1. Introduction
Transforming growth factor (TGF)-β is a central
regulator in chronic liver disease contributing to fibrogenesis
through inflammation (1). Within
inflammatory microenvironment, TGF-β is secreted by platelets and
Kupffer cells (2). A significant
increase in TGF-β expression is observed in the activated hepatic
stellate cell (HSC), thus indicating that TGF-β acts as an
autocrine positive regulator for ECM production. Responsiveness of
ECM production to TGF-β is transient in the process of tissue
repair such as liver regeneration after acute liver injury
(3,4), thus suggesting that some regulatory
mechanisms for the TGF-β signal are present in the activated HSC.
In contrast, persistent TGF-β signal associated with the
accelerated ECM accumulation is a common finding in human chronic
liver diseases of different etiologies (5), indicating that HSC lose their
negative regulation for ECM accumulation.
TGF-β inhibits hepatocyte proliferation, but it also
promotes hepatocellular carcinoma (HCC). TGF-β has been shown to
play both tumor-suppressive and tumor promoting roles (6–8). As
disease progresses toward malignancy, HCC gains advantage by
selective reduction of the tumor-suppressive activity of TGF-β
together with augmentation of TGF-β oncogenic activity (7). In concert with mitogens, TGF-β
induces accumulation of extracellular matrix (ECM), while mitogenic
signaling antagonizes cytostatic TGF-β function (9,10).
Recent studies have emphasized the possibility of the Smad family
involvement in the pathogenesis of fibrosis and carcinogenesis
(fibro-carcinogenesis).
Because TGF-β is involved in a variety of
physiologic processes such as liver regeneration, unraveling the
molecular mechanisms of TGF-β signal in a pathologic condition is
critical to our understanding of its role in disease and the
development of its therapies (1).
In this review, we first summarize cell-type specific and
context-dependent TGF-β signaling, especially focusing on dynamism
of phosphorylated Smad mediators. We next discuss differential
regulation of TGF-β/Smad signaling after acute or chronic liver
injuries. We then consider how chronic inflammation associated with
hepatitis virus infection promotes hepatic fibro-carcinogenesis.
Finally, we show reversibility of Smad phospho-isoform signaling
after anti-viral therapy.
2. TGF-β signaling
Linker phosphorylation can modify
COOH-terminally phosphorylated Smad2/3 signaling
TGF-β pathway involves the receptor-activated Smads
(Smad2 and Smad3) through direct serine phosphorylation of COOH
termini by TβRI upon TGF-β binding (11). TβRI mediated phosphorylation of
Smad2 and Smad3 induces their association with the shared partner
Smad4, followed by translocation into the nucleus where these
complexes activate transcription of specific genes (12). Smad2 and Smad3 proteins contain a
conserved Mad homology (MH)1 domain that binds DNA, and a conserved
MH2 domain that binds to receptors, Smad4, and transcription
co-activators (11). More
divergent linker regions separate the two domains (12). The linker domain undergoes
regulatory phosphorylation by Ras/mitogen-activated protein kinase
(MAPK) pathways including extracellular signal-regulated kinase
(ERK), c-Jun N-terminal kinase (JNK), p38 MAPK, and
cyclin-dependent kinase (CDK)-2/4, as well as glycogen synthase
kinase 3-β, Ca(2+)-calmodulin-dependent protein kinase II, and G
protein-coupled receptor kinase-2 (13–22).
Antibodies (Abs) reactive with structurally-related
phosphorylated peptides are emerging as valuable tools for
determining phosphorylation sites, and for investigating distinct
signals via the phosphorylated domains. To elucidate how linker
phosphorylation modulates Smad signaling through COOH-tail
phosphorylation, we generated several types of Abs, which
selectively react with individual phosphorylated domains in Smad2/3
(23). Domain-specific
phospho-Smad2/3 Abs have allowed us to reveal that TβRI and
JNK/CDK4 differentially phosphorylate Smad2/3 to create 3
phosphorylated forms (phospho-isoforms): COOH-terminally
phosphorylated Smad2/3 (pSmad2C and pSmad3C), linker phosphorylated
Smad2/3 (pSmad2L and pSmad3L), and dually phosphorylated Smad2/3
(pSmad2L/C and pSmad3L/C) (23).
Except for pSmad2L with cytoplasmic localization (13,24),
the other phospho-isoforms are localized to cell nuclei (15,18,19,
22,24–32).
Linker phosphorylation can modify COOH-terminally phosphorylated
Smad2/3 signaling (13–15,19,20,25).
Differential localization of kinases and phosphatases in the
cytoplasm or nucleus raises the intriguing possibility of different
temporal dynamics for cytoplasmic or nuclear Smad phospho-isoforms,
and adds to the repertoire of signaling responses that determine
cell-fate decisions (23).
Immunohistochemical and immunofluorescence analyses using specific
Abs in human tissues can examine clinical significance of
context-dependent and cell type-specific signaling mediated by Smad
phospho-isoforms, by comparison of tissue/cellular localization of
these phospho-isoforms in various pathologic specimens (23).
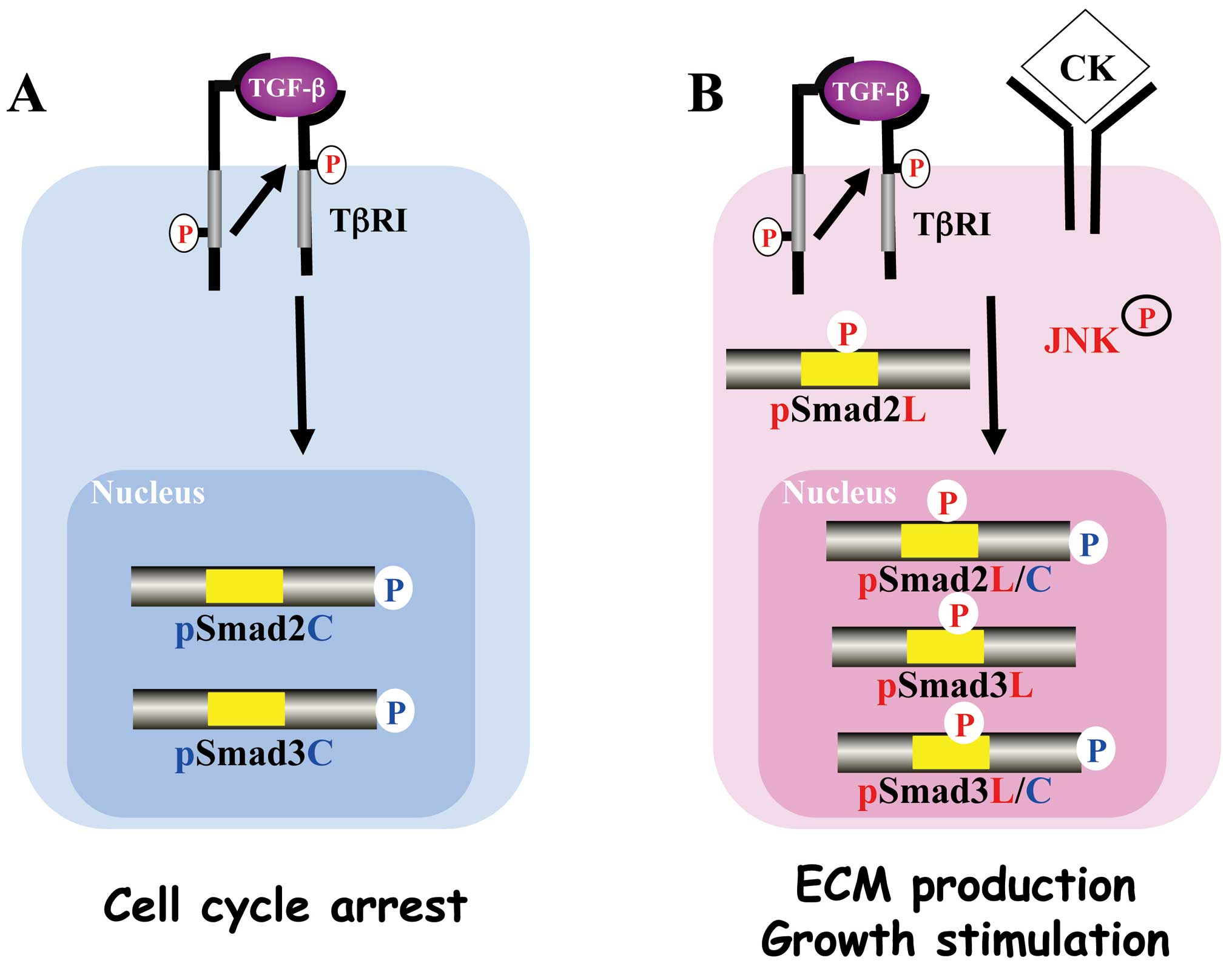
Canonical cytostatic TβRI/pSmad3C
signaling pathway
In canonical Smad signaling pathway, the activated
TβRI is well established as being starting point for signal
propagation to Smad3 (33). After
Smad3 is phosphorylated by the activated TβR1 on the C-terminal SXS
motif (Fig. 1A), pSmad3C forms the
complex with the common partner Smad4 (11). The complex translocates to the
nucleus, where they regulate target gene expression both direct DNA
binding and by interaction with other transcription factors,
co-activators, and co-repressors (34). This pathway is regulated by several
auto-inhibitory feedback loops (35). In particular, Smad7 interacts
stably with activated TβR1 receptor to inhibit TGF-β-mediated COOH
tail phosphorylation of Smad2 and Smad3 (36,37).
