) and tumor
necrosis factor receptor-associated factor 6 (TRAF6), may serve a
role in activation of two phosphorylation signaling pathways, NF‑κB
and MAPK (60–63). Both of the latter signaling
pathways are considered to regulate both the expression of the
member proteins of the NLRP3 inflammasome complex, and the
expression of the precursors of IL-1β and IL-18 during the
inflammatory response to specific cellular stress (64–66).
As aforementioned, after the activating priming
signal, ASC, an adaptor protein, recruits NLRP3 proteins and forms
the NLRP3 inflammasome complex. Of all the intracellular and
extracellular factors, the best-studied stimulus, K+
efflux, has been demonstrated to have a role in activation of the
NLRP3 inflammasome (67). This
non-selective K+ cation channel on the cell surface is
able to change intracellular ionic contents depending on ATP
binding and to activate downstream signals, inducing maturation and
secretion of IL-1β (68).
Previous studies have demonstrated that downregulated levels of
intracellular K+ are indispensable for activation of the
NLRP3 inflammasome pathway when compared with other known stimuli,
which highlighted the role of K+ efflux in this process.
P2X purinoceptor 7 (P2X7R) is one of the best‑characterized
receptors associated with K+ efflux, whose activation
might induce release of pro‑inflammatory cytokines and amplify
ischemic injury via ATP. Experimental evidence from P2X7R knockdown
mice confirmed the role of this receptor in NLRP3 inflammasome
response (69).
Mitochondrial damage is another important activation
mechanism of the NLRP3 inflammasome. The mitochondrion is a
double-membrane-bound intracellular organelle and the predominant
location in the cell that produces both energy and reactive oxygen
species (ROS) (70). Previous
research has indicated that high levels of ROS under multiple kinds
of cellular stress, particularly those produced by mitochondria
(mtROS), activates the NLRP3 inflammasome signal pathway (71‑74). In detail, high levels of ROS
induce the ROS scavenging protein thioredoxin resolving from
thioredoxin interacting/inhibiting protein (TXNIP), which then
directly binds with NLRP3 proteins and modulates its assembly via
oligomerization (75-77). Although several investigations
have demonstrated that TXNIP is necessary for NLRP3 inflammasome
assembly, a cell type‑specific modulation of TXNIP occurs, which
limits its mediation of inflammatory response ROS signaling pathway
activation to particular cell types (78-80). In addition to mtROS, dysfunctional
mitochondria also release mitochondrial DNA (mtDNA) into the
cytoplasm, directly inducing the assembly of NLRP3 inflammasome
complex via molecular self-association (81-83). Of note, appropriate amount of
nitric oxide, another kind of free radical, is able to trigger a
cascade to modulate mitochondrial permeability, limit mtROS
releases and suppress NLRP3 inflammasome activation (84,85).
Accumulating evidence indicates that the NLRP3
inflammasome has an important role in ischemic brain injury.
Targeting upstream or downstream of the NLRP3 inflammasome pathway
at the molecular level, including modulating protein expression,
assembly, activation and/or secretion, may have a promising
prospect in developing novel therapeutic agents for ischemic stroke
(Table I). Core proteins involved
in the NF-κB and MAPK pathways, proteins of the NLRP3 inflammasome
complex, plasma membrane receptors or channels, cytokines (IL-1β
and IL-18) and their receptors may serve as potential therapeutic
targets for ischemic stroke (19). A previous study focused on the
effect of Bay‑11‑7082 (an NF-κB inhibitor), SB 203580 (a P38‑MAPK
inhibitor), JNK Inhibitor V (a JNK inhibitor), and U‑0126 (an ERK
inhibitor) on a mice tMCAO model and in neurons undergoing OGD, and
demonstrated that all of these inhibitors protected neurons during
simulated ischemia, via attenuating the levels of NLRP3
inflammasome complex, and via inhibiting activation of NLRP3
inflammasome and maturation of IL‑1β and IL-18 (100). Probenecid, a pannexin 1
inhibitor, has also been found to induce astrocyte death and ROS
generation, attenuate expression levels of NLRP3 and inhibit the
extracellular release of IL-1β (101). MCC950 and glyburide, both NLRP3
oligomerization inhibitors, reduce infarction volume, neuronal
apoptosis, and neurological impairment, and have anti‑oxidative
stress and anti‑inflammatory effects, respectively (102-105). Recently, several natural
compounds, including resveratrol, paeoniflorin and sinomenine, were
also demonstrated to reduce cerebral infarction volume, decrease
brain water content, improve neurological scores and grip strength,
and prevent neuronal cell death following ischemic stroke, through
downregulating the expression of the components of the NLRP3
inflammasome and their downstream proteins and attenuating its
activation (106‑108). However, there is little clinical
data on the effects of these agents to date. Further studies are
required to examine the efficiency and safety of these agents in
the clinic in the future.
Although targeting the NLRP3 inflammasome pathway
rather than anti-ischemic systems may develop promising therapeutic
strategies to treat ischemic stroke, several aspects of this
potential treatment must be clarified. Firstly, the specific
expression pattern of NLRP3 activation in specific cell types and
brain regions following ischemic injury require further
investigation. Additionally, whether the upstream pathways are
different among various cell types remains to be elucidated.
