Introduction
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL), a member of the tumor necrosis factor cytokine
family, selectively induces cancer cell death by binding to two
death domain-containing receptors, TRAIL-receptor 1
(TRAIL-R1)/death receptor (DR) 4 and TRAIL-R2/DR5 (1,2).
Binding of TRAIL to TRAIL-R1 or TRAIL-R2 expressed on the cell
surface initiates the extrinsic apoptotic pathway, in which
caspase-8 plays a key role (3,4).
Active caspase-8 directly activates the effector caspase-3, -6 and
-7 that execute the apoptotic process. Caspase-8 can also engage
the intrinsic (mitochondrial) apoptotic pathway by cleaving and
activating the pro-apoptotic Bcl-2-family molecule Bid (5). Truncated Bid activates Bax and Bak,
leading to their oligomerization and pore formation in the outer
mitochondrial membrane through which cytochrome c is
released into the cytosol. The released cytochrome c binds
to another pro-apoptotic protein Apaf-1, leading to assembly of the
apoptosome and the resulting activation of caspase-9 (6). Caspase-9 also activates caspase-3, -6
and -7, thereby providing a positive feedback loop to the
caspase-8-induced apoptotic events. In a few type I cancer cells,
the extrinsic pathway is sufficient to commit the cells to
apoptosis, while in most type II cancer cells, the activation of
caspase-8 is low and amplification by the intrinsic mitochondrial
pathway is necessary to evoke substantial apoptosis (7).
Since TRAIL induces apoptosis in a variety of
transformed and cancer cells, but not in normal cells, it is
promising for cancer treatment. However, some cancer cell types,
such as malignant melanoma, glioma, osteosarcoma and non-small cell
lung cancer cells, are resistant to TRAIL-induced apoptosis despite
expression of the death-inducing TRAIL-Rs on their cell surface
(8). Moreover, TRAIL-responsive
tumors acquire a resistant phenotype that renders TRAIL therapy
ineffective. Therefore, overcoming the TRAIL-resistance of cancer
cells is necessary for effective TRAIL therapy, and drugs that can
potentiate TRAIL effectiveness are urgently required.
Depolarization has been shown to be an early event
in the apoptosis induced by diverse agents, including Fas (9), rotenone (ROT) (10) and arsenic trioxide (11), and is considered to play an
important pro-apoptotic role. On the contrary, depolarization has
also been shown to exhibit anti-apoptotic effects. Various
membrane-depolarizing agents, including ouabain, tetraethylammonium
(TEA) and veratridine, protect Purkinje cells against apoptosis
(12). These observations suggest
that depolarization can act in both pro-apoptotic and
anti-apoptotic manners depending on the cell types and apoptotic
stimuli involved. However, the cellular and molecular mechanisms
underlying these dual functions are unclear. Compared with other
DRs, the role of depolarization in TRAIL-induced apoptosis is
poorly documented. We previously showed that robust depolarization
is an early event during TRAIL-induced apoptosis in human melanoma
cells. Moreover, membrane-depolarizing agents including
K+ and ATP-sensitive potassium (KATP) channel
inhibitors such as glibenclamide (GLB) and U37883A (U37) markedly
potentiated TRAIL-induced apoptosis (13). This depolarization-mediated
potentiation of apoptosis was associated with upregulation of the
mitochondrial death pathway and endolasmic reticulum (ER)
stress-mediated death pathway involving caspase-12. Strikingly,
melanocytes were insensitive to TRAIL-induced depolarization and
apoptosis as well as the potentiation by membrane-depolarizing
drugs (13). These observations
suggest a tumor-selective role of depolarization in regulating
apoptosis. However, it remains to be elucidated whether this effect
of depolarization is characteristic of melanoma cells or a general
feature of different tumor cell types and how depolarization
affects these two death pathways. In the present study, we
addressed these questions by performing similar experiments in
human Jurkat leukemia cells and A549 lung cancer cells. In
addition, we examined the possible role of mitochondria-derived
reactive oxygen species (mROS) in the potentiation of apoptosis,
since our previous study showed that mROS mediated mitochondrial
and ER dysfunctions in Jurkat cells during TRAIL-induced apoptosis
(14). The results showed that the
previous observations in melanoma cells can essentially be expanded
to other tumor cells with different origins. Moreover, we found
that depolarization and mROS mutually control one another.
Importantly, our results suggest a positive loop between
depolarization and mROS through DR5 expression.
Materials and methods
Reagents
Soluble recombinant human TRAIL and the
K+ channel inhibitors GLB, U37, TEA, 5-hydroxydecanoate
(HD), α-dendrotoxin (DTX) and charybdotoxin (CTX) were obtained
from Enzo Life Sciences (San Diego, CA, USA). Throughout this
study, TRAIL was generally used at concentrations of 6.3–100 ng/ml
and the K+ channel inhibitors were used at 100
μM. ROT, antimycin A, oligomycin and carbonylcyanide
p-trifluoromethoxyphenylhydrazone (FCCP) were obtained from
Sigma-Aldrich (St. Louis, MO, USA). Mn(III) tetrakis (4-benzoic
acid) porphyrin chloride (MnTBaP), pan-caspase inhibitor
z-VAD-fluoromethylketone (FMK), caspase-3/7-specific inhibitor
z-DEVD-FMK, caspase-8-specific inhibitor z-IETD-FMK and
caspase-9-specific inhibitor z-LEHD-FMK were purchased from Merck
Japan (Tokyo, Japan). The caspase-12-specific inhibitor z-ATAD-FMK
and caspase-4-specific inhibitor z-LEVD-FMK were purchased from
BioVision (Mountain View, CA, USA). The reagents were dissolved in
dimethylsulfoxide and diluted with Hank’s balanced salt solution
(HBSS) to a final dimethylsulfoxide concentration of <0.1%
before use. Antimycin A was used with 0.5 μg/ml oligomycin
to inhibit complex III activity of the electron transport chain
(referred as to AM).
Cell culture
Human Jurkat leukemia cells were obtained from RIKEN
BioResource Center Cell Bank (Tsukuba, Japan) and cultured in high
glucose-containing RPMI-1640 medium (Sigma-Aldrich) supplemented
with 10% fetal bovine serum (FBS; Sigma-Aldrich) in a 5%
CO2-containing atmosphere. Human A549 lung cancer cells
and human fetal fibroblast-like lung cell WI-38-40 were obtained
from Health Science Research Resource Bank (Osaka, Japan) and grown
in low glucose-containing Dulbecco’s modified Eagle’s medium
supplemented with 10% FBS in a 5% CO2-containing
atmosphere. The cells were harvested by incubation in 0.25%
trypsin-EDTA medium (Gibco-Invitrogen, Carlsbad, CA, USA) for 5 min
at 37°C.
Measurement of depolarization
Depolarization was measured by flow cytometry using
bis-oxonol (Enzo Life Sciences), an anionic dye that shows
increased fluorescence intensity upon membrane depolarization, as
previously described (13).
Briefly, 4×105 cells suspended in 500 μl of HBSS
were incubated with 100 nM dye for 15 min at 37°C, and then
incubated with the agents to be tested for 2–4 h at 37°C in a 5%
CO2-containing atmosphere. Subsequently,
1×104 cells were counted for their fluorescence using
the FL-2 channel of a FACSCalibur (BD Biosciences, San Jose, CA,
USA) and analyzed using CellQuest software (BD Biosciences).
Determination of surface DR4/DR5
expression
The expression levels of DR4 and TDR5 on the cell
surface were determined by flow cytometry as previously described
(14). Briefly, 5×105
cells/100 μl were incubated with monoclonal anti-human DR4
and DR5 antibodies or mouse isotype-matched control antibodies
(R&D Systems; Minneapolis, MN, USA) for 30 min at 4°C. The
cells were then centrifuged into a pellet, resuspended in
phosphate-buffered saline, and incubated with
phycoerythrin-conjugated goat F(ab′)2 anti-mouse IgG
(R&D Systems) for 30 min at 4°C. The fluorescence was measured
using the FL-2 channel of the FACSCalibur and analyzed using
CellQuest software.
Determination of apoptotic cell
death
Apoptotic cell death was quantitatively assessed by
double-staining with fluorescein isothiocyanate (FITC)-conjugated
Annexin V and propidium iodide (PI) as previously described
(13). Briefly, 2×105
cells/ml in 24-well plates were incubated with the agents to be
tested for 20 h in 10% FBS-containing medium at 37°C. Subsequently,
the cells were stained with FITC-conjugated Annexin V and PI using
a commercially available kit (Annexin V FITC Apoptosis Detection
Kit I; BD Pharmingen, San Diego, CA, USA) according to the
manufacturer’s instructions. The stained cells were evaluated in
the FACSCalibur and analyzed using the CellQuest software. Four
cellular subpopulations were evaluated: viable cells (Annexin
V−/PI−); early apoptotic cells (Annexin
V+/PI−); late apoptotic cells (Annexin
V+/PI+); and necrotic/damaged cells (Annexin
V−/PI+). Annexin V+ cells were
considered to be apoptotic cells.
Measurement of mROS
mROS was measured by flow cytometry using MitoSOX
Red [(3,8-phenanthridinediamine,
5-(6′-triphenyl-phosphoniumhexyl)-5,6-dihydro-6-phenyl);
Invitrogen, Carlsbad, CA, USA] as previously described (14). Briefly, 5×105 cells
suspended in 500 μl of HBSS were incubated with the agents
to be tested for various times at 37°C, followed by incubation with
5 μM MitoSOX for 15 min at 37°C. The cells were then washed,
resuspended in HBSS on ice, and centrifuged at 4°C. The red
fluorescence was measured using the FL-2 channel of the FACSCalibur
and analyzed using CellQuest software. The data were expressed as
F/F0, where F0 was the
fluorescence in unstimulated cells and F was the fluorescence in
stimulated cells.