TGF-β represents a major growth inhibitory signal in
normal epithelial cells such as hepatocytes (6). In the context of cell cycle control,
the most important targets of action by TGF-β are the genes
encoding the two CDK inhibitors p15INK4B and
p21Cip1, that inhibit CDKs and downregulate c-Myc
expression (38). As the
cytostatic effects of TGF-β are reversible, pSmad3C is negatively
regulated by a number of phosphatases, such as protein phosphatase
1A, magnesium-dependent (PPM1A) (39). PPM1A overexpression abolishes, and
PPM1A depletion enhances TGF-β-induced anti-proliferative responses
(39). Furthermore, PPM1A binds
with pSmad3C to facilitate nuclear export of dephosphorylated Smad3
to the cytosol (22). Thus, C-tail
dephosphorylation mediates Smad recycling for further signaling and
thereby links duration of signaling to the presence of activated
TβRI (40).
Non-canonical Smad signaling
pathways
Mitogenic JNK/pSmad3L signaling
JNK is a serine/threonine kinase affecting
proliferation, differentiation, survival, and migration. JNK can
phosphorylate Smad3 at linker region (26). In contrast to cytoplasmic retention
of pSmad2L, pSmad3L is not retained in the cytoplasm (Fig. 1B). Both pSmad3C and pSmad3L can
form hetero-complexes with Smad4, and Smad complex move to the
nucleus (19). Because nuclear
hetero-oligomerization is essential to assembly of target-specific
transcriptional complexes (34),
Smad3 can utilize 2 different phospho-domains to transmit different
signals, as both a tumor suppressor and a tumor promoter (23).
JNK-mediated pSmad3L and TβRI-mediated pSmad3C
signals oppose each other; most importantly, the balance can shift
between cell growth and growth inhibition. Linker phosphorylation
of Smad3 indirectly inhibits its COOH-terminal phosphorylation and
subsequently suppresses tumor-suppressive pSmad3C signaling. Linker
phosphorylation can modify COOH-terminally phosphorylated Smad2/3
signaling (13–15,19,20,25).
By using genetic as well as pharmacologic approaches, we showed
that blockade of linker phosphorylation abolished oncogenic
properties in Ras-transformed cells and restored the
TβRI/pSmad3Cmediated tumor-suppressive function present in parental
epithelial cells (25).
Invasive/fibrogenic TβRI/JNK/pSmad2L/C
signaling
Activated JNK retains most Smad2 proteins in the
cytoplasm (13,25). Smad2 can accumulate in the nucleus
only if its C-terminus is phosphorylated under conditions of
sustained linker phosphorylation by JNK (Fig. 1B). Smad2 or Smad3 deficient mouse
embryo-derived fibroblasts suggest that both Smad2 and Smad3 are
required for induction of plasminogen activator inhibitor (PAI)-1
(41). Smad3 cooperates with Smad4
to activate the PAI-1 promoter in a TGF-β independent manner
(42). Smad3 mutant (Smad3SD), in
which the C-terminal serines are replaced by aspartic acids,
localized in the nucleus to activate PAI-1 transcription in TGF-β
independent fashion (43).
Importantly, Smad3SD mutant lacks induction of target genes
required for growth inhibition (43). Moreover, Smad3 phospho-mimetic
mutation in the linker domain enhance PAI-1 mRNA and protein
(44). pSmad2L/C undergoes
translocation to the nucleus, where it binds to pSmad3L and Smad4
complex (18,15), which in turn stimulates PAI-1
transcription (18). PAI-1
facilitates cell invasion (45)
and induces ECM deposition (46).
Mitogenic TβRI/CDK/pSmad2/3L/C
signaling
Liu group previously reported that Smad3 was
phosphorylated by CDK4 in vivo and in vitro (47). CDK4-mediated phosphorylation of
Smad3 at its linker region inhibits its transcriptional activity
and the anti-proliferative activity of TGF-β in fibroblasts
(14,48). We have confirmed that the nuclear
cyclin D1/CDK4 complex of fibroblasts activated by TGF-β and PDGF
signaling directly phosphorylates the linker segment of pSmad2C to
produce pSmad2L/C (15). The
expression of c-Myc in fibroblasts is initially repressed by TGF-β,
but subsequent cyclin D1/CDK4 undergoes a complete functional
change to stimulate c-Myc (15).
TGF-β inhibits cell growth by downregulating the c-Myc via the
pSmad2C and pSmad3C pathways (Fig.
2A, left). Moreover, Hayashida et al reported that
pSmad3L/C increases collagen I synthesis in human mesangial cells
(49) (Fig. 2A, right).
 | Figure 2Differential regulation of TGF-β/Smad
signaling after acute or chronic liver injuries. (A) After acute
liver injury, loss of hepatocytes rapidly induces a wave of cell
proliferation. TGF-β plays important roles during liver
regeneration. TGF-β inhibits HSC growth by downregulating c-Myc
expression by pSmad2C and pSmad3C pathways (left); TGF-β signaling
in turn enhances HSC growth and collagen synthesis via the
CDK4-dependent pSmad2L/C and pSmad3L/C pathways induced by cytokine
(CK) signal (right). However, Smad7 induced by pSmad3L/C signal
terminates the fibrogenic phospho-Smad signaling. This
negative-feedback mechanism of the fibrogenic TGF-β/CK signal
results in a transient collagen synthesis in the activated HSC,
which may thus contribute to tissue repair. (B) Several conditions
in chronically damaged livers favor human hepatocarcinogenesis,
mostly resulting from recurrent cycles of cellular proliferation,
inflammation and fibrosis. In MFB and pre-neoplastic hepatoycytes,
CK activates JNK, which phosphorylates Smad2L and Smad3L (left).