Secondly, it is well known that whether inflammation will have
destructive or beneficial effects depends on the severity,
frequency, and duration of ischemic injury. It is possible that
early immune responses may aggravate ischemic injury, while late
responses may help to repair the damaged region. Therefore, early
inhibition of the NLRP3 inflamma-some pathway may be a beneficial
and reasonable choice. On the other hand, using animal models, some
anti‑neuroinflammation agents have been demonstrated to protect
ipsilateral brain against ischemic injury up to 7 days following
ischemic induction (109‑111),
which highlights the long‑term effect and application prospect of
immunotherapy. Of note, intermittent fasting has been reported to
attenuate NLRP3 pathway activity and repair damaged tissues up to 4
months following ischemic stroke (112). Future studies should focus on
the optimal timing of NLRP3 inhibitor administration and explain
the mechanisms behind damaging and protective actions of the immune
system in ischemic stroke.
Finally, after glyburide was first found to
selectively inhibit activation of the NLRP3 inflammasome without
interfering with other inflammasome pathways (113), two other selective and direct
NLRP3 inhibitors, CY-09 (114)
and CP‑456,773 (115,116), were recently identified. These
drugs might provide a novel avenue toward therapeutic inhibition of
the NLRP3 inflammasome, although their effect in ischemic stroke
remains to be determined. Many NLRP3 inhibitors found to have
benefits following ischemic stroke were identified as natural
compounds, and further research should elucidate the relationship
between molecular structure and anti‑NLRP3 inflammasome function.
Furthermore, several microRNAs have been demonstrated to influence
the NLRP3 inflamma-some pathway, including miR‑22 (117), miR‑132 (118), and long non-coding RNA
XLOC_000647 (119). Further
studies should consider the modulation of the NLRP3 inflammasome
post-stroke via epigenetic approaches.
In conclusion, illuminating the role of NLRP3
inflammasome signaling pathways in ischemic injury will provide
significant knowledge and opportunities to clarify the relationship
between the innate immune system and the nervous system in the
pathophysiology of ischemic stroke. Above all, more and larger
clinical studies examining the associations between clinical
symptoms and signs and the expression levels of NLRP3 inflammasome
network proteins in brain parenchyma and body fluids are urgently
needed. Research to identify and design compounds and therapeutic
strategies to target the NLRP3 inflammasome and modulate the
detrimental inflammatory processes without excessively disturbing
the immune defense progress may be essential for the treatment of
ischemic stroke. Extensive clinical trials are required to develop
safer and more effective agents targeting the NLRP3 inflammasome
pathway in humans.
Not applicable.
This review was supported by the National Natural
Science Foundation of China (grant nos. 81430102, 81774122,
81774030, 81373886 and 81303260) and the Beijing University of
Traditional Chinese Medicine Independent Subject Selection Project
(grant no. 2017‑JYB‑XS‑014).
Not applicable.
FC, XW, CM, SL, SZ, TX made substantial
contributions to the conception of the present study and wrote the
manuscript and critically reviewed it. XY, YG, CZ, CLe, CLi, SF,
HT, YC, QW contributed to searching related articles and reviewing
the article. All authors read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Feigin VL, Krishnamurthi RV, Parmar P,
Norrving B, Mensah GA, Bennett DA, Barker‑Collo S, Moran AE, Sacco
RL, Truelsen T, et al: Update on the global burden of ischemic and
hemorrhagic stroke in 1990‑2013: The GBD 2013 study.
Neuroepidemiology. 45:161–176. 2015. View Article : Google Scholar :
|
|
2
|
Wang W, Jiang B, Sun H, Ru X, Sun D, Wang
L, Jiang Y, Li Y, Wang Y, Chen Z, et al: Prevalence, incidence, and
mortality of stroke in chinaclinical perspective: Results from a
nationwide population-based survey of 480687 adults. Circulation.
135:759–771. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu L, Wang D, Wong KL and Wang Y: Stroke
and stroke care in china: Huge burden, significant workload, and a
national priority. Stroke. 42:3651–3654. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman
M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C,
et al: Heart disease and stroke statistics-2017 update: A report
from the american heart association. Circulation. 135:e146–e603.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goldstein LB, Adams R, Alberts MJ, Appel
LJ, Brass LM, Bushnell CD, Culebras A, Degraba TJ, Gorelick PB,
Guyton JR, et al: Primary prevention of ischemic stroke: A
guideline from the american heart association/american stroke
association stroke council: Cosponsored by the atherosclerotic
peripheral vascular disease interdisciplinary working group;
cardiovascular nursing council; clinical cardiology council;
nutrition, physical activity, and metabolism council; and the
quality of care and outcomes research interdisciplinary working
group: The amer-ican academy of neurology affirms the value of this
guideline. Stroke. 37:1583–1633. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Furie KL and Jayaraman MV: 2018 guidelines
for the early management of patients with acute ischemic stroke.
Stroke. 49:509–510. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Astrup J, Siesjö BK and Symon L:
Thresholds in cerebral ischemia-the ischemic penumbra. Stroke.