Measurements of caspase-3/7 activation
and mitochondrial membrane potential (Δψm)
Activation of caspase-3/7 and changes in Δψm in
Jurkat cells were simultaneously measured as previously described
(13). Briefly, 2×105
cells/ml in 24-well plates were treated with the agents to be
tested for 20 h in 10% FBS-containing RPMI-1640 medium at 37°C, and
then stained with the dual sensor MitoCasp™ (Cell Technology Inc.,
Mountain View, CA, USA) according to the manufacturer’s protocol.
Caspase-3/7 activation and Δψm were evaluated using the FACSCalibur
and the data were analyzed using CellQuest software. Changes in Δψm
after a short TRAIL treatment were measured using the lipophilic
cation JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,
3′-tetraethylbenzimidazolylcarbocyanine iodide; Molecular Probes,
Eugene, OR, USA) as previously described (13). Briefly, 5×105 cells/500
μl were loaded with 2 μM JC-1 at 37°C for 15 min,
washed, and resuspended in HBSS. After cell stimulation for 2 or 4
h, the green fluorescence (monomeric JC-1) and red fluorescence
(J-aggregates) were measured using the FL-1 and FL-2 channels,
respectively, of the FACSCalibur and analyzed using CellQuest
software.
Measurement of caspase-12 activation
Caspase-12 activation in living cells was measured
using the caspase-12 inhibitor ATAD-FMK conjugated to FITC
(FITC-ATAD-FMK) as previously described (13). FITC-ATAD-FMK is cell-permeable and
non-toxic, and binds irreversibly to active caspase-12, but not
inactive caspase-12, in apoptotic cells. Briefly, 2×105
cells/ml in 24-well plates were treated with the agents to be
tested for 20 h in 10% FBS-containing medium at 37°C and then
stained with a CaspGLOW™ Fluorescein Active Caspase-12 Staining Kit
(BioVision) according to the manufacturer’s protocol. The
fluorescence was determined using the FL-1 channel of the
FACSCalibur and analyzed using CellQuest software.
Measurement of cardiolipin oxidation
Oxidation of cardiolipin was measured by flow
cytometry using the fluorescent dye 10-N-nonyl acridine
orange (NAO; Invitrogen), which binds to non-oxidized cardiolipin,
but not to oxidized cardiolipin, as previously described (14). Briefly, 5×105 cells
suspended in 500 μl of HBSS were incubated with the agents
to be tested for 4 h at 37°C, and then incubated with 100 nM NAO
for 15 min at 37°C. The harvested cells were washed and resuspended
in HBSS on ice. Their fluorescence was measured using the FL-1
channel of the FACSCalibur and analyzed using CellQuest software.
The data were expressed as F/F0, where
F0 was the fluorescence in unstimulated cells and
F was the fluorescence in stimulated cells.
Western blot analysis
Western blot analysis was performed as previously
described (14). Briefly,
1×106 cells/ml in 6-well plates were treated with the
agents to be tested for 20 h in 10% FBS-containing medium at 37°C,
washed, and lysed with SDS-sample buffer. Whole cell lysates (30
μg protein) were subjected to SDS-PAGE using a 10%
separation gel under reducing conditions and then transferred to
polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA).
The membranes were incubated with BlockAce (Dainippon Sumitomo
Pharma, Osaka, Japan) for 1 h at room temperature, washed,
incubated with polyclonal antibodies against X-box-binding protein
(XBP)-1 or caspase-3 (Cell Signaling Technology Japan, Tokyo,
Japan) overnight at 4°C, washed again, and incubated with
horseradish peroxidase-conjugated species-specific anti-rabbit Ig
(GE Healthcare Japan, Tokyo, Japan) for 1 h at room temperature.
After extensive washing, the immunoreactive proteins on the
membranes were detected using an Enhanced ChemiLuminescence (ECL)
Prime Kit (GE Healthcare Japan) according to the manufacturer’s
recommendations. To verify equal loading, the membranes were
re-probed with a monoclonal anti-GAPDH antibody (Santa Cruz
Biotechnology). The signal intensities were quantified relative to
the GAPDH signal intensity using NIH Image software (NIH, Bethesda,
MD, USA).
Statistical analysis
The statistical significance of differences among
multiple groups was analyzed by one-way analysis of variance
(ANOVA) followed by the Tukey’s test. The significance of
differences between two individual groups was analyzed by Student’s
t-test. Values of P<0.05 were considered to indicate statistical
significance.
Results
K+-mediated depolarization
potentiates TRAIL-induced apoptosis in human tumor cells with
different origins
To determine whether the modulation of TRAIL-induced
apoptosis by membrane-depolarizing agents is a general feature of
tumor cells with different origins, we examined the effect of high
K+ loading on TRAIL-induced apoptosis in Jurkat leukemia
cells. The cells were treated with TRAIL in the presence or absence
of 50 mM KCl for 20 h, and then double-stained with Annexin V/PI.
TRAIL at concentrations of ≥6.3 ng/ml increased apoptotic (Annexin
V+/PI−) cells, but not necrotic (Annexin
V−/PI+) cells, in a dose-dependent manner.
KCl alone caused minimal cell death, but significantly potentiated
TRAIL-induced apoptosis (Fig. 1A).
Measurement of membrane potential changes using bis-oxonol
showed that KCl induced rapid (within 5 min) membrane
depolarization that peaked at 2 h and declined thereafter (Fig. 1B). TRAIL evoked robust
depolarization in a dose- and time-dependent manner, but the effect
was observed after a considerable time lag. The effect was
initially observed at 2 h for 100 ng/ml TRAIL (1.3-fold) and
developed during another 2 h to reach 1.3- and 1.6-fold for 25 and
100 ng/ml TRAIL, respectively (Fig.
1B). K+ loading also enhanced TRAIL-induced
apoptosis, but not necrosis, in A549 lung cancer cells (Fig. 1C). In contrast, TRAIL and KCl alone
or in combination caused minimal cell death in WI-38-40 fibroblasts
despite their substantial DR5 expression (Fig. 1D). Collectively, these findings
show that K+-mediated depolarization potentiates
TRAIL-induced apoptosis in human tumor cells with different
origins, but not in non-transformed cells.
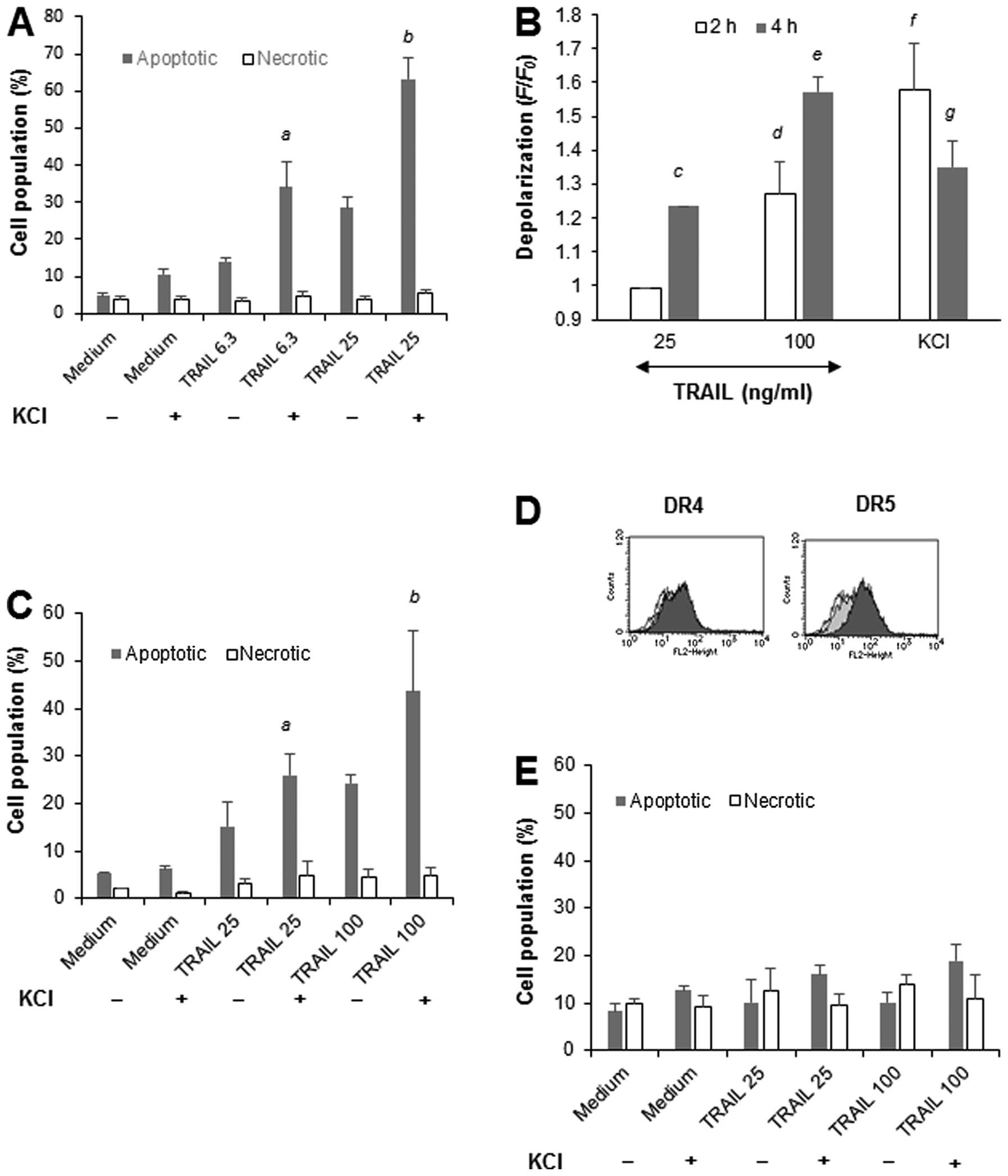 | Figure 1.K+-mediated depolarization
potentiates TRAIL-induced apoptosis in human tumor cells with
different origins, but not in non-transformed cells. (A) Jurkat
cells were treated with TRAIL at the indicated concentrations in
the presence or absence of 50 mM KCl for 20 h, stained with Annexin
V-FITC and PI, and analyzed by flow cytometry. Annexin V+ cells and
Annexin V−/PI+ cells were considered to be
apoptotic cells and necrotic cells, respectively. The data
represent means ± SE (n=4). Letters a and b, indicate significance
vs TRAIL alone. (B) Jurkat cells that had been loaded with
bis-oxonol were treated with 25 or 100 ng/ml TRAIL or KCl for 4 h,
and analyzed for their fluorescence by flow cytometry. The data are
expressed as F/F0, where F0 is the
fluorescence in unstimulated cells and F is the fluorescence in
stimulated cells, and represent means ± SE (n=4). Letters c to g,
indicate significance vs control. (C) A549 lung cancer cells and
(E) WI-38-40 cells were treated with TRAIL at the indicated
concentrations in the presence or absence of KCl for 20 h, stained
with Annexin V-FITC and PI, and analyzed by flow cytometry. Annexin
V+ cells and Annexin V−/PI+ cells
were considered to be apoptotic cells and necrotic cells,
respectively. The data represent means ± SE (n=3 and n=4,
respectively). Letters a and b, indicate significance vs TRAIL
alone. (D) Surface DR4/DR5 expression levels in WI-38-40 cells. The
cells were analyzed for their expression of DR4 and DR5 levels by
indirect immunofluorescence and flow cytometry. In the panels, the
black lines represent specific staining, the gray lines represent
IgG isotype control staining, and the solid lines represent the
unstained control. |
KATP inhibitors specifically
potentiate TRAIL-induced apoptosis in human tumor cells with
different origins
To verify the role of depolarization, we examined
the effects of the KATP channel inhibitors GLB and U37.
