Introduction
Many cancer therapies effectively kill proliferating
tumor cells by causing DNA damage. However, a major limitation of
current therapies is the emergence of resistance following the
initial treatment. Various mechanisms, including, altered drug
transport, increased expression of enzymes that the drugs target,
enhanced catabolization of the drug by an enzyme, and/or increased
tolerance of the cells to genotoxic stress via cell cycle
checkpoints, DNA repair, and/or apoptosis, have been implicated in
chemoresistance (1).
For the treatment of major solid tumors,
particularly colorectal cancers, 5-FU has long been a mainstay
chemotherapeutic antimetabolite drug. The response ratios of 5-FU
monotherapy, combination therapy with irinotecan or oxaliplatin,
and the newly developed combination therapy with bevacizumab and
cetuximab are 15% (2), 40%
(3), and 60–70% (4), respectively. However, the current
therapy causes severe side-effects because it is not tumor-specific
and it injures normal organs. Therefore, the development of
cancer-specific therapies is urgently required.
The major mechanism of the cytotoxicity of 5-FU is
the inhibition of nucleotide synthesis. This drug rapidly enters
tumor cells, and one of the principal intracellular derivatives of
5-FU, fluorodeoxyuridine monophosphate (FdUMP), forms a covalent
complex with thymidylate synthetase (TS), thereby inhibiting the
catalytic activity of TS (5),
leading to depletion of the intracellular pools of deoxythymidine
mono- and tri-phosphate (dTMP and dTTP) and an increase in the
relative levels of the normal precursor dUMP and its anabolic
derivative dUTP (6). In addition
to the nucleotide pool perturbations, UTP and FdUTP incorporate
into DNA, resulting in the induction of stalled replication forks
and S-phase arrest in cells treated with 5-FU (6,7).
FdUTP and dUTP misincorporation is potentially mutagenic and
miscoding, but both can be excised through the action of base
excision repair (BER) and mismatch repair (MMR). DNA strand breaks
are generated as byproducts of the repair processes, and such DNA
damage can induce apoptosis if left unrepaired (8–10).
In response to DNA damage, cells activate a complex
signaling network that mediates cell cycle arrest to allow time for
DNA repair or, when the damage is extensive, to trigger apoptosis.
The DNA damage response is initiated by activation of the ataxia
telangiectasia mutated (ATM) and ATM- and Rad-related (ATR) kinases
and the DNA-dependent protein kinase catalytic subunit. These
kinases recruit the repair machinery to sites of damaged DNA while
halting cell cycle progression by activating the effector kinases
checkpoint kinase (Chk) 1 and 2. The activation of checkpoints
controlled by ATM/ATR-Chk1/Chk2 stalls cell cycle progression in
G1, S, or G2 phase (11). G1
arrest is mediated by p53 through p21CIP1/WAF1 upregulation and, if
the DNA damage is extensive, triggers apoptosis (12). However, many cancer cells show a
loss of function of p53 or its regulatory pathways; therefore,
chemotherapy-induced DNA damage fails to arrest the cancer cells in
G1 phase and promote apoptosis. These cells are completely
dependent on the S and G2/M checkpoints to arrest the cell cycle
and facilitate DNA repair before entry into mitosis (M phase) after
genotoxic exposure. The ATR/Chk1 kinases are involved in the
intra-S and G2/M checkpoints (13), function in the regulation of cell
cycle arrest following genotoxic stress, and prevent new
replication origins from firing during S phase.
Checkpoint kinase 1 (Chk1) inhibition induces the
premature entry of cells with DNA damage into M phase and leads to
the promotion of abnormal mitotic spindles, altered chromosome
segregation, abnormal cell division, the formation of multiple
nuclei and apoptosis (14). This
synergistic cytotoxicity of Chk1 inhibitors in combination with
anticancer, DNA-damaging agents is especially effective against
p53-deficient cancer cells compared with p53-proficient cells,
including normal cells (15).