12:723–725. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu X, De Silva TM, Chen J and Faraci FM:
Cerebral vascular disease and neurovascular injury in ischemic
stroke. Circ Res. 120:449–471. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Famakin BM: The immune response to acute
focal cerebral isch-emia and associated post-stroke
immunodepression: A focused review. Aging Dis.
5:3073262014.PubMed/NCBI
|
|
10
|
Cai W, Zhang K, Li P, Zhu L, Xu J, Yang B,
Hu X, Lu Z and Chen J: Dysfunction of the neurovascular unit in
ischemic stroke and neurodegenerative diseases: An aging effect.
Ageing Res Rev. 34:77–87. 2017. View Article : Google Scholar :
|
|
11
|
Cuartero MI, Ballesteros I, Lizasoain I
and Moro MA: Complexity of the cell-cell interactions in the innate
immune response after cerebral ischemia. Brain Res. 1623:53–62.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hoseini Z, Sepahvand F, Rashidi B,
Sahebkar A, Masoudifar A and Mirzaei H: NLRP3 inflammasome: Its
regulation and involvement in atherosclerosis. J Cell Physiol.
233:2116–2132. 2018. View Article : Google Scholar
|
|
13
|
Elliott EI and Sutterwala FS: Initiation
and perpetuation of NLRP3 inflammasome activation and assembly.
Immunol Rev. 265:35–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martinon F, Burns K and Tschopp J: The
inflammasome: A molecular platform triggering activation of
inflammatory caspases and processing of proIL-beta. Mol Cell.
10:417–426. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Medzhitov R: Origin and physiological
roles of inflammation. Nature. 454:428–435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cassel SL and Sutterwala FS: Sterile
inflammatory responses mediated by the NLRP3 inflammasome. Eur J
Immunol. 40:607–611. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu SB, Mi WL and Wang YQ: Research
progress on the NLRP3 inflammasome and its role in the central
nervous system. Neurosci Bull. 29:7797872013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liao KC and Mogridge J: Expression of
Nlrp1b inflammasome components in human fibroblasts confers
susceptibility to anthrax lethal toxin. Infect Immun. 77:4455–4462.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fann DY, Lee SY, Manzanero S, Chunduri P,
Sobey CG and Arumugam TV: Pathogenesis of acute stroke and the role
of inflammasomes. Ageing Res Rev. 12:941–966. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Levinsohn JL, Newman ZL, Hellmich KA,
Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH and Moayeri M:
Anthrax lethal factor cleavage of Nlrp1 is required for activation
of the inflam-masome. PLoS Pathog. 8:e10026382012. View Article : Google Scholar
|
|
21
|
Gambin Y, Giles N, O'Carroll A,
Polinkovsky ME, Hunter DJ and Sierecki E: Single‑molecule
fluorescence reveals the oligomerisation and folding steps driving
the prion-like behaviour of ASC. J Mol Biol. 430:12632018.
View Article : Google Scholar
|
|
22
|
Srinivasula SM, Poyet JL, Razmara M, Datta
P, Zhang Z and Alnemri ES: The PYRIN-CARD protein ASC is an
activating adaptor for caspase‑1. J Biol Chem. 277:21119–21122.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi J, Gao W and Shao F: Pyroptosis:
Gasdermin-mediated programmed necrotic cell death. Trends Biochem
Sci. 42:245–254. 2017. View Article : Google Scholar
|
|
24
|
Inohara N and Nuñez G: Cell death and
immunity: NODs: Intracellular proteins involved in inflammation and
apoptosis. Nat Rev Immunol. 3:371–382. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Agostini L, Martinon F, Burns K, McDermott
MF, Hawkins PN and Tschopp J: NALP3 forms an IL-1beta‑processing
inflamma-some with increased activity in Muckle‑Wells
autoinflammatory disorder. Immunity. 20:319–325. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanneganti TD: Inflammatory bowel disease
and the NLRP3 inflammasome. N Engl J Med. 377:694–696. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ozaki E, Campbell M and Doyle SL:
Targeting the NLRP3 inflammasome in chronic inflammatory diseases:
Current perspectives. J Inflamm Res. 8:15–27. 2015.PubMed/NCBI
|
|
29
|
Alcocer‑Gómez E, Castejón‑Vega B and
Cordero MD: Stress‑induced NLRP3 inflammasome in human diseases.
Adv Protein Chem Struct Biol. 108:127–162. 2017. View Article : Google Scholar
|
|
30
|
Song L, Pei L, Yao S, Wu Y and Shang Y:
NLRP3 inflammasome in neurological diseases, from functions to
therapies. Front Cell Neurosci. 11:632017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Toldo S and Abbate A: The NLRP3
inflammasome in acute myocardial infarction. Nat Rev Cardiol.
15:203–214. 2018. View Article : Google Scholar
|
|
32
|
Pradillo JM, Denes A, Greenhalgh AD,
Boutin H, Drake C, McColl BW, Barton E, Proctor SD, Russell JC,
Rothwell NJ, et al: Delayed administration of interleukin-1
receptor antagonist reduces ischemic brain damage and inflammation
in comorbid rats. J Cereb Blood Flow Metab. 32:1810–1819. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iyer SS, Pulskens WP, Sadler JJ, Butter
LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA,
Leemans JC and Sutterwala FS: Necrotic cells trigger a sterile
inflammatory response through the Nlrp3 inflammasome. Proc Natl
Acad Sci USA. 106:20388–20393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shigeoka AA, Mueller JL, Kambo A, Mathison
JC, King AJ, Hall WF, Correia Jda S, Ulevitch RJ, Hoffman HM and
McKay DB: An inflammasome-independent role for epithelial-expressed
Nlrp3 in renal ischemia-reperfusion injury. J Immunol.