As shown in Fig. 2A and B, each
drug markedly potentiated the TRAIL-induced apoptosis in Jurkat
cells, although they caused minimal cell death on their own. In
contrast, TEA, which mainly inhibits voltage-dependent potassium
(Kv) channel and Ca2+-dependent potassium
(KCa) channels, had no such effect (Fig. 2B), suggesting a specific role of
KATP channels in the potentiation. In support of this
view, treatment of the cells with the Kv
channel-specific inhibitor DTX and KCa channel-specific
inhibitor CTX for 20 h had minimal effects on the apoptosis
(Fig. 2C). The mitochondrial
KATP channel inhibitor HD had no effect either. All of
these drugs had minimal effects on the apoptosis for at least
another 48 h (data not shown). As shown in Fig. 2D, GLB or U37 alone increased the
depolarization by 1.4- and 2.6-fold, respectively, and higher
degrees of depolarization were observed in the cells treated with
TRAIL in the presence of each drug compared with the cells treated
with TRAIL alone. However, unlike the case for apoptosis, their
effects were less than additive. In contrast, TEA induced marginal
depolarization (maximum of 1.2-fold) and had a minimal effect on
TRAIL-induced depolarization. GLB and U37 also potentiated the
TRAIL-induced apoptosis in A549 cells, while TEA, DTX and CTX had
no such effects (data not shown). Again, GLB and U37 alone or in
combination with TRAIL had minimal effects on the survival of
WI-38-40 fibroblasts (data not shown). These results show that
KATP channel inhibitors specifically potentiate
TRAIL-induced apoptosis in human tumor cells with different
origins, but not in non-transformed cells.
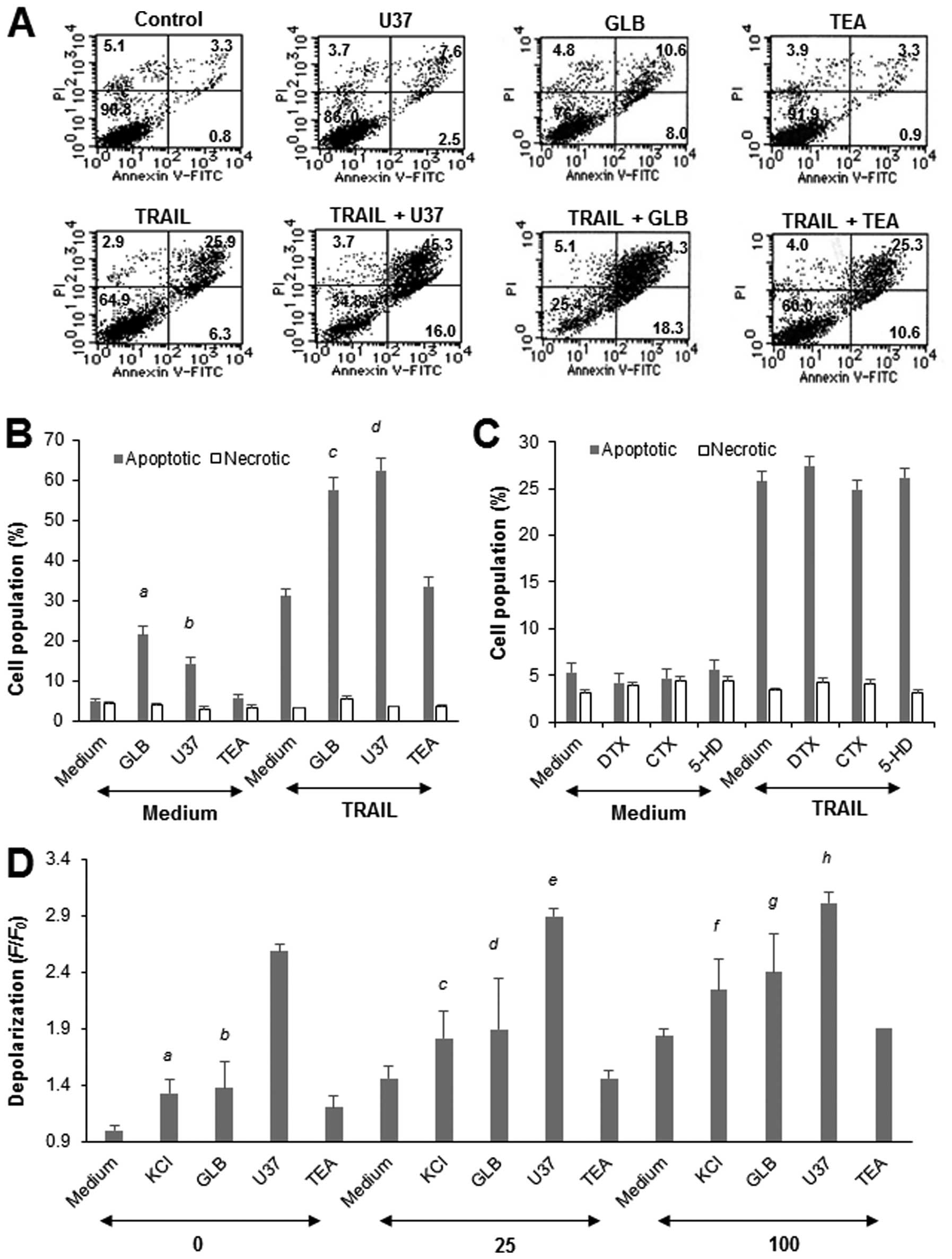 | Figure 2.KATP channel inhibitors
specifically potentiate TRAIL-induced apoptosis in Jurkat cells. (A
and B) Jurkat cells were treated with 25 ng/ml TRAIL and 100
μM U37, GLB or TEA alone or in combination for 20 h, stained
with Annexin V-FITC and PI, and analyzed by flow cytometry. Annexin
V+ cells and Annexin V−/PI+ cells
were considered to be apoptotic cells and necrotic cells,
respectively. (A) A typical histogram is shown. (B) The data
represent means ± SE (n=4). Letters a to d indicate significance vs
control. (C) The cells were treated with 25 ng/ml TRAIL and 100
μM DTX, CTX or HD alone or in combination for 24 h, and
apoptotic cell death was measured by flow cytometry using Annexin
V-FITC and PI staining. Annexin V+ cells and Annexin
V−/PI+ cells were considered to be apoptotic
cells and necrotic cells, respectively. The data represent means ±
SE (n=3 and n=4, respectively). (D) Cells that had been loaded with
bis-oxonol were treated with TRAIL at the indicated
concentrations and KCl, U37, GLB or TEA alone or in combination for
4 h, and analyzed for their fluorescence by flow cytometry. The
data are expressed as F/F0, where
F0 is the fluorescence in unstimulated cells and
F is the fluorescence in stimulated cells, and represent
means ± SE (n=4). Letters a to h indicate significance vs
control. |
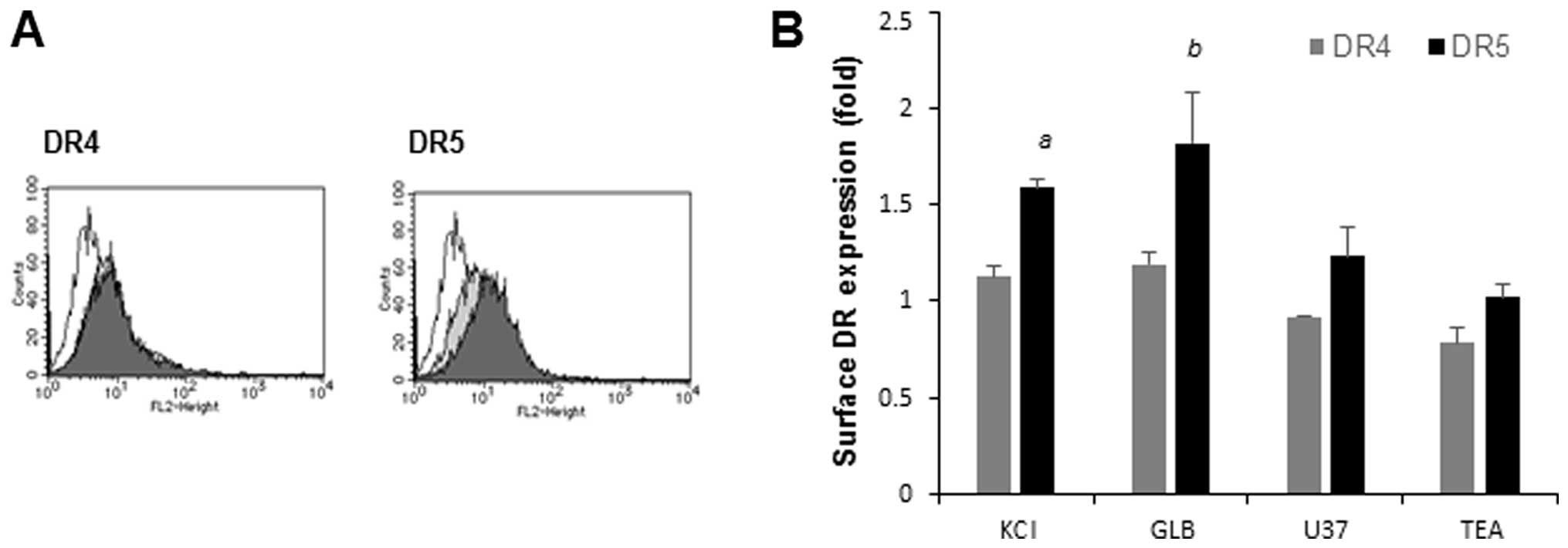
Potentiation of apoptosis is associated
with upregulation of cell surface DR5 expression
Since amplification of TRAIL-induced apoptosis is
often associated with upregulated expression of DR5 (15–17),
we examined the effects of the membrane-depolarizing agents on the
expressions of DR4 and DR5. Jurkat cells were exposed to KCl for
various times and analyzed for their expression levels of DR4 and
DR5 on the cell surface using specific antibodies. Until 4 h after
K+ loading, the cell surface DR4 and DR5 expression
levels were minimally changed compared with their basal levels. On
the other hand, K+ loading for a longer time (20 h)
increased the cell surface DR5 expression by 1.6-fold, but
minimally increased the cell surface DR4 expression (Fig. 3A). Similarly, GLB and U37 increased
the DR5 expression levels by 1.8- and 1.2-fold, respectively, while
the former, but not the latter, marginally increased the DR4
expression (maximum of 1.2-fold). In contrast, TEA had minimal
effects on the DR4 and DR5 expression levels (Fig. 3A). These results show that the
potentiation of apoptosis is associated with upregulation of cell
surface DR5 expression.