These cancer-specific therapies have been considered examples of
synthetic lethality, and many Chk1 inhibitors in combination with a
variety of anticancer DNA-damaging agents are at various stages of
preclinical and clinical development (16). In colorectal cancer, ATR, one of
the regulators of Chk1 activation, is activated by DNA-damaging
agents, and inhibition of ATR selectively sensitizes p53-deficient
cells to cisplatin (17). It has
also been reported that selective Chk1 inhibitors abrogate cell
cycle checkpoints and potentiate the cytotoxicity of topoisomerase
inhibitors in p53-deficient, but not in p53-proficient, colon
cancer cells (18). 5-FU is the
most frequently used chemotherapeutic agent for colorectal cancer.
It has been reported that 5-FU activates Chk1 and that Chk1
downregulation abrogates S-phase arrest (19). Judging from these results, it is
expected that the synergistic antitumor effects of Chk1 inhibition
with 5-FU are more effective in p53-deficient cells than in
p53-proficient colorectal cancer cells. However, these p53
status-dependent, synergistic antitumor effects of 5-FU and Chk
inhibition in colorectal cancer are still unclear. Moreover, thus
far, no therapy has reached the bedside, even though a highly
selective Chk1 inhibitor would theoretically synergize with
chemotherapy, suggesting the limitation of these therapies.
In this study, we investigated the effect of 5-FU
treatment in p53-proficient and -deficient GI-tract cancer cells to
develop tumor-specific anticancer therapy. In addition, we
hypothesized that Chk1 inhibition might be effective in sensitizing
5-FU-resistant (5FUR) cancer cells to 5-FU because Chk1 activation
is reported to be related to the resistance to chemotherapy
(20). Therefore, we also
investigated the synergistic cytotoxic potential of Chk1 inhibition
on 5-FU treatment in p53-deficient colon cancer cells with/without
5-FU resistance. We observed that 5-FU specifically induced S-phase
arrest in p53-deficient cancer cells, that 5-FU induced Chk1
activation and that Chk1 inhibition produced a synergistic effect
on 5-FU cytotoxicity. We also found that in p53-deficient, 5FUR
cancer, 5-FU did not induce S-phase arrest or Chk1 activation,
although 5-FU induced significant DNA damage, and Chk1 inhibition
did not sensitize the cells to 5-FU cytotoxicity.
Materials and methods
Cell lines and culture conditions
LS-174T and MKN45, which are wild-type p53 human
colorectal and gastric cancer cell lines, respectively, and HT29,
WiDr, and KATO III, which are p53-mutant human colorectal and
gastric cancer cell lines, were obtained from the American Type
Culture Collection (Rockville, MD, USA) and Riken Cell Bank
(Ibaraki, Japan).
To prepare the 5FUR cancer cell line, HT29 was
exposed to increasing doses of 5-FU, up to the clinically relevant
plasma concentration of 2 μg/ml. The surviving resistant cells were
named 5FUR cells. All cells were cultured in RPMI-1640 medium
containing 10% fetal bovine serum and 1% antibiotics and
antimycotics at 37°C in a humidified atmosphere of 5%
CO2.
Drugs and antibodies
5-FU was purchased from Sigma-Aldrich (St. Louis,
MO, USA). SB218078, a Chk1 inhibitor, was obtained from Calbiochem
(San Diego, CA, USA). The antibodies used for western blotting were
as follows: rabbit polyclonal antibodies to phospho-ATR (Ser428),
phospho-Chk1 (Ser296), and β-actin; rabbit monoclonal antibodies to
phospho-ATM (Ser1981) and phospho-Chk1 (Ser345); and mouse
monoclonal antibodies to Chk1 and TS (Cell Signaling Technology,
Inc., Danvers, MA, USA).