185:6277–6285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Leemans JC, Cassel SL and Sutterwala FS:
Sensing damage by the NLRP3 inflammasome. Immunol Rev.
243:1521622011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fann DY, Lee SY, Manzanero S, Tang SC,
Gelderblom M, Chunduri P, Bernreuther C, Glatzel M, Cheng YL,
Thundyil J, et al: Intravenous immunoglobulin suppresses NLRP1 and
NLRP3 inflammasome‑mediated neuronal death in ischemic stroke. Cell
Death Dis. 4:e7902013. View Article : Google Scholar
|
|
37
|
Dong Y, Fan C, Hu W, Jiang S, Ma Z, Yan X,
Deng C, Di S, Xin Z, Wu G, et al: Melatonin attenuated early brain
injury induced by subarachnoid hemorrhage via regulating NLRP3
inflammasome and apoptosis signaling. J Pineal Res. 60:253–262.
2016. View Article : Google Scholar
|
|
38
|
Yang F, Wang Z, Wei X, Han H, Meng X,
Zhang Y, Shi W, Li F, Xin T, Pang Q, et al: NLRP3 deficiency
ameliorates neurovascular damage in experimental ischemic stroke. J
Cereb Blood Flow Metab. 34:660–667. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koizumi J: Experimental studies of
ischemic brain edema 1 A new experimental model of cerebral
embolism in rats in which recirculation can be introduced in the
ischemic area. Jpn J Stroke. 8:1–8. 1986. View Article : Google Scholar
|
|
40
|
Denes A, Coutts G, Lénárt N, Cruickshank
SM, Pelegrin P, Skinner J, Rothwell N, Allan SM and Brough D: AIM2
and NLRC4 inflammasomes contribute with ASC to acute brain injury
independently of NLRP3. Proc Natl Acad Sci USA. 112:4050–4055.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Benchoua A, Guégan C, Couriaud C, Hosseini
H, Sampaïo N, Morin D and Onténiente B: Specific caspase pathways
are activated in the two stages of cerebral infarction. J Neurosci.
21:7127–7134. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Abulafia DP, de Rivero Vaccari JP, Lozano
JD, Lotocki G, Keane RW and Dietrich WD: Inhibition of the
inflammasome complex reduces the inflammatory response after
thromboembolic stroke in mice. J Cereb Blood Flow Metab.
29:534–544. 2009. View Article : Google Scholar
|
|
43
|
Friedlander RM, Gagliardini V, Hara H,
Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA
and Yuan J: Expression of a dominant negative mutant of
interleukin-1beta converting enzyme in transgenic mice prevents
neuronal cell death induced by trophic factor withdrawal and
ischemic brain injury. J Exp Med. 185:933–940. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schielke GP, Yang GY, Shivers BD and Betz
AL: Reduced ischemic brain injury in interleukin-1beta converting
enzyme-deficient mice. J Cereb Blood Flow Metab. 18:180–185. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ray AM, Owen DE, Evans ML, Davis JB and
Benham CD: Caspase inhibitors are functionally neuroprotective
against oxygen glucose deprivation induced CA1 death in rat
organotypic hippocampal slices. Brain Res. 867:62–69. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mathiesen T, Edner G, Ulfarsson E and
Andersson B: Cerebrospinal fluid interleukin-1 receptor antagonist
and tumor necrosis factor-α following subarachnoid hemorrhage. J
Neurosurg. 87:215–220. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Iadecola C and Anrather J: The immunology
of stroke: From mechanisms to translation. Nat Med. 17:796–808.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mathiesen T, Andersson B, Loftenius A and
von Holst H: Increased interleukin‑6 levels in cerebrospinal fluid
following subarachnoid hemorrhage. J Neurosurg. 78:562–567. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mizuma A and Yenari MA: Anti‑inflammatory
targets for the treatment of reperfusion injury in stroke. Front
Neurol. 8:4672017. View Article : Google Scholar
|
|
50
|
Creagh EM: Caspase crosstalk: Integration
of apoptotic and innate immune signalling pathways. Trends Immunol.
35:631–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Galea J and Brough D: The role of
inflammation and interleukin‑1 in acute cerebrovascular disease. J
Inflamm Res. 6:121–128. 2013.PubMed/NCBI
|
|
52
|
Yin Y, Yan Y, Jiang X, Mai J, Chen NC,
Wang H and Yang XF: Inflammasomes are differentially expressed in
cardiovascular and other tissues. Int J Immunopathol Pharmacol.
22:311–322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gao L, Dong Q, Song Z, Shen F, Shi J and
Li Y: NLRP3 inflammasome: A promising target in ischemic stroke.