Membrane-depolarizing agents potentiate
the mitochondrial death pathway
We previously showed that TRAIL-induced apoptosis in
Jurkat cells and A375 cells was caspase-dependent (13,14).
Therefore, an array of caspase-specific inhibitors (10 μM)
were tested for their abilities to affect the potentiation of
apoptosis. The pan-caspase inhibitor z-VAD-FMK almost completely
blocked apoptosis. The caspase-8-specific inhibitor z-IETD-FMK,
caspase-9-specific inhibitor z-LEHD-FMK, and/or
caspase-3/7-specific inhibitor z-DEVD-FMK significantly abolished
the effects of KCl or U37 (Fig. 4A and
B), indicating that both the extrinsic and intrinsic
(mitochondrial) apoptotic pathways are involved in the
potentiation. Consistent with the role of caspase-12 in the
TRAIL-induced apoptosis in Jurkat cells (14), the caspase-12-specific inhibitor
z-ATAD-FMK also completely abrogated the effects of KCl or U37
(Fig. 4A and B). In support of the
role of the mitochondrial death pathway, TRAIL induced robust
mitochondrial membrane potential depolarization and activation of
caspase-3/7. The MMP depolarization and caspase-3/7 activation were
markedly potentiated by KCl or KATP channel inhibitors,
although each drug alone, except for GLB, had minimal effects on
the two events (Fig. 4C and D).
Consistent with the role of caspase-3, TRAIL induced robust
caspase-3 cleavage, as shown by the appearance of a truncated form,
i.e., caspase-3 (19 kDa) (Fig.
4E). KCl potentiated the effect, resulting in the new
appearance of an even smaller form of caspase-3 (17 kDa). The
appearance of caspase-3 (17 kDa) was completely abrogated by the
pan-caspase inhibitor and the specific inhibitors of caspase-8, -9,
-3 and-12, but not by that of caspase-4. The caspase-8 and
caspase-12 inhibitors also abolished the appearance of caspase-3
(19 kDa), while the caspase-9 inhibitor did not (Fig. 4E). These results show that in the
presence or absence of depolarization, TRAIL induces caspase-3
cleavage in a different manner.
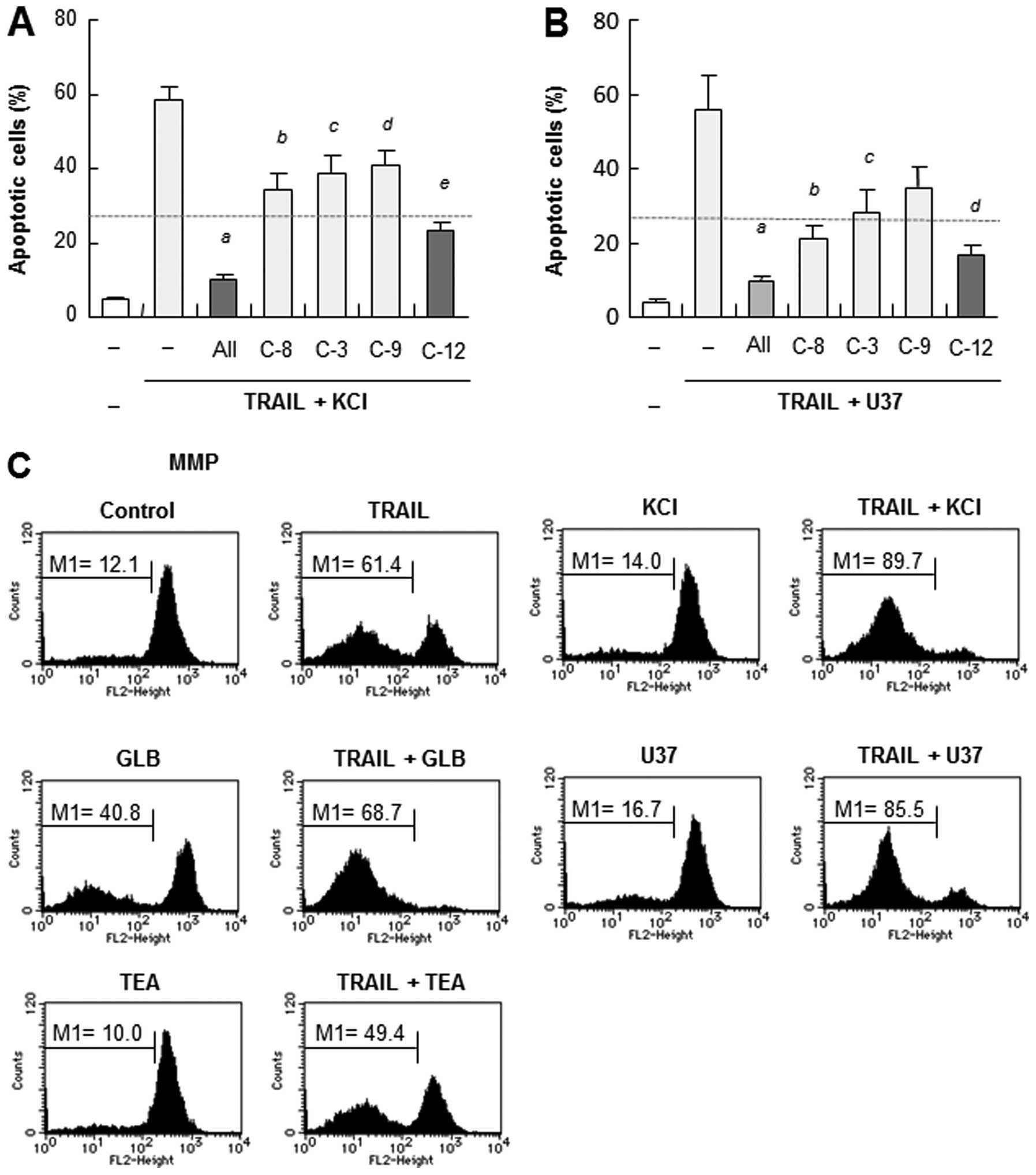 | Figure 4.Membrane-depolarizing agents
potentiate the mitochondrial death pathway in Jurkat cells. (A and
B) Jurkat cells were treated with 25 ng/ml TRAIL and KCl or U37
alone or in combination in the presence or absence of 10 μM
z-VAD-FMK (All), z-IETD-FMK (C-8), z-DEVD-FMK (C-3), z-LEHD-FMK
(C-9) or z-ATAD-FMK (C-12) for 20 h, and apoptotic cell death was
measured by flow cytometry using Annexin V-FITC and PI staining.