Cell cycle analysis
Cells were seeded at 2.0×105 cells/well
in 6-well plates and treated with or without 5-FU (2 μg/ml) and
SB218078 (1 μM) for the indicated time periods. The cell cycle
profile was determined by the propidium iodide staining of nuclei
isolated using the BD Cycletest Plus kit (BD Biosciences, San Jose,
CA, USA) according to the supplier’s directions. Fluorescence was
quantitated using a FACSCanto™ flow cytometer with FACSDiva 6.1.3
software (BD Biosciences).
Cell survival assay
The rates of drug resistance were assessed using the
WST assay with the Cell Count Reagent SF (Nacalai Tesque, Kyoto,
Japan) according to the manufacturer’s protocol. Briefly, 8,000
cells of each cell line were plated in each well of a 96-well plate
in 100 μl medium with or without 5-FU (2 μg/ml) for 48 h. After
treatment, 10 μl WST reagent was added to each well, and the plate
was incubated for 1 h at 37°C. Colorimetric analysis was then
performed at a wavelength of 450 nm using a standard microplate
reader. Cell survival was calculated by dividing the surviving cell
number estimated by WST in the presence of the drug by the number
in the absence of the drug.
Alkaline comet assay
The alkaline comet assay was performed according to
the method described by Singh et al (21), with slight modifications (22). After staining with 20 μg/ml
ethidium bromide for 1 min, we quantified the DNA damage of 100
randomly selected cells using the Comet Assay IV software
(Perceptive Instruments Ltd., Suffolk, UK) on a computer attached
to a fluorescence microscope (Olympus, Tokyo, Japan). We used the
tail moment (the product of the relative intensity of the tail and
the distance from the center of the nucleus to the center of
gravity of the tail) to evaluate the degree of DNA damage.
Intracellular concentrations of 5-FU
The intracellular concentrations of 5-FU were
measured by gas chromatography/mass spectrometry (GC/MS).
Initially, cells were seeded at 1.2×106 cells/10-cm
tissue culture dish and treated with 5-FU (2 μg/ml) for 48 h. The
GC/MS system consisted of a Trace GC gas chromatograph separation
module and a Trace MS mass spectrometer (both from Thermo Fisher
Scientific, Inc., Waltham, MA, USA). A DB-5 column (length, 30 m;
inside diameter, 0.32 mm; film thickness, 0.25 μm; J&W
Scientific, Folsom, CA, USA) was used for the peak separation of
5-FU. The detector was used in SIM mode using the selected
ion-monitoring procedure at m/z=309 for 5-FU and at m/z=311 for
5-FU-15N2. An internal standard solution (including 5-FU-15N2) was
added to each sample, and the solution was then extracted using
ethyl acetate. The reaction product was extracted using a solution
of mixed ethyl acetate and n-hexane, which was then evaporated to
dryness under a stream of nitrogen. The residue was dissolved in
ethyl acetate, and a 1-μl aliquot was injected into the GC/MS
system.
Western blotting
For all western blotting, cells were seeded at
2.0×105 cells/well in 6-well plates and treated with or
without 5-FU (2 μg/ml) and SB218078 (1 μM) for 48 h. The cells were
lysed in lysis buffer [1% Nonidet P-40, 0.5% sodium deoxycholate,
0.1% sodium dodecyl sulfate, 1x protease inhibitor cocktail
(Nacalai Tesque), and phosphate-buffered saline, pH 7.4]. Next, the
lysates were cleared by centrifugation at 10,000 g at 4°C for 15
min. The protein concentrations were determined using a
bicinchoninic acid protein assay kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Equal amounts (10 μg) of the protein lysates
were then electrophoretically separated on SDS-polyacrylamide gels
and transferred onto a polyvinylidene fluoride membrane. For
immunodetection, the above-mentioned primary antibodies were
used.