Inflamm Res. 66:17–24. 2017. View Article : Google Scholar
|
|
54
|
Gustin A, Kirchmeyer M, Koncina E, Felten
P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P and Dostert C:
NLRP3 inflammasome is expressed and functional in mouse brain
microglia but not in astrocytes. PLoS One. 10:e01306242015.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Abais JM, Xia M, Zhang Y, Boini KM and Li
PL: Redox regulation of NLRP3 inflammasomes: ROS as trigger or
effector? Antioxid Redox Signal. 22:1111–1129. 2015. View Article : Google Scholar :
|
|
56
|
Sun HS and Feng ZP: Neuroprotective role
of ATP-sensitive potassium channels in cerebral ischemia. Acta
Pharmacol Sin. 34:24–32. 2013. View Article : Google Scholar
|
|
57
|
He J, Gao Y, Wu G, Lei X, Zhang Y, Pan W
and Yu H: Bioinformatics analysis of microarray data to reveal the
pathogenesis of brain ischemia. Mol Med Rep. 18:333–341.
2018.PubMed/NCBI
|
|
58
|
Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu
W, Bennett MVL and Chen J: Oxidative stress and DNA damage after
cerebral ischemia: Potential therapeutic targets to preserve the
genome and improve stroke recovery. Neuropharmacology. 134:208–217.
2018. View Article : Google Scholar
|
|
59
|
Buendia I, Tenti G, Michalska P,
Méndez‑López I, Luengo E, Satriani M, Padín-Nogueira F, López MG,
Ramos MT, García AG, et al: ITH14001, a CGP37157-nimodipine hybrid
designed to regulate calcium homeostasis and oxidative stress,
exerts neuroprotection in cerebral ischemia. ACS Chem Neurosci.
8:67–81. 2017. View Article : Google Scholar
|
|
60
|
Burm SM, Zuiderwijk‑Sick EA, 't Jong AE,
van der Putten C, Veth J, Kondova I and Bajramovic JJ:
Inflammasome‑induced IL-1β secretion in microglia is characterized
by delayed kinetics and is only partially dependent on inflammatory
caspases. J Neurosci. 35:678–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Frank MG, Weber MD, Watkins LR and Maier
SF: Stress sounds the alarmin: The role of the danger-associated
molecular pattern HMGB1 in stress‑induced neuroinflammatory
priming. Brain Behav Immun. 48:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lee HM, Kang J, Lee SJ and Jo EK:
Microglial activation of the NLRP3 inflammasome by the priming
signals derived from macrophages infected with mycobacteria. Glia.
61:441–452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nagyőszi P, Nyúl‑Tóth Á, Fazakas C,
Wilhelm I, Kozma M, Molnár J, Haskó J and Krizbai IA: Regulation of
NOD-like receptors and inflammasome activation in cerebral
endothelial cells. J Neurochem. 135:551–564. 2015. View Article : Google Scholar
|
|
64
|
Bauernfeind F, Bartok E, Rieger A, Franchi
L, Núñez G and Hornung V: Cutting edge: Reactive oxygen species
inhibitors block priming, but not activation, of the NLRP3
inflammasome. J Immunol. 187:613–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
He Q, You H, Li XM, Liu TH, Wang P and
Wang BE: HMGB1 promotes the synthesis of pro-IL-1β and pro-IL-18 by
activation of p38 MAPK and NF-κB through receptors for advanced
glycation end-products in macrophages. Asian Pac J Cancer Prev.
13:1365–1370. 2012. View Article : Google Scholar
|
|
66
|
Liu HD, Li W, Chen ZR, Hu YC, Zhang DD,
Shen W, Zhou ML, Zhu L and Hang CH: Expression of the NLRP3
inflammasome in cerebral cortex after traumatic brain injury in a
rat model. Neurochem Res. 38:2072–2083. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Muñoz‑Planillo R, Kuffa P, Martínez‑Colón
G, Smith BL, Rajendiran TM and Núñez G: K+ efflux is the
common trigger of NLRP3 inflammasome activation by bacterial toxins
and particulate matter. Immunity. 38:1142–1153. 2013. View Article : Google Scholar
|
|
68
|
Adinolfi E, Giuliani AL, De Marchi E,
Pegoraro A, Orioli E and Di Virgilio F: The P2X7 receptor: A main
player in inflammation. Biochem Pharmacol. 151:234–244. 2018.
View Article : Google Scholar
|
|
69
|
Pétrilli V, Papin S, Dostert C, Mayor A,
Martinon F and Tschopp J: Activation of the NALP3 inflammasome is
triggered by low intracellular potassium concentration. Cell Death
Differ. 14:1583–1589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Peng TI and Jou MJ: Oxidative stress
caused by mitochondrial calcium overload. Ann NY Acad Sci.