The data represent means ± SE (n=4). Letters a to e in (A) and a to
d in (B), indicate significance vs control without inhibitors. (C)
The cells were treated with TRAIL and KCl, U37, GLB or TEA alone or
in combination, and MMP depolarization was determined by flow
cytometry (n=3). (D) The cells were treated with TRAIL and KCl,
U37, GLB or TEA alone or in combination, and caspase-3/7 activation
was determined by flow cytometry (n=3). (E) The cells were treated
with TRAIL and KCl alone or in combination, in the presence or
absence of z-VAD-FMK (All), z-IETD-FMK (C-8), z-DEVD-FMK (C-3),
z-LEHD-FMK (C-9), z-ATAD-FMK (C-12) or z-LEVD-FMK for 20 h. The
cells were then washed, lysed with SDS-sample buffer and analyzed
for their contents of full-length and cleaved caspase-12 by western
blot analysis with a specific antibody. To verify equal loading,
the blots were re-probed with an anti-GAPDH antibody. |
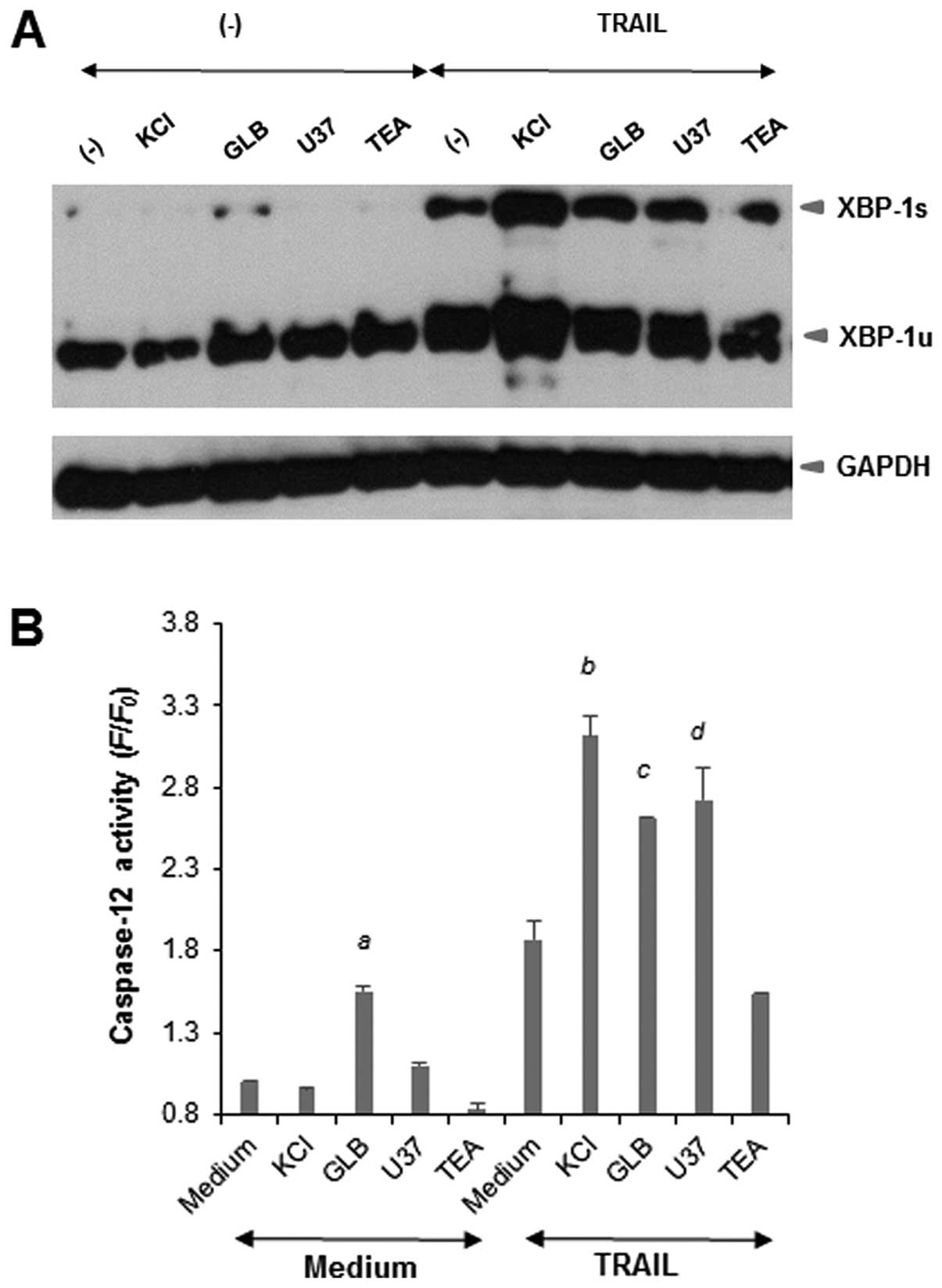
Membrane-depolarizing agents potentiate
ER stress responses
We previously showed that TRAIL induced ER stress in
Jurkat cells (14). The
ER-mediated death pathway is another pathway for apoptosis that is
independent of the extrinsic and intrinsic pathways (18–20).
To examine the possible role of this pathway, we analyzed the
effects of the membrane-depolarizing drugs on XBP-1 activation, a
cellular response to ER stress. Western blot analyses revealed that
TRAIL dose-dependently increased the expression levels of both the
inactive unspliced form of XBP-1 (XBP-1u) and the active spliced
form of XBP-1 (XBP-1s) by about 2-fold, indicating activation of
XBP-1. Although each drug alone caused minimal activation of XBP-1,
KCl and KATP channel inhibitors, but not TEA, markedly
potentiated the effects of TRAIL (Fig.
5A). Next, we examined the effects of the membrane-depolarizing
agents on TRAIL-induced caspase-12 activation. The activation of
caspase-12 was evaluated by measuring the conversion of a
cell-permeable substrate, FITC-ATAD-FMK. As shown in Fig. 5B, each drug alone except for GLB,
caused minimal caspase-12 activation, while KCl and KATP
channel inhibitors, but not TEA, markedly potentiated TRAIL-induced
caspase-12 activation. Taken together, these results show that the
membrane-depolarizing agents potentiate ER stress responses
including caspase-12 activation.
Functional coupling of mROS and
depolarization during TRAIL-induced apoptosis
Previously we showed that TRAIL treatment resulted
in mROS accumulation that mediated mitochondrial and ER
dysfunctions during TRAIL-induced apoptosis (14). Therefore, we investigated the
possible role of mROS in the potentiation of apoptosis by
depolarization. To explore the possibility that mROS mediate the
depolarization, we examined the effects of membrane-depolarizing
drugs on mROS generation. MitoSOX Red localizes to mitochondria and
serves as a fluoroprobe for selective detection of superoxide in
these organelles (21,22). TRAIL induced mROS generation in a
dose-dependent manner, and KCl or U37 markedly potentiated this
effect, while each drug alone minimally increased the generation
(Fig. 6A). Oxidation of
cardiolipin serves as another biochemical hallmark of mitochondrial
oxidative stress, because this phospholipid exists in association
with cytochrome c on the outer surface of the inner
mitochondrial membrane. Because the fluorescent dye NAO binds to
the non-oxidized form, but not to the oxidized form, of
cardiolipin, independently of Δψm, measurements of NAO fluorescence
enable us to monitor the oxidation of cardiolipin in mitochondria
(23). Consistent with our
previous study (14), TRAIL
treatment resulted in a dose-dependent decrease in NAO
fluorescence, indicating the induction of cardiolipin oxidation.
Agonistic antibodies against DR4 and DR5, which trigger the
formation of multimeric complexes containing only specific TRAIL-Rs
(24–26) also induced robust cardiolipin
oxidation in a dose-dependent manner (Fig. 6B), indicating that this oxidation
is mediated by DR4/DR5. Collectively, these results show that TRAIL
induces mROS accumulation and that depolarization potentiates this
process. Mitochondria serve a major source of ROS under
physiological conditions and generate large amounts of ROS when
their metabolism is impaired under pathological conditions. Indeed,
we previously showed that mitochondrial metabolic inhibitors, such
as the complex I inhibitor ROT, complex III inhibitor AM and
mitochondrial uncoupling agent FCCP, considerably increased the
mROS levels in Jurkat cells, thereby enhancing the TRAIL-induced
mitochondrial and ER dysfunctions and apoptosis (14). In agreement with these previous
observations, FCCP considerably increased the cardiolipin oxidation
(Fig. 6B). To obtain further
evidence for the functional coupling between mROS and
depolarization, we examined the ability of these metabolic
inhibitors to provoke depolarization. As expected, among these
metabolic inhibitors, FCCP was the most potent at provoking
depolarization (Fig. 6C). This
effect (1.7-fold) was comparable to that of 100 ng/ml of TRAIL,
while ROT and AM had marginal effects (maximum of 1.2-fold), in
parallel with their effects on MitoSOX Red signals (14). The coincident induction of
depolarization and mROS led us to hypothesize the presence of
another biochemical consequence between them. i.e., that mROS
mediate the depolarization. To test this hypothesis, we examined
the effect of MnTBaP on the TRAIL-induced depolarization, since
this antioxidant can block TRAIL-induced mROS generation in the
cells (14). As shown in Fig. 6D, treatment with non-toxic
concentrations of MnTBaP ranging from 3 to 30 μM
dose-dependently reduced the TRAIL-induced depolarization. The
effectiveness of this antioxidant varied considerably depending on
the concentration of TRAIL applied. MnTBaP (30 μM) almost
completely reduced the depolarization induced by 25 ng/ml TRAIL,
while it reduced the depolarization induced by 100 ng/ml TRAIL by a
maximum of 50% (Fig. 6D).
Consequently, the levels of depolarization became comparable to
those induced by 25 ng/ml TRAIL. On the other hand, MnTBaP reduced
the depolarization induced by FCCP by only 20% even when used at
the highest concentration. Collectively, these findings show a
closed functional coupling of mROS and depolarization during
TRAIL-induced apoptosis.