The JNK-mediated Smad3L phosphorylation leads to a hetero-complex
of Smad3 with Smad4 in the nucleus where the complex stimulates MFB
and pre-neoplastic hepatycyte growth by upregulation of c-Myc
transcription. After COOH-tail phophorylation of cytoplasmic
pSmad2L by TGF-β signal, pSmad2L/C translocates to the nucleus
where it binds to the pSmad3L and Smad4 complex, which then
stimulates plasminogen activator inhibitor type I (PAI-1) gene
transcription (right). In contrast of Smad7 induction in HSC via
pSmad3C pathway, pSmad3L cannot induce Smad7 in MFB and
pre-neoplastic hepatoycyte (left). Under a low level of Smad7, the
fibrogenic phospho-Smad signaling can constitutively promote ECM
deposition by MFB, which may eventually develop into accelerated
liver fibro-carcinogenesis. |
Non-Smad pathway
TGF-β also uses non-Smad signaling pathways
including JNK and p38 MAPK pathways to convey the same invasive/
fibrogenic signals (50). Tumor
necrosis factor (TNF)-receptor-associated factor 6 (TRAF6) and
TGF-β associated kinase 1 (TAK1) have recently been shown to be
crucial for the activation of the MAPK (51–53).
TAK1 pathway is known to regulate cell survival, migration and
invasion.
Especially important among genes induced by JNK
pathway are the 2 immediate early genes encoding the Fos and c-Jun
transcription factors. Once synthesized, these proteins can
associate with one another to form activator protein (AP)-1, a
widely acting heterodimeric transcription factor that is often
found in hepatocarcinogenesis and liver fibrosis (54). TGF-β and pro-inflammatory cytokines
elicit signaling responses through JNK/non-Smad pathway (50). In JNK1−/− mice, both
fibrosis and HCC development are prevented. Collagen deposition is
marked in wild-type and JNK2−/− mice, but is less dense
in JNK1−/− mice, suggesting the importance of JNK1 in
development of liver fibrosis (55). JNK1−/− mice exhibit
impaired liver carcinogenesis, with smaller and fewer tumor masses
(56). Importantly,
JNK1−/− mice displayed decreased HCC proliferation in a
carcinogenic model and decreased hepatocytic growth in a model of
liver regeneration. In both instances, impaired proliferation is
caused by increased expression of p21WAF1, a cell-cycle
inhibitor, and reduced expression of c-Myc, a negative regulator of
p21WAF1.
These observations suggest cross-talk between TGF-β
induced non-Smad signaling and non-canonical Smad pathway in the
nucleus appear to play an important role during the liver fibrosis
and carcinogenesis. The recognition of non- Smad and non-canonical
Smad pathway as a potent driver of fibrocarcinogenesis makes it
urgent to investigate in more detail the molecular mechanisms by
which TGFβ promotes its oncogenic effects.
3. Differential regulation of the Smad
phospho-isoform signaling between acute and chronic liver
diseases
Acute liver injury
Compensatory growth of the liver to regain mass lost
by partial hepatectomy and chemical damage is orchestrated by
interplay of positive and negative polypeptide cytokines and growth
factors (57). After acute liver
damage, transient release of inflammatory cytokines participate in
the restoration (57). Patches of
quiescent cells are stimulated by cytokines to move into a primed
state (G0→G1), when growth factors can stimulate DNA synthesis and
cellular replication (58). If
hepatocytes are damaged so that this response is impaired,
hepatocytes may be derived from progenitor/stem cells located in
the vicinity of the canals of the Hering (58).
We reported that plasma TGF-β levels increased after
acute liver injury, and the anti-proliferative response to TGF-β
decreased in hepatocytes by downregulation of TGF-β receptor
expression in rat livers (3,4). In
HSC, whenever TGF-β is increased, TGF-β could transduce its signal
for ECM production via its receptor because signaling receptors
were expressed constantly (4).
From the available evidence, examples of acute liver hepatitis are
followed by complete or near-completed resolution and return of the
liver to normal (58).
We further examined in more detail TGF-β signaling
in hepatocytes and HSC during acute liver injury, focusing on
pSmad2L/C and pSmad3L/C pathways in chemically injured rat livers
(18,26). These phospho-isoforms are involved
in collagen synthesis and transmit a proliferative, invasive TGF-β
signal in mesenchymal cells (18,26).
Nuclear localization of pSmad2L/C and pSmad3L/C is seen in the
activated HSC (26). In
particular, strong Smad2/3 phosphorylation at the COOH-tail and
threonine residues in the linker regions is observed in the
activated HSC (unpublished data). Because TGF-β, pro-inflammatory
cytokines and PDGF activate JNK pathway in HSC (26), pro-inflammatory cytokines and PDGF
can convert a cytostatic TGF-β signal to a collagen-producing
character in activated HSC under the influence of inflammatory
microenvironments (Fig. 2A,
right). Collectively, pSmad2L/C and pSmad3L/C signaling may
mobilize HSC from the space of Disse to sites of damage, where the
activated HSC contribute to tissue repair by producing large
amounts of collagens.
In HSC after acute liver injury, TβRI activated by
endogenous TGF-β signal phosphorylated Smad3C, further upregulating
Smad7 transcription (Fig. 2A,
right) (59). Subsequently, Smad7
terminates fibrogenic signals mediated by pSmad2L/C and pSmad3L/C,
and could be involved in transient response to the autocrine TGF-β
signal after acute liver injury (26,59).
In the same way, the activation of Smad2/3 was tightly restricted
in primary cultured HSC (26,59).
Taken together, Smad7 is involved in this tight restriction of the
non-canonical Smad signaling in HSC and regulates the intensity and
duration of the TGF-β responses (60).
Chronic liver injury
Chronic inflammation causes progressive liver
fibrosis (Fig. 2B). Fibrogenesis
is a mechanism of wound healing and repair (61). However, prolonged injury causes
deregulation of the normal processes and results in extensive
deposition of ECM proteins and fibrosis (62). Activation of HSC is a key step in
liver fibrogenesis (62). When
freshly isolated and cultured, quiescent HSC have a low
proliferative rate and very modest fibrogenic potential and lack of
contractile properties (63).
Therefore, the main function of these quiescent HSC is considered
to be the storage and metabolism of vitamin A (2). However, following liver injury of any
etiology, HSC undergo activation. Activated HSC show increased
proliferation, motility and ECM production (64,65).
A number of cytokines, continuously released by damaged Kupffer
cells and endothelial cells, can change activated HSC to
myofibroblasts (MFB) (66). These
include TGF-β, PDGF and ET-1, which stimulate transcription factors
such as Sp1, c-jun, STAT-1 and Smad proteins that regulate gene
expression (67–70). MFB perpetuate their own activation
through several autocrine loops, including the secretion of TGF-β
and upregulation of its receptors (59). Following chronic liver injury,
there is a marked accumulation of α-smooth muscle actin (α-SMA)
positive cells at the sites of active liver fibrosis (71,72).
The most powerful growth factor for MFB is PDGF (68). Moreover, following cell activation,
there is upregulation of PDGF receptors in MFB, which in turn can
secrete this potent mitogen (73).