1201:183–188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Heid ME, Keyel PA, Kamga C, Shiva S,
Watkins SC and Salter RD: Mitochondrial reactive oxygen species
induces NLRP3‑dependent lysosomal damage and inflammasome
activation. J Immunol. 191:5230–5238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sorbara MT and Girardin SE: Mitochondrial
ROS fuel the inflammasome. Cell Res. 21:558–560. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
74
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar
|
|
75
|
Yin Y, Zhou Z, Liu W, Chang Q, Sun G and
Dai Y: Vascular endothelial cells senescence is associated with
NOD‑like receptor family pyrin domain-containing 3 (NLRP3)
inflammasome activation via reactive oxygen species
(ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int J
Biochem Cell Biol. 84:22–34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ye X, Zuo D, Yu L, Zhang L, Tang J, Cui C,
Bao L, Zan K, Zhang Z, Yang X, et al: ROS/TXNIP pathway contributes
to thrombin induced NLRP3 inflammasome activation and cell
apoptosis in microglia. Biochem Biophys Res Commun. 485:499–505.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yang SJ, Shao GF, Chen JL and Gong J: The
NLRP3 inflammasome: An important driver of neuroinflammation in
hemorrhagic stroke. Cell Mol Neurobiol. 38:595–603. 2018.
View Article : Google Scholar
|
|
78
|
Wang W, Wang C, Ding XQ, Pan Y, Gu TT,
Wang MX, Liu YL, Wang FM, Wang SJ and Kong LD: Quercetin and
allopurinol reduce liver thioredoxininteracting protein to
alleviate inflammation and lipid accumulation in diabetic rats. Br
J Pharmacol. 169:1352–1371. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
El-Azab MF, Baldowski BR, Mysona BA,
Shanab AY, Mohamed IN, Abdelsaid MA, Matragoon S, Bollinger KE,
Saul A and El‑Remessy AB: Deletion of thioredoxininteracting
protein preserves retinal neuronal function by preventing
inflammation and vascular injury. Br J Pharmacol. 171:1299–1313.
2014. View Article : Google Scholar :
|
|
80
|
Mohamed IN, Hafez SS, Fairaq A, Ergul A,
Imig JD and El‑Remessy AB: Thioredoxin‑interacting protein is
required for endothelial NLRP3 inflammasome activation and cell
death in a rat model of high-fat diet. Diabetologia. 57:413–423.
2014. View Article : Google Scholar
|
|
81
|
Ip WE and Medzhitov R: Macrophages monitor
tissue osmolarity and induce inflammatory response through NLRP3
and NLRC4 inflammasome activation. Nat Commun. 6:69312015.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
West AP and Shadel GS: Mitochondrial DNA
in innate immune responses and inflammatory pathology. Nat Rev
Immunol. 17:363–375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Gurung P, Lukens JR and Kanneganti TD:
Mitochondria: Diversity in the regulation of the NLRP3
inflammasome. Trends Mol Med. 21:193–201. 2015. View Article : Google Scholar
|
|
84
|
Bogdan C: Nitric oxide synthase in innate
and adaptive immunity: An update. Trends Immunol. 36:161–178. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Man SM and Kanneganti TD: Regulation of
inflammasome activation. Immunol Rev. 265:6–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Brough D, Le Feuvre RA, Wheeler RD,
Solovyova N, Hilfiker S, Rothwell NJ and Verkhratsky A:
Ca2+ stores and Ca2+ entry differentially
contribute to the release of IL-1β and IL-1α from murine
macrophages. J Immunol. 170:3029–3036. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Murakami T, Ockinger J, Yu J, Byles V,
McColl A, Hofer AM and Horng T: Critical role for calcium
mobilization in activation of the NLRP3 inflammasome. Proc Natl
Acad Sci USA. 109:11282–11287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Horng T: Calcium signaling and
mitochondrial destabilization in the triggering of the NLRP3
inflammasome. Trends Immunol. 35:253–261. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Clapham DE: Calcium signaling. Cell.
131:1047–1058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Humeau J, Bravo‑San Pedro JM, Vitale I,
Nuñez L, Villalobos C, Kroemer G and Senovilla L: Calcium signaling
and cell cycle: Progression or death. Cell Calcium. 70:3152018.
View Article : Google Scholar
|
|
91
|
Lee GS, Subramanian N, Kim AI,
Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner
DL and Chae JJ: The calcium‑sensing receptor regulates the NLRP3
inflammasome through Ca2+ and cAMP. Nature. 492:123–127.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Rizzuto R, Brini M, Murgia M and Pozzan T:
Microdomains with high Ca2+ close to IP3-sensitive
channels that are sensed by neighboring mitochondria. Science.
262:744–747. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Duchen MR: Mitochondria and calcium: From
cell signalling to cell death. J Physiol. 529:57–68. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Eisenbarth SC, Colegio OR, O'Connor W,
Sutterwala FS and Flavell RA: Crucial role for the Nalp3
inflammasome in the immunostimulatory properties of aluminium
adjuvants. Nature. 453:1122–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Deng D, Jiang N, Hao SJ, Sun H and Zhang
GJ: Loss of membrane cholesterol influences lysosomal permeability
to potassium ions and protons. Biochim Biophys Acta. 1788:470–476.
2009. View Article : Google Scholar
|
|
97
|
Compan V, Baroja‑Mazo A, López‑Castejón G,
Gomez AI, Martínez CM, Angosto D, Montero MT, Herranz AS, Bazán E,
Reimers D, et al: Cell volume regulation modulates NLRP3
inflammasome activation. Immunity. 37:487–500. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Okada M, Matsuzawa A, Yoshimura A and
Ichijo H: The lysosome rupture‑activated TAK1‑JNK pathway regulates
NLRP3 inflammasome activation. J Biol Chem. 289:32926–32936. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yaron JR, Gangaraju S, Rao MY, Kong X,
Zhang L, Su F, Tian Y, Glenn HL and Meldrum DR: K+
regulates Ca2+ to drive inflammasome signaling: Dynamic
visualization of ion flux in live cells. Cell Death Dis.