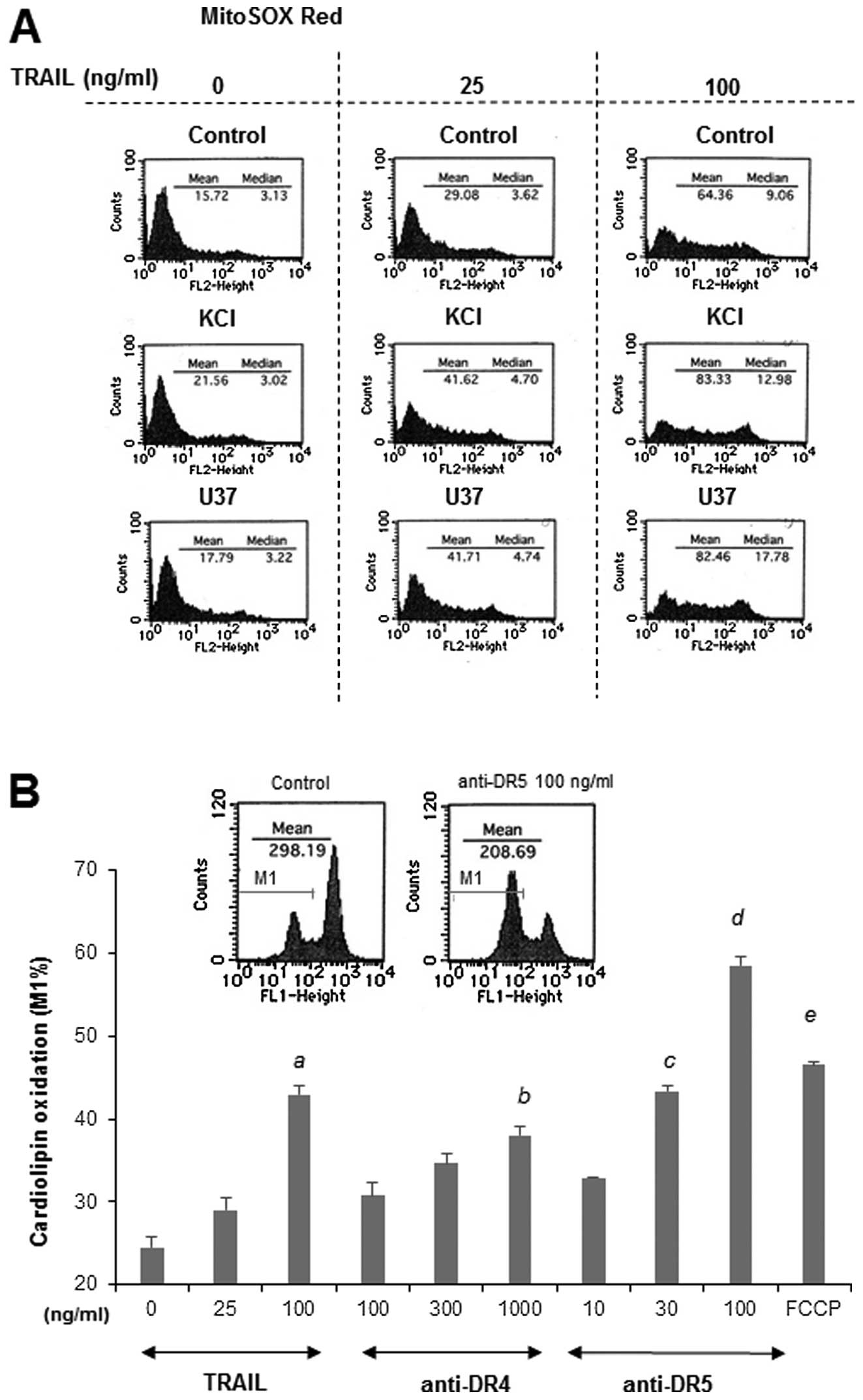 | Figure 6.Functional coupling of mitochondrial
ROS and depolarization in Jurkat cells. (A) Jurkat cells were
treated with 25 or 100 ng/ml TRAIL and KCl or U37 alone or in
combination for 4 h, and then incubated with 5 μM MitoSOX
Red for 15 min at 37°C. After washing, the cells were immediately
resuspended in HBSS on ice. The fluorescence was measured using the
FL-1 or FL-2 channel of the FACSCalibur and analysed using the
CellQuest software. A representative histogram is shown (n=3). (B)
The cells were loaded with 100 nM NAO for 15 min at 37°C, washed
and resuspended in HBSS. The NAO-loaded cells were treated with
TRAIL, anti-DR4 or anti-DR5 antibodies at the indicated
concentrations or 5 μM FCCP at 37°C for 4 h. After washing,
the cells were immediately resuspended in HBSS on ice. The NAO
fluorescence was measured using the FL-1 channel of the
FACSCalibur. Representative histograms are shown in the upper
panel. The data shown are expressed as percentages of the cell
population with cardiolipin oxidation (M1 in the upper panel)
relative to the whole cell population (set at 100%), and represent
means ± SE (n=3). Letters a to e, indicate statistical significance
vs control. (C) Cells loaded with bis-oxonol were treated with 100
ng/ml TRAIL, 5 μM ROT, 5 μg/ml AM or 5 μM FCCP
for 4 h, and analyzed for their fluorescence by flow cytometry. The
data are expressed as F/ F0, where F0 is the
fluorescence in unstimulated cells and F is the fluorescence in
stimulated cells, and represent means ± SE (n=2–6). Letters a and
b, indicate statistical significance vs control. (D) Cells loaded
with bis-oxonol were treated with 25 or 100 ng/ml TRAIL in the
presence or absence of MnTBaP at the indicated concentrations, and
analyzed for their fluorescence by flow cytometry. A representative
histogram is shown in the upper panel. In the panel, the dark gray
lines represent the fluorescence of 25 ng/ml TRAIL alone and the
light gray lines represents the fluorescence of TRAIL in the
presence of 30 μM MnTBaP, while the solid lines represent
the fluorescence of the medium control. (E) Cells loaded with
bis-oxonol were treated with 25 or 100 ng/ml or 5 μM
FCCP in the presence or absence of MnTBaP at the indicated
concentrations, and analyzed for their fluorescence by flow
cytometry. The data are expressed as F/F0, where
F0 is the fluorescence in unstimulated cells and
F is the fluorescence in stimulated cells, and represent
means ± SE (n=4). Letters a to c indicate statistical significance
vs TRAIL alone. |
Role of mROS in the potentiation of
TRAIL-induced apoptosis by depolarization in human A375 melanoma
cells
Since depolarization potentiates TRAIL-induced
apoptosis in several human melanoma cell lines (13), we investigated whether similar
biochemical pathways involving mROS underlie this potentiation
using A375 melanoma cells as a model. First, we examined the
effects of the membrane-depolarizing drugs on the cell surface DR
expression levels. Until 4 h after exposure to each drug, the DR4
and DR5 expression levels were minimally changed compared with
their basal levels. On the other hand, treatment with KCl or U37
for 20 h increased the DR5 expression levels by 1.3 and 1.6-fold,
respectively, while minimally increasing the DR4 expression levels
(Fig. 7A). In contrast, GLB and
TEA had minimal effects on the DR4 and DR5 expression levels, in
parallel with their ineffectiveness at potentiating apoptosis
(13). Similar to Jurkat cells
(14), ROT, AM and FCCP increased
the mROS levels. Among these agents, AM was the most powerful
(7.8-fold) and the effects of ROT and FCCP were comparable (1.9-
and 2.1-fold, respectively). These drugs also potentiated
TRAIL-induced apoptosis in the cells (Fig. 7B). For 25 ng/ml TRAIL, necrotic
cell death was also substantially increased. Finally, we examined
whether these mitochondrial metabolic inhibitors affected the cell
membrane potential. AM and FCCP, but not ROT, alone caused robust
depolarization (1.2-1.4-fold), and potentiated TRAIL-induced
depolarization (Fig. 7C),
indicating that mROS accumulation potentiates depolarization.
Conversely, MnTBaP treatment, which abolishes mROS generation in
melanoma cells (27), considerably
reduced TRAIL-induced depolarization in a dose-dependent manner and
this effect was more pronounced for lower concentrations of TRAIL
(e.g. 30 μM MnTBaP reduced 25 and 100 ng/ml TRAIL-induced
depolarization by 62% and 48%, respectively). These results show
that similar biochemical pathways including upregulation of surface
DR5 expression and mROS accumulation regulate the
depolarization-mediated potentiation of TRAIL-induced apoptosis in
the cells.
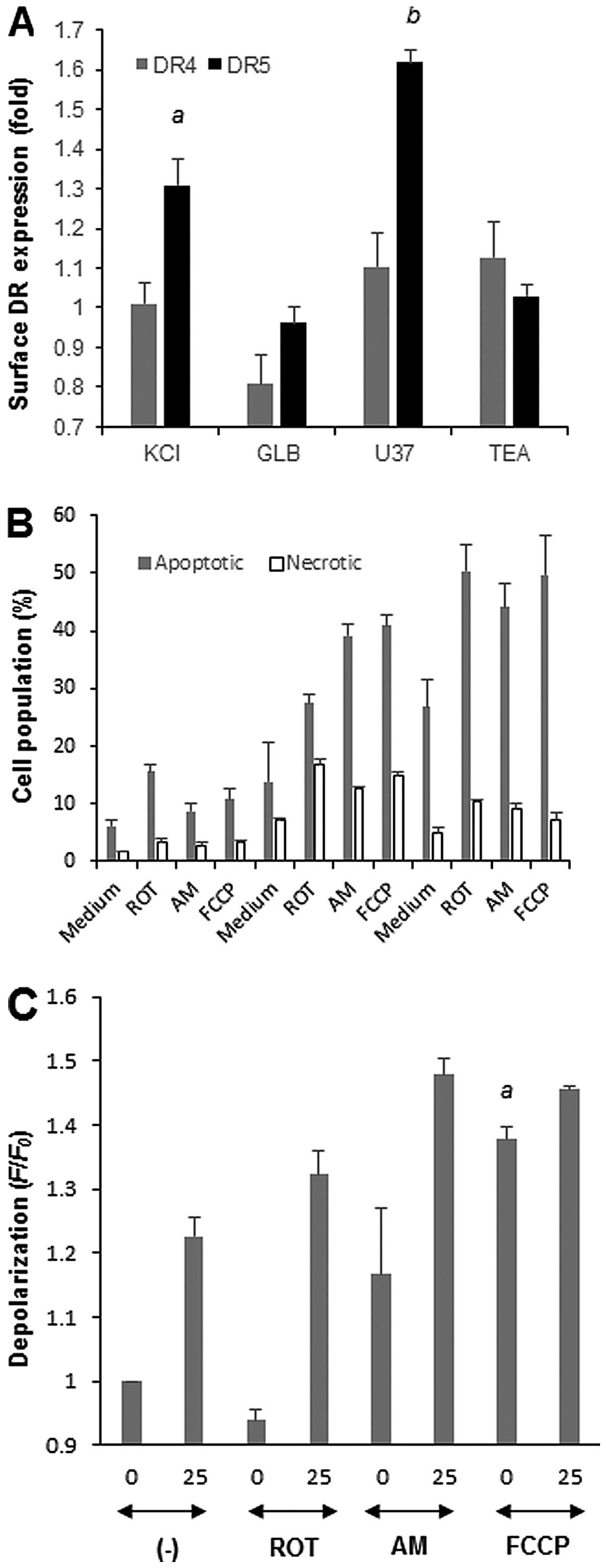 | Figure 7.Role of ROS in the
depolarization-mediated potentiation of TRAIL-induced apoptosis in
human A375 melanoma cells. (A) A375 cells were treated with KCl,
U37, GLB or TEA for 20 h, and analyzed for their DR4/DR5 expression
levels on the cell surface by indirect immunofluorescence followed
by flow cytometry. The fluorescence was measured using the FL-2
channel of the FACSCalibur and analyzed using CellQuest software.