Results
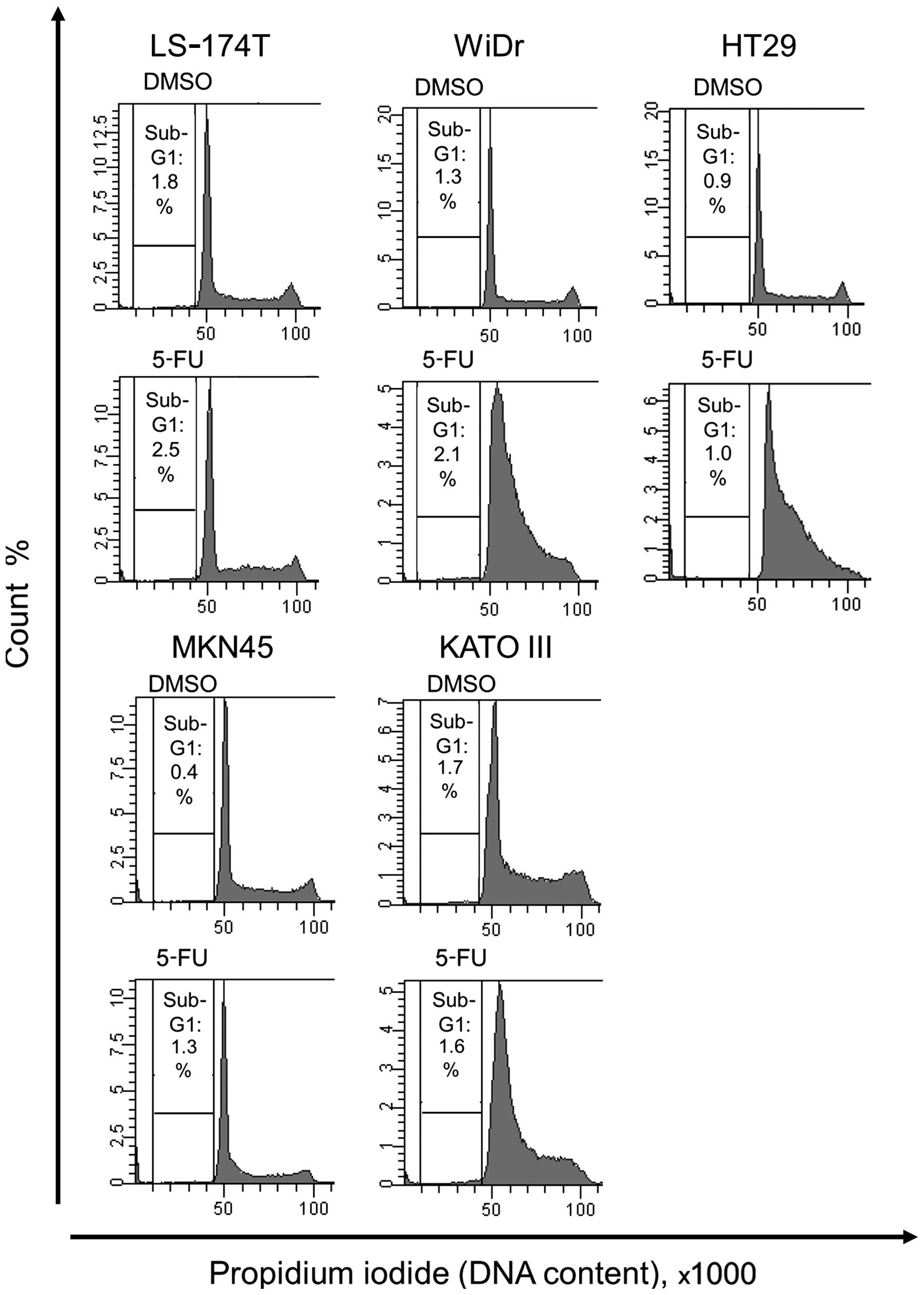
Cell cycle analysis of wild-type and
mutant p53 human gastrointestinal cancer cells treated with
5-FU
At first, we investigated the effect of 5-FU
treatment on the cell cycle of human colorectal and gastric cancer
cell lines expressing wild-type p53 or mutant p53. In neither
LS-174T nor MKN45 cells, which express wild-type p53, treated with
5-FU, no change was observed in S- or G2/M-phase compared with
control cells. In contrast, 5-FU treatment induced S-phase arrest
in HT29, WiDr, and KATO III cells, which express mutant p53
(Fig. 1).