During transdifferentiation from HSC to MFB in
culture, pSmad3C-mediated signal decreases while pSmad3L pathway
predominates (18). The
observations fully support the finding of pSmad3L rather than
pSmad3C in nuclei of α-SMA-immunoreactive MFB in portal tracts of
chronically HCV-infected liver specimens (27). The presence of α-SMA is associated
with transdifferentiation of HSC into scar-forming MFB, an event
that is considered pivotal in the fibrogenic response (2).
In contrast to a transient increase in Smad7 in the
activated HSC after acute liver injury, Smad7 remains at a low
level in MFB throughout chronic liver injury (59). Because Smad7 cannot be induced by
the pSmad3L pathway (unpublished data), the lack of Smad7 induction
in MFB during chronic liver disease might lead to constitutive
fibrogenic TGF-β (59,74,75).
Accordingly, Smad7 overexpression results in less accumulation of
interstitial collagens and improves liver fibrosis (76). Moreover, interferon (IFN)-γ
displays antifibrotic effects by upregulation of Smad7 expression
(77).
Although MFB have been considered the primary cells
involved in development of liver fibrosis, possible direct
involvement of hepatocytes in fibrosis has not been examined. In
parallel with emergence of epithelial-to-mesenchymal (EMT) paradigm
in fibro-carcinogenesis, a large body of work has established roles
for epithelial cells as important mediators of progressive fibrosis
(27,62). During progression of HCV-related
chronic liver disorders, our current data indicated that
hepatocytes affected by chronic inflammation undergo transition
from the tumor-suppressive pSmad3C pathway, which is characteristic
of mature hepatocytes, to the JNK/pSmad3L pathway, which appears to
favor the state of flux shown by MFB, accelerating liver fibrosis.
These phenomena were also observed in HBV-related liver disease
(28).
Hepatocarcinognesis
HCC is the sixth most common cancer and third most
frequent cause of cancer-related death worldwide (78,79).
Although there is a growing understanding of the molecular
mechanisms that induce hepatocarcinogenesis, the mechanisms have
not been completely elucidated. Chronic infections with HBV or HCV
appear to be the most significant causes of HCC (80). Recent studies reveal that the
development and progression of HCC are caused by the accumulation
of genetic changes, thus resulting in altered expression of
cancer-related genes (81).
As HBV contains partially double stranded-DNA, it
can directly cause HCC by integrating its DNA into the host genome.
HBV genomic integration is present in 85 to 90% of livers
developing HBV-related HCC, usually even before the development of
HCC (82). Integration of HBV DNA
is not restricted to HCC, but is also found in non-tumor tissue in
patients with chronic HBV infection (83,84).
HBV integration induces a wide range of genetic alterations within
the host genome, including chromosomal deletions, translocations,
production of fusion transcripts, amplification of cellular DNA and
generalized genomic instability (85,86).
HBx protein encoded by the X gene has been suspected as a viral
oncoprotein participating in hepatocarcinogenesis (87). HBx was shown to potentiate
c-Myc-induced liver carcinogenesis in transgenic mice (88).
Unlike HBV, HCV is a positive, single-strand RNA
virus, apparently incapable of integration into the host’s genome.
The HCV components modulate a number of cellular regulatory
functions by targeting a wide spectrum of cellular signaling
pathways (89,90–96).
HCV core expression has been shown to induce activation of the JNK
pathway in regulation of vascular endothelial growth factor
(96). NS5A acts as a positive
regulator of the JNK signaling pathway by interacting with TNF
receptor-associated factor 2, which may be highly important in HCV
pathogenesis (97). In an HCV
infection model, Lin et al demonstrated that HCV directly
induced TGF-β release from hepatocytes in a reactive oxygen species
(ROS)-dependent and JNK-dependent manner (98). Moreover, recent studies using
transgenic mouse models indicated that HCV is involved directly in
hepatocarcinogenesis. Three different HCV core transgenic lines
develop liver steatosis and HCC (99–101).
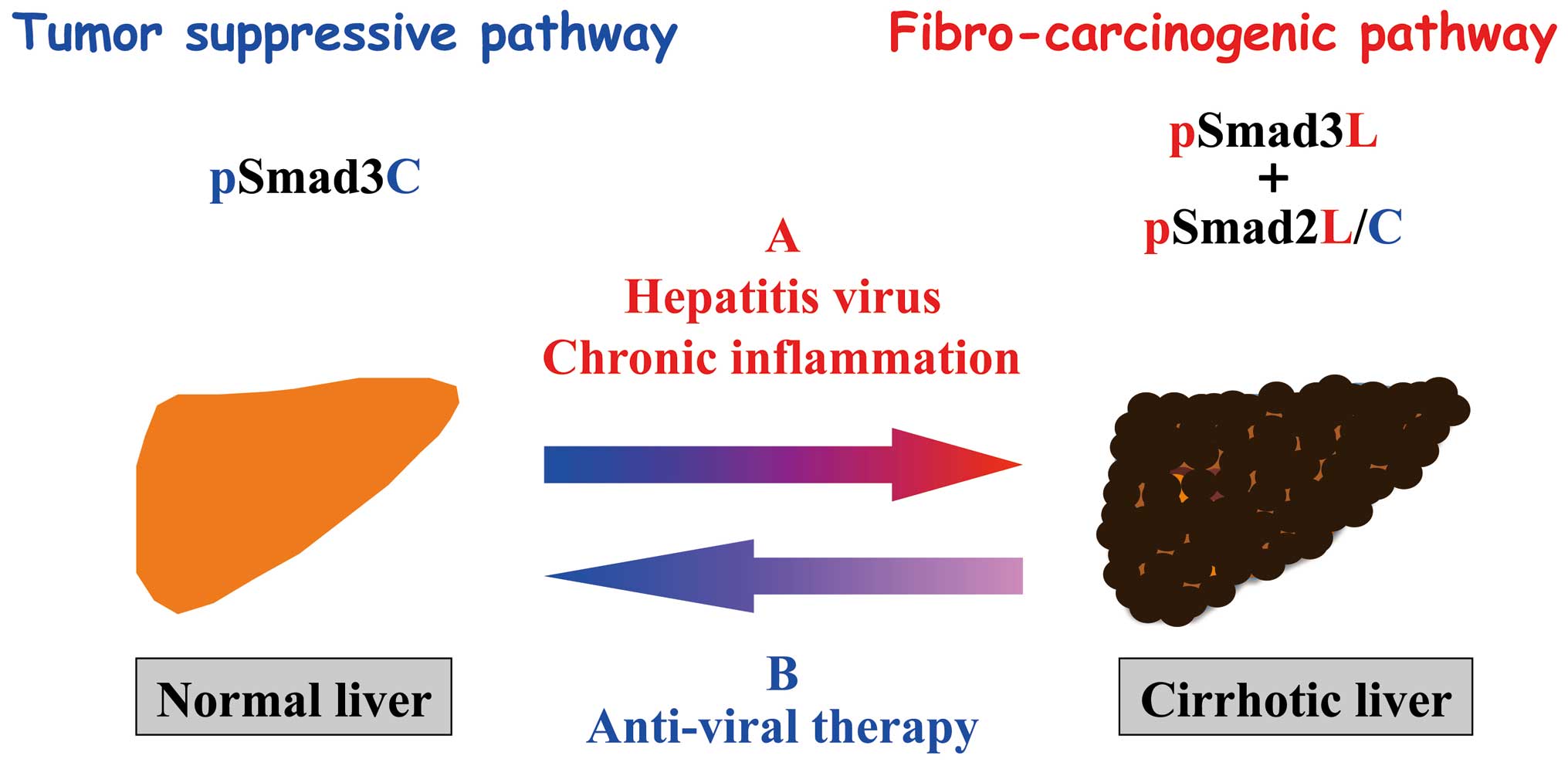
We have shown that in patients with chronic liver
disease progression, HBV or HCV components and pro-inflammatory
cytokine additively activate JNK to shift Smad phospho-isoform
signaling from tumor-suppressive TβRI/pSmad3C pathway to
carcinogenic JNK/pSmad3L pathway and fibrogenic pSmad2L/C pathway,
accelerating liver fibrosis and increasing the risk of HCC
(Fig. 3A). To support this notion,
high level of linker Smad3 phosphorylation is reported both in HCC
specimens and human HCC cell lines (102). Moreover, specimens from patients
with chronic hepatitis B who develop HCC show abundant hepatocytic
Smad3L but limited Smad3C phosphorylation in hepatocytic nuclei,
whereas other patients with abundant heptocytic pSmad3C but limited
pSmad3L do not develop HCC (28).