The data represent means ± SE (n=3). Letters a and b indicate
statistical significance vs control. (B) Cells were treated with 25
or 100 ng/ml TRAIL and ROT, AM or FCCP alone or in combination for
20 h, stained with Annexin V-FITC and PI, and analyzed by flow
cytometry. Annexin V+ cells and Annexin
V−/PI+ cells were considered to be apoptotic
cells and necrotic cells, respectively. (B) The data represent
means ± SE (n=3). (C) Cells loaded with bis-oxonol were treated
with 25 ng/ml TRAIL and ROT, AM or FCCP alone or in combination for
4 h, and analyzed for their fluorescence by flow cytometry. The
data are expressed as F/F0, where F0 is the
fluorescence in unstimulated cells and F is the fluorescence in
stimulated cells, and represent means ± SE (n=3 or 4). Letter a
indicates statistical significance vs control. |
Discussion
This study was undertaken to examine whether
depolarization plays a general role in TRAIL-induced tumor cell
apoptosis, and can therefore serve as a common target for treatment
of tumor cells with different origins. The data presented in this
paper taken together with our previous data show that
membrane-depolarizing agents, such as K+ and
KATP channel inhibitors, potentiate TRAIL-induced
apoptosis in human tumor cells with different origins, including
Jurkat leukemia cells, and A549 lung cancer cells, but not in
non-transformed melanocytes and fibroblsts. Plasma membrane
KATP channels appear to be specifically associated with
the apoptosis, since inhibitors of other potassium channels, such
as KCa and Kv and mitochondrial
KATP channels had no such effect. The findings expand
our previous findings for melanoma cells to various types of human
malignant cells, and indicate that depolarization may be a
tumor-selective target for potentiating apoptosis. This may
strengthen the therapeutic potential of membrane-depolarizing
agents in cancer treatment. Our previous study showed that
membrane-depolarizing agents potentiate TRAIL-induced apoptosis in
melanoma cells by upregulating mitochondrial and ER-associated
death pathways (13). The present
study indicates that this is a common mechanism at least among
certain cell types, including Jurkat cells. In addition, this study
provides new insight into the mechanisms by which depolarization
potentiates these two death pathways. First, we found that
K+ and KATP channel inhibitors commonly
upregulated surface DR5 expression in Jurkat cells and A375
melanoma cells, similar to the effects of diverse chemicals such as
thapsigargin, tunicamycin and 2-deoxy-D-glucose on melanoma cells
(15–17). In contrast, inhibitors of other
potassium channels such as TEA had no such effects. Essentially
similar results were obtained in A375 melanoma cells. Thus, the
upregulation of surface DR5 expression may be relevant to
KATP channels function and play a role in the
potentiation of apoptosis. Second, we found that the potentiation
of death signals is not only caused by quantitative changes but
also by qualitative changes. Western blot analyses revealed that
depolarization modulated the manner of caspase-3 cleavage, a
molecular hallmark of the enzyme activation. TRAIL alone induced
cleavage of caspase-3 (35 kDa) to caspase-3 (19 kDa), while under
depolarization conditions, TRAIL caused a higher degree of
caspase-3 cleavage, resulting in new appearance of an even smaller
form, caspase-3 (17 kDa). The appearance of caspase-3 (19/17 kDa)
was completely blocked by specific inhibitors of caspase-8 and
caspase-9, as well as caspase-3, consistent with the conventional
view that caspase-3 activation occurs downstream of extrinsic
(caspase-8) and intrinsic (caspase-9) pathways. Caspase-12 is
ubiquitously expressed, localized to the ER membrane, and
specifically activated by ER stress to play a key role in
stress-induced apoptosis (28–30)
The caspase-12 inhibitor prevented the cleavage of caspase-3,
suggesting that caspase-12 is also involved in the activation of
caspase-3. Strikingly, however, the caspase-9 inhibitor prevented
the appearance of caspase-3 (17 kDa), but not that of caspase-3 (19
kDa), suggesting that different sets of caspases are involved in
the two different manners of caspase-3 cleavage. Thus,
depolarization may modulate the caspase cascade pathways involved
in caspase-3 activation. Caspase-4, another ER-associated caspase,
has also been shown to play a role in ER stress-mediated apoptosis
in melanoma cells (31,32). However, the caspase-4-specific
inhibitor had minimal effects on the cleavage of caspase-3,
suggesting that if caspase-4 does play a role in the potentiation,
it may have another target. Further studies investigating the roles
of these two ER-associated caspases in the potentiation are under
way.
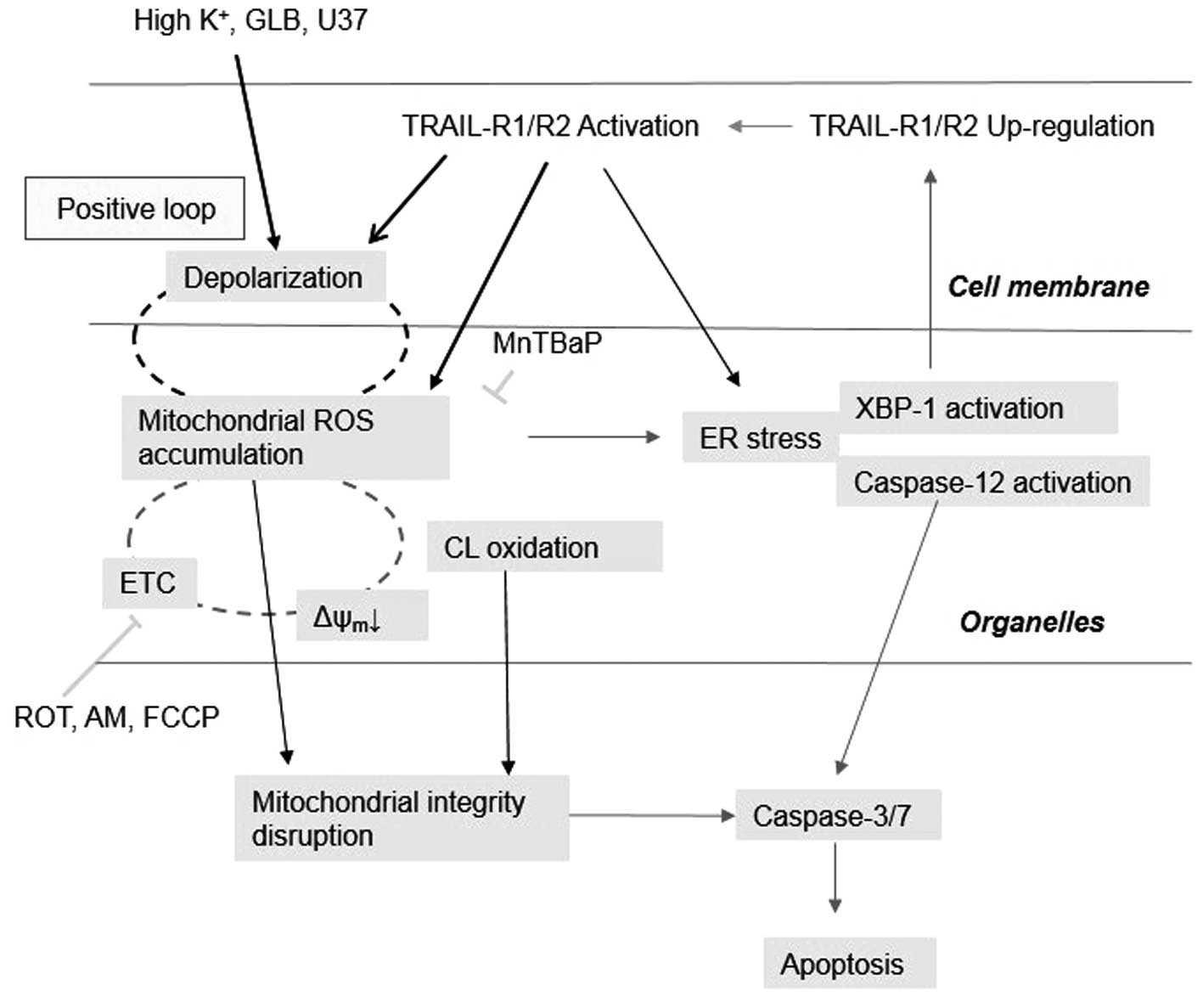
Our previous findings that mROS mediate
mitochondrial and ER dysfunctions in Jurkat cells (14) led us to investigate the possible
role of mROS in the potentiation of apoptosis. The
membrane-depolarizing agents by themselves increased mROS and
potentiated TRAIL-induced mROS generation, indicating that
depolarization controls mROS. It is notable that depolarization
increased the surface expression of DR5, the triggering of which
increases mROS. Since depolarization potentiated the TRAIL-induced
activation of the transcription factor XBP-1, which is engaged in
the regulation of surface DR5 expression (17), it is possible to speculate that the
upregulation of surface DR5 expression results in increased mROS
accumulation, thereby causing mitochondrial and ER dysfunctions. On
the other hand, our data showed that scavenging of mROS by the
antioxidant MnTBaP reduced depolarization, while mROS accumulation
caused by metabolic dysfunction potentiated the depolarization.