Characteristics of 5FUR cells
To prepare 5FUR HT29 cells, dubbed 5FUR cells, HT29
cells were treated with increasing concentrations of 5-FU for 1
year. Fig. 2A shows the 5-FU
sensitivity assay of the parental and 5FUR cells. The clinical 5-FU
concentration in plasma is reported to be <1.5 μg/ml (23); the survival of the 5FUR cells was
significantly higher than that of the parental cells in the
presence of 2 μg/ml 5-FU (p<0.05). There was no significant
difference in the cellular 5-FU concentrations of the 5-FU-treated
parental HT29 cells and the 5-FU-treated 5FUR cells (Fig. 2B). In certain parts of the patient
population, an elevated TS level is linked to 5-FU resistance, so
we compared the amount of free TS between parental cells and 5FUR
cells treated with or without 5-FU for 48 h. 5-FU treatment formed
complexes of FdUMP and TS, and the residual 5-FU in the 5FUR cells
formed a small amount of the complex. However, the amounts of free
TS in the 5-FU-treated parental HT29 cells and the 5-FU-treated
5FUR cells were not different (Fig.
2C). We then used alkaline comet assays to analyze the levels
of damaged DNA in the HT29 and 5FUR cells 48 h after treatment with
2 μg/ml 5-FU. In the comet assays, both the parental and the 5FUR
cells treated with 5-FU had longer tail moments than did the
5-FU-untreated parental and 5FUR cells (Fig. 2D). We also investigated the effect
of 5-FU treatment on the cell cycle phase distribution of both cell
types (Fig. 2E). The sub-G1
fraction was not detected in the parental HT29 or 5-FU-treated 5FUR
cells, indicating that 5-FU did not induce apoptosis in either cell
type. Additionally, 5-FU induced S-phase arrest in the parental
HT29 cells, but in the 5FUR cells, 5-FU induced no change in the
cell cycle compared with DMSO treatment, indicating that the
acquisition of 5-FU resistance abrogated the 5-FU-induced S-phase
arrest.
Acquisition of 5-FU resistance abrogates
5-FU-induced Chk1 phosphorylation
Chk1 regulates S-phase arrest; therefore, we
determined whether 5-FU activated the Chk1 pathway in both parental
and 5FUR cells (Fig. 3). Chk1
activation is associated with phosphorylation at Ser345 (the site
of Chk1 phosphorylation by ATR/ATM) and autophosphorylation at
Ser296. The assessment of Chk1 activation (using a DNA-damaging
agent or antimetabolite) and Chk1 inhibition (by a Chk1 inhibitor)
is most likely best accomplished by monitoring the phosphorylation
of Ser296 (15). In particular,
SB218078 inhibits Chk1 autophosphorylation (Ser296) and increases
the phosphorylation of ATM (Ser1981) and Chk1 (Ser345) (24). In this study, the phosphorylation
of Chk1 and ATM (Ser1981) was not enhanced in 5FUR cells, whereas
the phosphorylation of Chk1 and ATM was enhanced in parental cells
treated with 5-FU. As a previous study reported (24), SB218078 significantly reduced the
phosphorylation of Chk1 (Ser296), whereas the phosphorylation of
Chk1 (Ser345) and ATM (Ser1981) was actually enhanced in parental
and 5FUR cells with or without 5-FU treatment. In contrast, the
phosphorylation of ATR (Ser428) was not enhanced by 5-FU treatment
in parental or 5FUR cells.