The same relationships are observed in human hepatitis C
virus-related hepatocarcinogenesis (27).
4. Reversible Smad signaling after
successful therapies against hepatitis viruses
Chronic hepatitis B and C are now treatable
diseases. Interferon therapy and nucleoside analogues are available
for HBV. Lamivudine and four other nucleoside and nucleoside
analogues have been licensed (103): adefovir (in 2002) (104), entecavir (in 2005) (105), telbivudine (in 2006) (106), and most recently, tenofovir
disoproxil fumarate (in 2008). These nucleoside analogues suppress
HBV replication through inhibition of reverse transcriptase and DNA
polymerase, and inhibit reverse transcription of pregenomic RNA to
HBV DNA (103). On the other
hand, the current treatment of hepatitis C is pegylated IFN
(PEG-IFN)-α, given by subcutaneous injection once weekly, and oral
ribavirin (RBV) daily. RBV is a guanosine nucleoside analogue. This
agent shows only modest activity against hepatitis C but it
increases the activity of IFN-α when the 2 agents are used in
combination (107). Efficacy of
PEG-IFN and RBV has been investigated in several controlled trials
that demonstrated an overall SVR rate of 40 to 50% (107). However, limitations of IFN and
RBV treatment have prompted a continuing search for improved
therapies. Various molecular targets are the focus of anti- HCV
drug development, several new NS3 protease inhibitors, NS5b
nucleoside polymerase inhibitors, and non-nucleoside polymerase
inhibitors are being assessed in phase III studies.
Previous studies have shown that successful
anti-viral therapy can improve biochemical liver function
parameters as well as histological findings (108). Patients with mild liver fibrosis
are likely to show histologically evident decreases in fibrosis and
inflammation after a sustained virological response (SVR) in
response to IFN treatment against HCV infection (108). Furthermore, treated patients show
marked reduction in decompensated liver disease and HCC occurrence
(109,110). Patients with advanced fibrosis,
however, retain relatively low but still considerable risks of HCC
occurrence and hepatic decompensation despite having attained SVR
(109).
Clinical analyses of pSmad3L and pSmad3C in liver
disease progression have provided substantial mechanistic insight.
After achievement of SVR, IFN or an oral nucleoside therapy could
restore Smad phospho-isoform signaling from oncogenic pSmad3L to
tumor-suppresssive pSmad3C pathway shown by normal hepatocytes both
in chronic hepatitis B and C (Fig.
3B) (31,32). In contrast, patients with advanced
liver fibrosis progressed to HCC despite improved inflammatory
activity, because hepatocytes maintained high pSmad3L and low
pSmad3C signaling (31). One
reason why pSmad3L level remains high may be that chronic HBV or
HCV infection and chronic inflammation no longer play critical
roles in HCC development in later cirrhotic livers after
pre-neoplastic hepatocytes have acquired oncogenic signaling caused
by genetic alteration and epigenetic changes. These clinical
observations support roles for pSmad3C as a tumor suppressor and
pSmad3L as a promoter during hepatic carcinogenesis.
Antiviral therapy can achieve recovery of liver
inflammation and fibrosis in HBV or HCV infected patients.
Moreover, we also demonstrated that hepatocytic tumor-suppressive
pSmad3C signaling shifted to fibro-carcinogenic pSmad3L signaling
as the livers progressed from chronic hepatitis B and C infection
to HCC, and suppression of liver inflammation and regression of
fibrosis by an anti-viral therapy resulted in the reduction of
pSmad3L and increase in pSmad3C signaling. Likewise, oncogenic
c-Myc and fibrogenic PAI-1 expression was significantly decreased
in the livers post-anti-HBV or HCV-treated patients. Both HBV and
HCV trigger changes in gene expression, which is mediated by
genetic or epigenetic alterations. The contribution of HBV to HCC
involves the expression of HBx; for HCV, the core protein, and
nonstructural protein NS3 and NS5A contribute to oncogenic
transformation (94,97). Suzuki et al reported a −0.6
improvement in HBe-antigen negative chronic hepatitis B patients
after one year lamivudine treatment (111). We also found that fibrosis
regressed −1 point after 52 weeks of treatment of anti- HBV
treatment in the livers of chronic HBV patients (32). On the contrary, fibrosis regression
rate was −0.28 point/year in HCV infected patients after SVR
(31). These results indicate that
treatment with nucleoside analogues resulted in a 3–4 times faster
fibrosis regression rate in HBV-infected patient livers compared
with that of IFN treated HCV-infected patient. Notably, the
fibro-carcinogenesis regression rate was much faster in HBV than
that of HCV-related liver disease (32). These data coincide with the
clinical observations that decompensated HBV-infected patients
often show disappearance of ascites or jaundice even in advanced
stages after an oral nucleoside therapies. As the hepatic
fibrocarcinogenesis seems to be a multistep process, the
differences of virus specific genetic changes and biological
consequences may cause the alternation of fibrosis regression rate
between HBV and HCV. During HCV-related carcinogenesis,
JNK-activated chronic inflammation confers a selective advantage on
preneoplastic hepatocytes by shifting Smad3 signaling from the
tumor-suppressive pSmad3C to the oncogenic pSmad3L pathway
(27). On the other hand, HBx
oncoprotein participates directly in hepatocarcinogenesis by
shifting hepatocytic Smad3-mediated signaling from tumor
suppression to oncogenesis in patients with chronic hepatitis B
infection (28). Such differences
may result in faster progression or regression rate of
fibrocarcinogenesis in HBV patients compared with that in HCV
patients during, before, and after anti-viral therapy.
5. Problems and future perspectives
In this review, we describe TGF-β/Smad
phospho-isoform signaling during acute and chronic liver diseases.
Anti-viral therapy has been shown to decrease the risk of HCC by
shifting fibro-carcinogenic pSmad3L signaling to tumor-suppressive
pSmad3C in hepatocytes. Therefore, understanding molecular
mechanisms of human fibro-carcinogenesis is of fundamental
importance in guiding development of effective prevention and
treatment for hepatic fibrosis and HCC. Additionally, Smad
phospho-isoform signaling can be used as a new predictive biomarker
for early assessment of pharmacologic interventions to suppress
human fibro-carcinogenesis.