These data indicate that mROS control the depolarization. However,
several lines of evidence suggest that this role is limited for
weak depolarization. First, depolarization became more resistant to
MnTBaP treatment as the concentration of TRAIL (magnitude of
depolarization) increased. Second, FCCP-induced depolarization was
quite resistant to MnTBaP. It is noteworthy that 5 μM FCCP
and 100 ng/ml TRAIL caused comparable levels of mROS accumulation
(14) and depolarization (this
study), although the time courses of these events were quite
different. TRAIL provoked depolarization and mROS accumulation
after a considerable time lag, while FCCP caused both responses
rapidly. It is noted that FCCP was much more powerful than TRAIL
for inducing MMP depolarization. TRAIL induced a moderate MMP
depolarization (28%) with a lag of 2 h, while FCCP caused strong
MMP depolarization (92%) immediately. Taken together with the
dose-dependent induction of MMP depolarization by TRAIL, these
observations suggest that mROS are responsible for weak
depolarization, while another event, probably MMP depolarization is
required for strong depolarization. Further studies are necessary
to prove this hypothesis. Collectively, our data suggest that
depolarization and mROS accumulation mutually regulated one another
and that a positive loop exists between the two events (Fig. 8). Although the precise mechanisms
underlying the mutual regulation remain to be elucidated, this
finding may provide a rationale for the tumor-selective
cytotoxicity and/or potentiation of TRAIL cytotoxicity of a wide
variety of ROS-producing substances such as wogonin (33,34)
and diallyl trisulfide (35,36)
in different types of cancer cells including leukemia and melanoma
cells.
Acknowledgements
The authors thank Dr M. Murai and Dr
T. Inoue for technical assistance. This study was supported in part
by a Grant-in-Aid from the Ministry of Education, Culture, Sports,
Science and Technology (KAKENHI 23591631; to Y.S-K.) and
Grant-in-Aid from Nihon University (to Y.S-K.).
References
|
1.
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy. Cell Death Differ. 10:66–75. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:612–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lavrik IN, Golks A and Krammer PH:
Caspases: pharmacological manipulation of cell death. J Clin
Invest. 15:2665–2662. 2005. View
Article : Google Scholar
|
|
4.
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2014. View Article : Google Scholar
|
|
5.
|
Green DR: Apoptotic pathways: paper wraps
stone blunts scissors. Cell. 102:1–4. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Korsmeyer SJ, Wei MC, Saito M, Weiler S,
Oh KJ and Schlesinger PH: Pro-apoptotic cascade activates BID,
which oligomerizes BAK or BAX into pores that result in the release
of cytochrome c. Cell Death Differ. 7:1166–1173. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Yan N and Shi Y: Mechanisms of apoptosis
through structural biology. Annu Rev Cell Dev Biol. 21:35–56. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25:4506–4507. 2007.PubMed/NCBI
|
|
9.
|
Bortner CD, Gomez-Angelats M and Cidlowski
JA: Plasma membrane depolarization without repolarization is an
early molecular event in anti-Fas-induced apoptosis. J Biol Chem.
276:4304–4314. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Yin W, Li X, Feng S, et al: Plasma
membrane depolarization and Na,K-ATPase impairment induced by
mitochondrial toxins augment leukemia cell apoptosis via a novel
mitochondrial amplification mechanism. Biochem Pharmacol.
78:191–202. 2009. View Article : Google Scholar
|
|
11.
|
Nolte F, Friedrich O, Rojewski M, Fink RH,
Schrezenmeier H and Körper S: Depolarisation of the plasma membrane
in the arsenic trioxide (As2O3)-and
anti-CD95-induced apoptosis in myeloid cells. FEBS Lett. 578:85–89.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Ghoumari AM, Piochon C, Tomkiewicz C, et
al: Neuroprotective effect of mifepristone involves neuron
depolarization. FASEB J. 20:1377–1386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Suzuki Y, Inoue T, Murai M,
Suzuki-Karasaki M, Ochiai T and Ra C: Depolarization potentiates
TRAIL-induced apoptosis in human melanoma cells: role for
ATP-sensitive K+ channels and endoplasmic reticulum
stress. Int J Oncol. 41:465–475. 2012.PubMed/NCBI
|
|
14.
|
Inoue T and Suzuki-Karasaki Y:
Mitochondrial superoxide mediates mitochondrial and endoplasmic
reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells.
Free Radic Biol Med. 61:273–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Chen LH, Jiang CC, Kiejda KA, et al:
Thapsigargin sensitizes human melanoma cells to TRAIL-induced
apoptosis by up-regulation of TRAIL-R2 through the unfolded protein
response. Carcinogenesis. 28:2328–2336. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Jiang CC, Chen LH, Gillespie S, et al:
Tunicamycin sensitizes human melanoma cells to tumor necrosis
factor-related apoptosis-inducing ligand-induced apoptosis by
up-regulation of TRAIL-R2 via the unfolded protein response. Cancer
Res. 67:5880–5888. 2007. View Article : Google Scholar
|
|
17.
|
Liu H, Jiang CC, Lavis CJ, et al:
2-Deoxy-D-glucose enhances TRAIL-induced apoptosis in human
melanoma cells through XBP-1-mediated up-regulation of TRAIL-R2.
Mol Cancer. 8:1222009. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Boyce M and Yuan J: Cellular response to
endoplasmic reticulum stress: a matter of life or death. Cell Death
Differ. 13:363–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Breckenridge DG, Germain M, Mathai JP,
Nguyen M and Shore GC: Regulation of apoptosis by endoplasmic
reticulum pathways. Oncogene. 22:8608–8618. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Groenendyk J and Michalak M: Endoplasmic
reticulum quality control and apoptosis. Acta Biochim Pol.
52:381–395. 2005.PubMed/NCBI
|
|
21.
|
Robinson KM, Janes MS, Pehar M, et al:
Selective fluorescencet imaging of superoxide in vivo using
ethidium-based probes. Proc Natl Acad Sci USA. 103:15038–15043.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Mukhopadhyay P, Rajesh M, Kashiwaya Y,
Haskó G and Pacher P: Simple quantitative detection of
mitochondrial superoxide production in live cells. Biochem Biophys
Res Commun. 358:203–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Petit JM, Maftah A, Ratinaud MH and Julien
R: 10N-nonyl acridine orange interacts with cardiolipin and allows
the quantification of this phospholipid in isolated mitochondria.
Eur J Biochem. 209:267–273. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Griffith TS, Rauch CT, Smolak PJ, et al:
Functional analysis of TRAIL receptors using monoclonal antibodies.
J Immunol. 162:2597–2605. 1999.PubMed/NCBI
|
|
25.
|
Pukac L, Kanakaraj P, Humphreys R, et al:
HGS-ETR1, a fully human TRAIL-receptor 1 monoclonal antibody,
induces cell death in multiple tumour types in vitro and in vivo.
Br J Cancer. 92:1430–1441. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Georgakis GV, Li Y, Humphreys R, et al:
Activity of selective fully human agonistic antibodies to the TRAIL
death receptors TRAIL-R1 and TRAIL-R2 in primary and cultured
lymphoma cells: induction of apoptosis and enhancement of
doxorubicin- and bortezomib-induced cell death. Br J Haematol.
130:501–510. 2005. View Article : Google Scholar
|
|
27.
|
Tochigi M, Inoue T, Suzuki-Karasaki M,
Ochiai T, Ra C and Suzuki-Karasaki Y: Hydrogen peroxide induces
cell death in human TRAIL-resistant melanoma through intracellular
superoxide generation. Int J Oncol. 42:863–872. 2013.PubMed/NCBI
|
|
28.
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Rutkowski DT and Kaufman RJ: A trip to the
ER: coping with stress. Trends Cell Biol. 14:20–28. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Jiang CC, Mao ZG, Avery-Kiejda KA, Wade M,
Hersey P and Zhang XD: Glucose-regulated protein 78 antagonizes
cisplatin and adriamycin in human melanoma cells. Carcinogenesis.
30:197–204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Mao ZG, Jiang CC, Yang F, Thorne RF,
Hersey P and Zhang XD: TRAIL-induced apoptosis of human melanoma
cells involves activation of caspase-4. Apoptosis. 15:1211–1222.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Fas SC, Baumann S, Zhu JY, et al: Wogonin
sensitizes resistant malignant cells to TNFalpha- and TRAIL-induced
apoptosis. Blood. 108:3700–3706. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Baumann S, Fas SC, Giaisi M, et al:
Wogonin preferentially kills malignant lymphocytes and suppresses
T-cell tumor growth by inducing PLCgamma1- and
Ca2+-dependent apoptosis. Blood. 111:2354–2363. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Powlny AA and Singh SV: Multitargeted
prevention and therapy of cancer by diallyl trisulfide and related
Allium vegetable-derived organosulfur compounds. Cancer
Lett. 269:305–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Murai M, Inoue T, Suzuki-Karasaki M,
Ochiai T, Ra C, Nishida S, et al: Diallyl trisulfide sensitizes
human melanoma cells to TRAIL-induced cell death by promoting
endoplasmic reticulum-mediated apoptosis. Int J Oncol.
41:2029–2037. 2012.
|