6:e19542015. View Article : Google Scholar
|
|
100
|
Fann DY, Lim YA, Cheng YL, Lok KZ,
Chunduri P, Baik SH, Drummond GR, Dheen ST, Sobey CG, Jo DG, et al:
Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome
activation in neurons following ischemic stroke. Mol Neurobiol.
55:1082–1096. 2018. View Article : Google Scholar
|
|
101
|
Jian Z, Ding S, Deng H, Wang J, Yi W, Wang
L, Zhu S, Gu L and Xiong X: Probenecid protects against
oxygen-glucose deprivation injury in primary astrocytes by
regulating inflammasome activity. Brain Res. 1643:123–129. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Ye X, Shen T, Hu J, Zhang L, Zhang Y, Bao
L, Cui C, Jin G, Zan K, Zhang Z, et al: Purinergic 2X7
receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic
stroke in the mouse. Exp Neurol. 292:46–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Murthy P, Durco F, Miller-Ocuin JL,
Takedai T, Shankar S, Liang X, Liu X, Cui X, Sachdev U, Rath D, et
al: The NLRP3 inflammasome and bruton's tyrosine kinase in
platelets co‑regulate platelet activation, aggregation, and in
vitro thrombus formation. Biochem Biophys Res Commun. 483:230–236.
2017. View Article : Google Scholar
|
|
104
|
Peng J, Deng X, Huang W, Yu JH, Wang JX,
Wang JP, Yang SB, Liu X, Wang L, Zhang Y, et al: Irisin protects
against neuronal injury induced by oxygen-glucose deprivation in
part depends on the inhibition of ROS-NLRP3 inflammatory signaling
pathway. Mol Immunol. 91:185–194. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ismael S, Zhao L, Nasoohi S and Ishrat T:
Inhibition of the NLRP3‑inflammasome as a potential approach for
neuroprotection after stroke. Sci Rep. 8:59712018. View Article : Google Scholar
|
|
106
|
He Q, Li Z, Wang Y, Hou Y, Li L and Zhao
J: Resveratrol alleviates cerebral ischemia/reperfusion injury in
rats by inhibiting NLRP3 inflammasome activation through
Sirt1-dependent autophagy induction. Int Immunopharmacol.
50:208–215. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
He YB, Nan LH, Huang M, Zheng YF, Yang L,
Xu W and Chu KD: Paeoniflorin down‑regulates the expression of
NLRP1 and NLRP3 inflammasomes in rat hippocampal slices after
oxygen-glucose deprivation. Int J Clin Exp Med. 9:10907–10914.
2016.
|
|
108
|
Qiu J, Wang M, Zhang J, Cai Q, Lu D, Li Y,
Dong Y, Zhao T and Chen H: The neuroprotection of Sinomenine
against ischemic stroke in mice by suppressing NLRP3 inflammasome
via AMPK signaling. Int Immunopharmacol. 40:492–500. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Rabuffetti M, Sciorati C, Tarozzo G,
Clementi E, Manfredi AA and Beltramo M: Inhibition of
caspase‑1‑like activity by Ac‑Tyr‑Val‑Ala‑Asp‑chloromethyl ketone
induces long‑lasting neuroprotection in cerebral ischemia through
apoptosis reduction and decrease of proinflammatory cytokines. J
Neurosci. 20:4398–4404. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ross J, Brough D, Gibson RM, Loddick SA
and Rothwell NJ: A selective, non‑peptide caspase‑1 inhibitor,
VRT‑018858, markedly reduces brain damage induced by transient
ischemia in the rat. Neuropharmacology. 53:638–642. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lu Y, Xiao G and Luo W: Minocycline
suppresses NLRP3 inflammasome activation in experimental ischemic
stroke. Neuroimmunomodulation. 23:230–238. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Fann DY, Santro T, Manzanero S,
Widiapradja A, Cheng YL, Lee SY, Chunduri P, Jo DG, Stranahan AM,
Mattson MP and Arumugam TV: Intermittent fasting attenuates
inflammasome activity in ischemic stroke. Exp Neurol. 257:114–119.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Lamkanfi M, Mueller JL, Vitari AC, Misaghi
S, Fedorova A, Deshayes K, Lee WP, Hoffman HM and Dixit VM:
Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Boil.
187:61–70. 2009. View Article : Google Scholar
|
|
114
|
Jiang H, He H, Chen Y, Huang W, Cheng J,
Ye J, Wang A, Tao J, Wang C, Liu Q, et al: Identification of a
selective and direct NLRP3 inhibitor to treat inflammatory
disorders. J Exp Med. 214:3219–3238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Primiano MJ, Lefker BA, Bowman MR, Bree
AG, Hubeau C, Bonin PD, Mangan M, Dower K, Monks BG, Cushing L, et
al: Efficacy and pharmacology of the NLRP3 inflammasome inhibitor
CP-456,773 (CRID3) in murine models of dermal and pulmonary
inflammation. J Immunol. 197:2421–2433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Coll RC, Robertson A, Butler M, Cooper M
and O'Neill LA: The cytokine release inhibitory drug CRID3 targets
ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS One.