References
|
1
|
Dooley S and Ten Dijke P: TGF-beta in
progression of liver disease. Cell Tissue Res. 347:245–256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pinzani M and Macias-Barragan J: Update on
the pathophysiology of liver fibrosis. Expert Rev Gastroenterol
Hepatol. 4:459–472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Date M, Matsuzaki K, Matsushita M, et al:
Differential expression of transforming growth factor-beta and its
receptors in hepatocytes and nonparenchymal cells of rat liver
after CCl4 administration. J Hepatol. 28:572–581. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Date M, Matsuzaki K, Matsushita M, Tahashi
Y, Furukawa F and Inoue K: Modulation of transforming growth factor
beta function in hepatocytes and hepatic stellate cells in rat
liver injury. Gut. 46:719–724. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kisseleva T and Brenner DA: Mechanisms of
fibrogenesis. Exp Biol Med (Maywood). 233:109–122. 2008. View Article : Google Scholar
|
|
6
|
Moses HL and Serra R: Regulation of
differentiation by TGF-beta. Curr Opin Genet Dev. 6:581–586. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roberts AB and Sporn MB: The transforming
growth factor-βs. Peptide Growth Factors and Their Receptors I
Berlin: Springer; pp. 419–472. 1990
|
|
8
|
Bellam N and Pasche B: Tgf-beta signaling
alterations and colon cancer. Cancer Treat Res. 155:85–103. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsuzaki K: Smad phosphoisoform signaling
specificity: the right place at the right time. Carcinogenesis.
32:1578–1588. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matsuzaki K: Smad phosphoisoform signals
in acute and chronic liver injury: similarities and differences
between epithelial and mesenchymal cells. Cell Tissue Res.
347:225–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Derynck R and Miyazono K: The TGF-β
Signaling. Cold Spring Harbor Laboratory Press; NY: 2008
|
|
12
|
Shi Y and Massague J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kretzschmar M, Doody J, Timokhina I and
Massague J: A mechanism of repression of TGFbeta/Smad signaling by
oncogenic Ras. Genes Dev. 13:804–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsuura I, Denissova NG, Wang G, He D,
Long J and Liu F: Cyclin-dependent kinases regulate the
antiproliferative function of Smads. Nature. 430:226–231. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsuzaki K, Kitano C, Murata M, et al:
Smad2 and Smad3 phosphorylated at both linker and COOH-terminal
regions transmit malignant TGF-beta signal in later stages of human
colorectal cancer. Cancer Res. 69:5321–5330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kamaraju AK and Roberts AB: Role of
Rho/ROCK and p38 MAP kinase pathways in transforming growth
factor-beta-mediated Smad-dependent growth inhibition of human
breast carcinoma cells in vivo. J Biol Chem. 280:1024–1036. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wicks SJ, Lui S, Abdel-Wahab N, Mason RM
and Chantry A: Inactivation of smad-transforming growth factor beta
signaling by Ca(2+)-calmodulin-dependent protein kinase II. Mol
Cell Biol. 20:8103–8111. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Furukawa F, Matsuzaki K, Mori S, et al:
p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation
in rat myofibroblasts. Hepatology. 38:879–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mori S, Matsuzaki K, Yoshida K, Furukawa
F, et al: TGF-beta and HGF transmit the signals through
JNK-dependent Smad2/3 phosphorylation at the linker regions.
Oncogene. 23:7416–7429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ho J, Cocolakis E, Dumas VM, Posner BI,
Laporte SA and Lebrun JJ: The G protein-coupled receptor kinase-2
is a TGFbeta-inducible antagonist of TGFbeta signal transduction.
EMBO J. 24:3247–3258. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Millet C, Yamashita M, Heller M, Yu LR,
Veenstra TD and Zhang YE: A negative feedback control of
transforming growth factor-beta signaling by glycogen synthase
kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol
Chem. 284:19808–19816. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alarcon C, Zaromytidou AI, Xi Q, et al:
Nuclear CDKs drive Smad transcriptional activation and turnover in
BMP and TGF-beta pathways. Cell. 139:757–769. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matsuzaki K: Smad phospho-isoforms direct
context-dependent TGF-beta signaling. Cytokine Growth Factor Rev.
24:385–399. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yamagata H, Matsuzaki K, Mori S, et al:
Acceleration of Smad2 and Smad3 phosphorylation via c-Jun
NH(2)-terminal kinase during human colorectal carcinogenesis.
Cancer Res. 65:157–165. 2005.PubMed/NCBI
|
|
25
|
Sekimoto G, Matsuzaki K, Yoshida K, et al:
Reversible Smad-dependent signaling between tumor suppression and
oncogenesis. Cancer Res. 67:5090–5096. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshida K, Matsuzaki K, Mori S, et al:
Transforming growth factor-beta and platelet-derived growth factor
signal via c-Jun N-terminal kinase-dependent Smad2/3
phosphorylation in rat hepatic stellate cells after acute liver
injury. Am J Pathol. 166:1029–1039. 2005. View Article : Google Scholar
|
|
27
|
Matsuzaki K, Murata M, Yoshida K, et al:
Chronic inflammation associated with hepatitis C virus infection
perturbs hepatic transforming growth factor beta signaling,
promoting cirrhosis and hepatocellular carcinoma. Hepatology.
46:48–57. 2007. View Article : Google Scholar
|
|
28
|
Murata M, Matsuzaki K, Yoshida K, et al:
Hepatitis B virus X protein shifts human hepatic transforming
growth factor (TGF)-beta signaling from tumor suppression to
oncogenesis in early chronic hepatitis B. Hepatology. 49:1203–1217.
2009. View Article : Google Scholar
|
|
29
|
Nagata H, Hatano E, Tada M, et al:
Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling
from oncogenesis to tumor-suppression in rat hepatocellular
carcinoma. Hepatology. 49:1944–1953. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kawamata S, Matsuzaki K, Murata M, et al:
Oncogenic Smad3 signaling induced by chronic inflammation is an
early event in ulcerative colitis-associated carcinogenesis.
Inflamm Bowel Dis. 17:683–695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamaguchi T, Matsuzaki K, Inokuchi R, et
al: Phosphorylated Smad2 and Smad3 signaling: Shifting between
tumor suppression and fibro-carcinogenesis in chronic hepatitis C.
Hepatol Res. 43:1327–1342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng YR, Yoshida K, Jin Q, et al:
Reversible phospho-Smad3 signaling between tumor-suppression and
fibro-carcinogenesis in chronic hepatitis B infection. Clin Exp
Immunol. 176:102–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signaling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng XH and Derynck R: Specificity and
versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev
Biol. 21:659–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miyazono K: Positive and negative
regulation of TGF-beta signaling. J Cell Sci. 113:1101–1109.
2000.PubMed/NCBI
|
|
36
|
Nakao A, Afrakhte M, Moren A, et al:
Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta
signalling. Nature. 389:631–635. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hayashi H, Abdollah S, Qiu Y, et al: The
MAD-related protein Smad7 associates with the TGFbeta receptor and
functions as an antagonist of TGFbeta signaling. Cell.
89:1165–1173. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Massague J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar
|
|
39
|
Lin X, Duan X, Liang YY, et al: PPM1A
functions as a Smad phosphatase to terminate TGFbeta signaling.
Cell. 125:915–928. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hill CS: Nucleocytoplasmic shuttling of
Smad proteins. Cell Res. 19:36–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Piek EJW, Heyer J, Escalante-Alcalde D, et
al: Functional characterization of transforming growth factor beta
signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem.
276:19945–19953. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang YFX, We R and Derynck R:
Receptor-associated Mad homologues synergize as effectors of the
TGF-beta response. Nature. 383:168–172. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu XSY, Constantinescu SN, Karam E,
Weinberg RA and Lodish HF: Transforming growth factor beta-induced
phosphorylation of Smad3 is required for growth inhibition and
transcriptional induction in epithelial cells. Proc Natl Acad Sci
USA. 94:10669–10674. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Velden JL AJ, Guala AS, Badura EC and
Janssen-Heininger YM: c-Jun N-terminal kinase 1 promotes
transforming growth factor-β1-induced epithelial-to-mesenchymal
transition via control of linker phosphorylation and
transcriptional activity of Smad3. Am J Respir Cell Mol Biol.