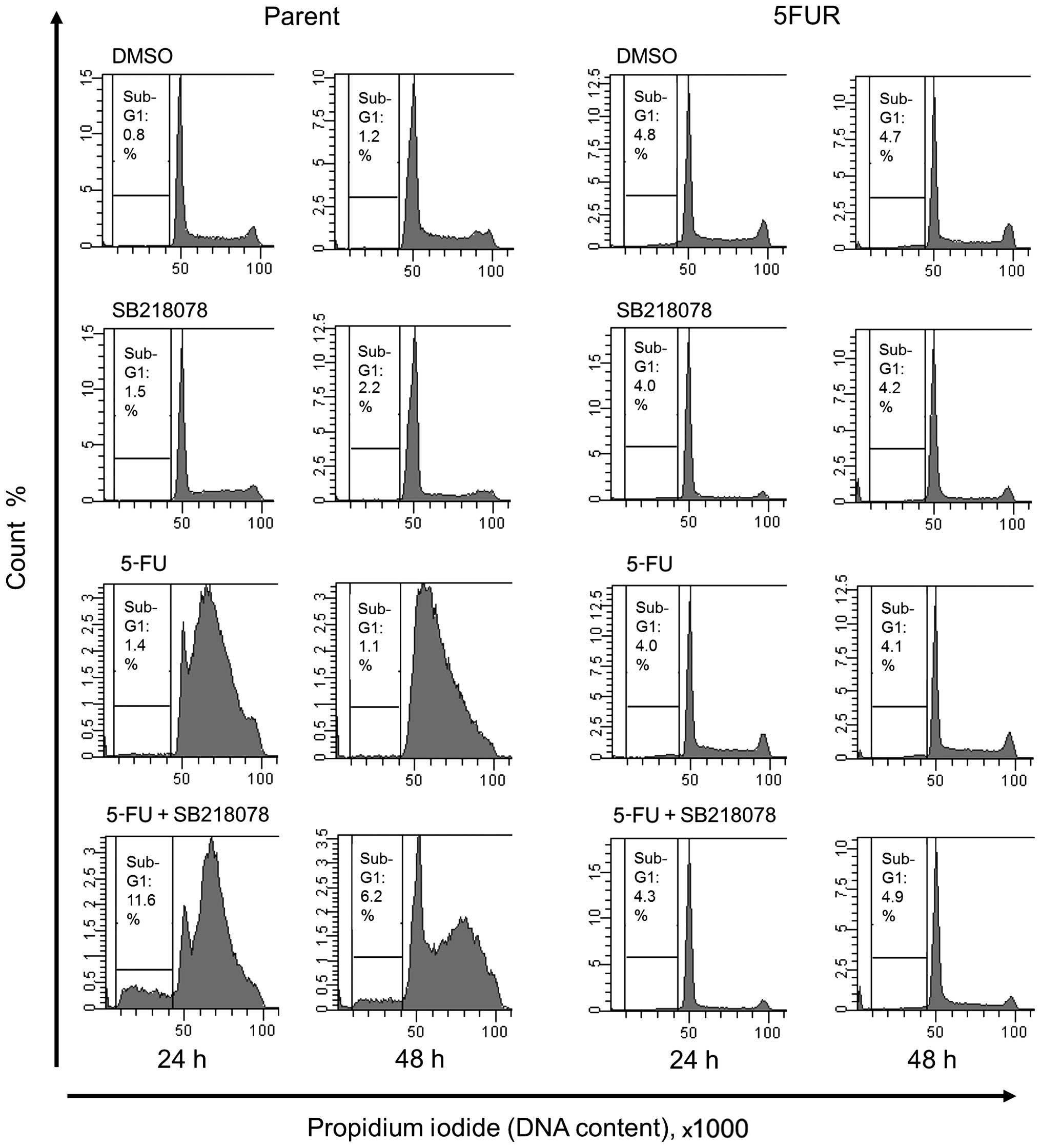
Effect of the acquisition of 5-FU
resistance on the synergistic effect of the Chk1 inhibitor SB218078
and 5-FU in HT29 cells
The synergistic effect of combined 5-FU and SB218078
treatment was evaluated by analyzing the cell cycle phase
distribution and cell death rates. SB218078 showed no effect on
cell viability in the parent cell line during monotherapy, but when
in combination with 5-FU, SB218078 reduced the early S-phase arrest
induced by 5-FU treatment and increased the sub-G1 population,
indicating induction of apoptotic cell death. In contrast, SB218078
exhibited no effect on cell death in 5FUR cell lines during
monotherapy or when in combination with 5-FU (Fig. 4).