6:e295392011. View Article : Google Scholar
|
|
117
|
Feng X, Luo Q, Wang H, Zhang H and Chen F:
MicroRNA-22 suppresses cell proliferation, migration and invasion
in oral squamous cell carcinoma by targeting NLRP3. J Cell Physiol.
233:6705–6713. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Byeon HE, Jeon JY, Kim HJ, Kim DJ, Lee KW,
Kang Y and Han SJ: MicroRNA-132 negatively regulates
palmitate-induced NLRP3 inflammasome activation through FOXO3
down‑regulation in THP-1 cells. Nutrients. 9:E13702017. View Article : Google Scholar
|
|
119
|
Hu H, Wang Y, Ding X, He Y, Lu Z, Wu P,
Tian L, Yuan H, Liu D, Shi G, et al: Long non-coding RNA
XLOC_000647 suppresses progression of pancreatic cancer and
decreases epithelial-mesenchymal transition-induced cell invasion
by down‑regulating NLRP3. Mol Cancer. 17:182018. View Article : Google Scholar
|
|
120
|
Qin YY, Li M, Feng X, Wang J, Cao L, Shen
XK, Chen J, Sun M, Sheng R, Han F and Qin ZH: Combined NADPH and
the NOX inhibitor apocynin provides greater anti-inflammatory and
neuroprotective effects in a mouse model of stroke. Free Radic Biol
Med. 104:333–345. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhao AP, Dong YF, Liu W, Gu J and Sun XL:
Nicorandil inhibits inflammasome activation and toll‑like
receptor‑4 signal transduction to protect against oxygen-glucose
deprivation-induced inflammation in BV‑2 cells. CNS Neurosci Ther.
20:147–153. 2014. View Article : Google Scholar
|
|
122
|
Li C, Wang J, Fang Y, Liu Y, Chen T, Sun
H, Zhou XF and Liao H: Nafamostat mesilate improves function
recovery after stroke by inhibiting neuroinflammation in rats.
Brain Behav Immun. 56:230–245. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Cao G, Jiang N, Hu Y, Zhang Y, Wang G, Yin
M, Ma X, Zhou K, Qi J, Yu B, et al: Ruscogenin attenuates cerebral
ischemia-induced blood-brain barrier dysfunction by suppressing
TXNIP/NLRP3 inflammasome activation and the MAPK pathway. Int J Mol
Sci. 17:E14182016. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Wang X, Li R, Wang X, Fu Q and Ma S:
Umbelliferone ameliorates cerebral ischemia-reperfusion injury via
upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3
inflammasome. Neurosci Lett. 600:182–187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Li Y, Li J, Li S, Wang X, Liu B, Fu Q and
Ma S: Curcumin attenuates glutamate neurotoxicity in the
hippocampus by suppression of ER stress‑associated TXNIP/NLRP3
inflamma-some activation in a manner dependent on AMPK. Toxicol
Appl Pharmacol. 286:53–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Zhang N, Zhang X, Liu X, Wang H, Xue J, Yu
J, Kang N and Wang X: Chrysophanol inhibits NALP3 inflammasome
activation and ameliorates cerebral ischemia/reperfusion in mice.
Mediators Inflamm. 2014:3705302014. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Thakkar R, Wang R, Sareddy G, Wang J,
Thiruvaiyaru D, Vadlamudi R, Zhang Q and Brann D: NLRP3
inflammasome activation in the brain after global cerebral ischemia
and regulation by 17β-estradiol. Oxid Med Cell Longev.
2016:83090312016. View Article : Google Scholar
|
|
128
|
Lammerding L, Slowik A, Johann S, Beyer C
and Zendedel A: Poststroke inflammasome expression and regulation
in the peri-infarct area by gonadal steroids after transient focal
ischemia in the rat brain. Neuroendocrinology. 103:460–475. 2016.
View Article : Google Scholar
|
|
129
|
Zhang S, Jiang L, Che F, Lu Y, Xie Z and
Wang H: Arctigenin attenuates ischemic stroke via SIRT1-dependent
inhibition of NLRP3 inflammasome. Biochem Biophys Res Commun.
493:821–826. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Yu C, He Q, Zheng J, Li LY, Hou YH and
Song FZ: Sulforaphane improves outcomes and slows cerebral
ischemic/reperfusion injury via inhibition of NLRP3 inflammasome
activation in rats. Int Immunopharmacol. 45:74–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Li M, Li H, Fang F, Deng X and Ma S:
Astragaloside IV attenuates cognitive impairments induced by
transient cerebral ischemia and reperfusion in mice via
anti‑inflammatory mechanisms. Neurosci Lett. 639:114–119. 2017.
View Article : Google Scholar
|
|
132
|
Ishrat T, Mohamed IN, Pillai B, Soliman S,
Fouda AY, Ergul A, El‑Remessy AB and Fagan SC:
Thioredoxin-interacting protein: A novel target for neuroprotection
in experimental thromboembolic stroke in mice. Mol Neurobiol.
51:766–778. 2015. View Article : Google Scholar
|