44:571–581. 2011.
|
|
45
|
Hirashima YKH, Suzuki M, Tanaka Y,
Kanayama N and Terao T: Transforming growth factor-beta1 produced
by ovarian cancer cell line HRA stimulates attachment and invasion
through an up-regulation of plasminogen activator inhibitor type-1
in human peritoneal mesothelial cells. J Biol Chem.
278:26793–26802. 2003. View Article : Google Scholar
|
|
46
|
Hu PF, Chen H, Zhong W, Lin Y, Zhang X,
Chen YX and Xie WF: Adenovirus-mediated transfer of siRNA against
PAI-1 mRNA ameliorates hepatic fibrosis in rats. J Hepatol.
51:102–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu F: Smad3 phosphorylation by
cyclin-dependent kinases. Cytokine Growth Factor Rev. 17:9–17.
2006. View Article : Google Scholar
|
|
48
|
Wang G, Matsuura I, He D and Liu F:
Transforming growth factor-{beta}-inducible phosphorylation of
Smad3. J Biol Chem. 284:9663–9673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hayashida T, Decaestecker M and Schnaper
HW: Cross-talk between ERK MAP kinase and Smad signaling pathways
enhances TGF-beta-dependent responses in human mesangial cells.
FASEB J. 17:1576–1578. 2003.PubMed/NCBI
|
|
50
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Landstrom M: The TAK1-TRAF6 signalling
pathway. Int J Biochem Cell Biol. 42:585–589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sorrentino A, Thakur N, Grimsby S, et al:
The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a
receptor kinase-independent manner. Nat Cell Biol. 10:1199–1207.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yamashita M, Fatyol K, Jin C, Wang X, Liu
Z and Zhang YE: TRAF6 mediates Smad-independent activation of JNK
and p38 by TGF-beta. Mol Cell. 31:918–924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kodama Y, Kisseleva T, Iwaisako K, et al:
c-Jun N-terminal kinase-1 from hematopoietic cells mediates
progression from hepatic steatosis to steatohepatitis and fibrosis
in mice. Gastroenterology. 137:1467–1477.e5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hui L, Zatloukal K, Scheuch H, Stepniak E
and Wagner EF: Proliferation of human HCC cells and chemically
induced mouse liver cancers requires JNK1-dependent p21
downregulation. J Clin Invest. 118:3943–3953. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Michalopoulos GK and DeFrances MC: Liver
regeneration. Science. 276:60–66. 1997. View Article : Google Scholar
|
|
58
|
Dooley JS, Lok ASF, Burroughs AK and
Heathcote EJ: Sherlock’s Disease of the Liver and Biliary System.
12th edition. Wiley-Blackwell; 2011
|
|
59
|
Tahashi Y, Matsuzaki K, Date M, et al:
Differential regulation of TGF-beta signal in hepatic stellate
cells between acute and chronic rat liver injury. Hepatology.
35:49–61. 2002. View Article : Google Scholar
|
|
60
|
Yoshida K and Matsuzaki K: Differential
regulation of TGF-beta/Smad signaling in hepatic stellate cells
between acute and chronic liver injuries. Front Physiol. 3:532012.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Friedman SL: Mechanisms of disease:
Mechanisms of hepatic fibrosis and therapeutic implications. Nat
Clin Pract Gastroenterol Hepatol. 1:98–105. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Friedman SL: Evolving challenges in
hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 7:425–436. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rockey DC, Housset CN and Friedman SL:
Activation-dependent contractility of rat hepatic lipocytes in
culture and in vivo. J Clin Invest. 92:1795–1804. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Brenner DA, Waterboer T, Choi SK, et al:
New aspects of hepatic fibrosis. J Hepatol. 32:32–38. 2000.
View Article : Google Scholar
|
|
66
|
Marra F: Chemokines in liver inflammation
and fibrosis. Front Biosci. 7:d1899–d1914. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Reimann T, Hempel U, Krautwald S, Axmann
A, Scheibe R, Seidel D and Wenzel KW: Transforming growth
factor-beta1 induces activation of Ras, Raf-1, MEK and MAPK in rat
hepatic stellate cells. FEBS Lett. 403:57–60. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pinzani M, Gesualdo L, Sabbah GM and
Abboud HE: Effects of platelet-derived growth factor and other
polypeptide mitogens on DNA synthesis and growth of cultured rat
liver fat-storing cells. J Clin Invest. 84:1786–1793. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rockey DC, Fouassier L, Chung JJ, Carayon
A, Vallee P, Rey C and Housset C: Cellular localization of
endothelin-1 and increased production in liver injury in the rat:
potential for autocrine and paracrine effects on stellate cells.
Hepatology. 27:472–480. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Marra F, Arrighi MC, Fazi M, et al:
Extracellular signal-regulated kinase activation differentially
regulates platelet-derived growth factor’s actions in hepatic
stellate cells, and is induced by in vivo liver injury in the rat.
Hepatology. 30:951–958. 1999.
|
|
71
|
Nouchi T, Tanaka Y, Tsukada T, Sato C and
Marumo F: Appearance of alpha-smooth-muscle-actin-positive cells in
hepatic fibrosis. Liver. 11:100–105. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Schmitt-Graff A, Kruger S, Bochard F,
Gabbiani G and Denk H: Modulation of alpha smooth muscle actin and
desmin expression in perisinusoidal cells of normal and diseased
human livers. Am J Pathol. 138:1233–1242. 1991.PubMed/NCBI
|
|
73
|
Pinzani M, Milani S, Herbst H, et al:
Expression of platelet-derived growth factor and its receptors in
normal human liver and during active hepatic fibrogenesis. Am J
Pathol. 148:785–800. 1996.PubMed/NCBI
|
|
74
|
Dooley S, Delvoux B, Lahme B,
Mangasser-Stephan K and Gressner AM: Modulation of transforming
growth factor beta response and signaling during
transdifferentiation of rat hepatic stellate cells to
myofibroblasts. Hepatology. 31:1094–1106. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Stopa M, Anhuf D, Terstegen L, Gatsios P,
Gressner AM and Dooley S: Participation of Smad2, Smad3, and Smad4
in transforming growth factor beta (TGF-beta)-induced activation of
Smad7. THE TGF-beta response element of the promoter requires
functional Smad binding element and E-box sequences for
transcriptional regulation. J Biol Chem. 275:29308–29317. 2000.
View Article : Google Scholar
|
|
76
|
Dooley S, Hamzavi J, Breitkopf K, et al:
Smad7 prevents activation of hepatic stellate cells and liver
fibrosis in rats. Gastroenterology. 125:178–191. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Weng HL, Liu Y, Chen JL, et al: The
etiology of liver damage imparts cytokines transforming growth
factor beta1 or interleukin- 13 as driving forces in fibrogenesis.
Hepatology. 50:230–243. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Parkin DM, Pisani P and Ferlay J: Global
cancer statistics. Cancer J Clin. 49:33–64. 1999. View Article : Google Scholar
|
|
80
|
Bosch FX, Ribes J and Borras J:
Epidemiology of primary liver cancer. Semin Liver Dis. 19:271–285.
1999. View Article : Google Scholar
|
|
81
|
Shiraha H, Yamamoto K and Namba M: Human
hepatocyte carcinogenesis (Review). Int J Oncol. 42:1133–1138.
2013.PubMed/NCBI
|
|
82
|
Jiang Z, Jhunjhunwala S, Liu J, et al: The
effects of hepatitis B virus integration into the genomes of
hepatocellular carcinoma patients. Genome Res. 22:593–601. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Brechot C, Pourcel C, Louise A, Rain B and
Tiollais P: Presence of integrated hepatitis B virus DNA sequences
in cellular DNA of human hepatocellular carcinoma. Nature.
286:533–535. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Shafritz DA, Shouval D, Sherman HI,
Hadziyannis SJ and Kew MC: Integration of hepatitis B virus DNA
into the genome of liver cells in chronic liver disease and
hepatocellular carcinoma. Studies in percutaneous liver biopsies
and post-mortem tissue specimens. N Engl J Med. 305:1067–1073.
1981. View Article : Google Scholar
|
|
85
|
Bonilla Guerrero R and Roberts LR: The
role of hepatitis B virus integrations in the pathogenesis of human
hepatocellular carcinoma. J Hepatol. 42:760–777. 2005.PubMed/NCBI
|
|
86
|
Feitelson MA and Lee J: Hepatitis B virus
integration, fragile sites, and hepatocarcinogenesis. Cancer Lett.
252:157–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Terradillos O, Billet O, Renard CA, Levy
R, Molina T, Briand P and Buendia MA: The hepatitis B virus X gene
potentiates c-myc-induced liver oncogenesis in transgenic mice.
Oncogene. 14:395–404. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Feitelson MA: c-myc overexpression in
hepatocarcinogenesis. Hum Pathol. 35:1299–1302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hayashi J, Aoki H, Kajino K, Moriyama M,
Arakawa Y and Hino O: Hepatitis C virus core protein activates the
MAPK/ERK cascade synergistically with tumor promoter TPA, but not
with epidermal growth factor or transforming growth factor alpha.
Hepatology. 32:958–961. 2000. View Article : Google Scholar
|
|
90
|
Erhardt A, Hassan M, Heintges T and
Haussinger D: Hepatitis C virus core protein induces cell
proliferation and activates ERK, JNK, and p38 MAP kinases together
with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line.
Virology. 292:272–284. 2002. View Article : Google Scholar
|
|
91
|
He Y, Nakao H, Tan SL, et al: Subversion
of cell signaling pathways by hepatitis C virus nonstructural 5A
protein via interaction with Grb2 and P85 phosphatidylinositol
3-kinase. J Virol. 76:9207–9217. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Qadri I, Iwahashi M, Capasso JM, Hopken
MW, Flores S, Schaack J and Simon FR: Induced oxidative stress and
activated expression of manganese superoxide dismutase during
hepatitis C virus replication: role of JNK, p38 MAPK and AP-1.
Biochem J. 378:919–928. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhao LJ, Wang L, Ren H, Cao J, Li L, Ke JS
and Qi ZT: Hepatitis C virus E2 protein promotes human hepatoma
cell proliferation through the MAPK/ERK signaling pathway via
cellular receptors. Exp Cell Res. 305:23–32. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Hassan M, Ghozlan H and Abdel-Kader O:
Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is
essential for the stimulation of hepatitis C virus (HCV)
non-structural protein 3 (NS3)-mediated cell growth. Virology.
333:324–336. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Choi SH and Hwang SB: Modulation of the
transforming growth factor-beta signal transduction pathway by
hepatitis C virus nonstructural 5A protein. J Biol Chem.
281:7468–7478. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hassan M, Selimovic D, Ghozlan H and
Abdel-Kader O: Hepatitis C virus core protein triggers hepatic
angiogenesis by a mechanism including multiple pathways.
Hepatology. 49:1469–1482. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Park KJ, Choi SH, Choi DH, Park JM, Yie
SW, Lee SY and Hwang SB: Hepatitis C virus NS5A protein modulates
c-Jun N-terminal kinase through interaction with tumor necrosis
factor receptor-associated factor 2. J Biol Chem. 278:30711–30718.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lin W, Tsai WL, Shao RX, Wu G, Peng LF,
Barlow LL, Chung WJ, et al: Hepatitis C virus regulates
transforming growth factor beta1 production through the generation
of reactive oxygen species in a nuclear factor kappaB-dependent
manner. Gastroenterology. 138:2509–2518. 2518.e12010. View Article : Google Scholar
|
|
99
|
Moriya K, Yotsuyanagi H, Shintani Y, Fujie
H, Ishibashi K, Matsuura Y, Miyamura T, et al: Hepatitis C virus
core protein induces hepatic steatosis in transgenic mice. J Gen
Virol. 78:1527–1531. 1997.PubMed/NCBI
|
|
100
|
Moriya K, Fujie H, Shintani Y, Yotsuyanagi
H, Tsutsumi T, Ishibashi K, Matsuura Y, et al: The core protein of
hepatitis C virus induces hepatocellular carcinoma in transgenic
mice. Nat Med. 4:1065–1067. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lerat H, Honda M, Beard MR, Loesch K, Sun
J, Yang Y, Okuda M, et al: Steatosis and liver cancer in transgenic
mice expressing the structural and nonstructural proteins of
hepatitis C virus. Gastroenterology. 122:352–365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Dzieran J, Fabian J, Feng T, et al:
Comparative analysis of TGF-beta/Smad signaling dependent
cytostasis in human hepatocellular carcinoma cell lines. PLoS One.
8:e722522013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Dienstag JL, Schiff ER, Wright TL, et al:
Lamivudine as initial treatment for chronic hepatitis B in the
United States. N Engl J Med. 341:1256–1263. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Marcellin P, Chang TT, Lim SG, et al:
Adefovir dipivoxil for the treatment of hepatitis B e
antigen-positive chronic hepatitis B. N Engl J Med. 348:808–816.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chang TT, Gish RG, de Man R, et al: A
comparison of entecavir and lamivudine for HBeAg-positive chronic
hepatitis B. N Engl J Med. 354:1001–1010. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lai CL, Gane E, Liaw YF, et al:
Telbivudine versus lamivudine in patients with chronic hepatitis B.
N Engl J Med. 357:2576–2588. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Khakoo S, Glue P, Grellier L, et al:
Ribavirin and interferon alfa-2b in chronic hepatitis C: assessment
of possible pharmacokinetic and pharmacodynamic interactions. Br J
Clin Pharmacol. 46:563–570. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Shiratori Y, Imazeki F, Moriyama M, et al:
Histologic improvement of fibrosis in patients with hepatitis C who
have sustained response to interferon therapy. Ann Intern Med.
132:517–524. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Morgan TR, Ghany MG, Kim HY, et al:
Outcome of sustained virological responders with histologically
advanced chronic hepatitis C. Hepatology. 52:833–844. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Yoshida H, Shiratori Y, Moriyama M, et al:
Interferon therapy reduces the risk for hepatocellular carcinoma:
national surveillance program of cirrhotic and noncirrhotic
patients with chronic hepatitis C in Japan. IHIT Study Group.
Inhibition of Hepatocarcinogenesis by Interferon Therapy. Ann
Intern Med. 131:174–181. 1999. View Article : Google Scholar
|
|
111
|
Suzuki Y, Kumada H, Ikeda K, et al:
Histological changes in liver biopsies after one year of lamivudine
treatment in patients with chronic hepatitis B infection. J
Hepatol. 30:743–748. 1999.PubMed/NCBI
|