Discussion
In this study, we investigated the synergistic
antitumor effect of a Chk1 inhibitor and 5-FU that was specific to
p53-deficient cells, and we analyzed the potential of Chk1
inhibition to sensitize 5FUR cancer cells to 5-FU to develop a
tumor-specific, effective therapy that overcame 5-FU
resistance.
The enhanced excretion or degradation of 5-FU and
the expression of TS are well-known mechanisms for 5-FU resistance.
In this study, however, equal levels of intracellular 5-FU and free
TS protein were detected in parental and 5FUR cells that were
treated with 5-FU. Chk1 is overexpressed in a variety of human
tumors, including breast, colon, liver, gastric, and nasopharyngeal
carcinoma (25–31). Remarkably, Chk1 expression often
positively correlates with tumor grade and disease recurrence and
may contribute to therapy resistance (29,30,32).
The enhanced activation of Chk1 is also related to the resistance
of cancer cells, including cancer stem cells from brain
glioblastoma, prostate, and lung NSCLC, to chemotherapy or
radiotherapy (33–38). Therefore, we hypothesized that 5FUR
cells might possess more Chk1 activity than control cells and that
Chk1 inhibition might exert a synergistic, cytotoxic effect and
sensitize chemo-resistant cells to 5-FU.
DNA repair systems promote the faithful transmission
of genomes in dividing cells by reversing extrinsic and intrinsic
DNA damage and are required for cell survival during replication.
Cancer cells are frequently found to be deficient in certain
aspects of DNA repair. The impairment of DNA repair systems
contributes not only to the initial mechanism of carcinogenesis but
also to its weakness, as these repair systems are required for the
cancer cells to maintain their own survival (39).
Ovarian and breast cancer patients with BRCA
mutations exhibit favorable responses to poly(ADP-ribose)
polymerase (PARP) inhibitors compared with patients without BRCA
mutations, as homologous recombination-deficient tumors can be
effectively targeted by DNA double-strand break-inducing agents
(40). This concept, that is,
synthetic lethality, is attracting attention in the development of
tumor-specific therapy. Synthetic lethal interactions are defined
as two genetic alterations that cause cell death when they occur
together, although neither mutation alone is lethal (41). The pharmacological inhibition of
one gene product can be synthetically lethal when it occurs in
combination with a pre-existing, cancer-related mutation,
especially when the mutated cancer cells have become dependent on
special pathways, leading to the ability to selectively target and
kill the cancer cells while sparing the normal cells (42).
The development of anticancer regimens that take
advantage of the molecular differences between normal and cancer
cells is highly desirable. TP53 is one of the most
frequently mutated genes in human cancers, so there is great
interest in finding anticancer regimens that selectively target
p53-deficient tumors (43). The
cancer cells showing a loss of function of p53 or its regulatory
pathways have a deficiency in the G1 checkpoint and are completely
dependent on the S and G2/M checkpoints to arrest the cell cycle
after genotoxic stress. Chk1 is critical for S and G2/M arrest via
downregulation of Cdc25A, cyclin A and CDK2 expression (44). Chk1 inhibition abolishes the S and
G2 checkpoints induced by 5-FU, causes excessive accumulation of
DNA damage, induces apoptosis, and ultimately selectively
potentiates the efficacy of 5-FU in p53-deficient cells. In
contrast, p53-dependent checkpoint(s) in p53-proficient cells allow
DNA repair and thereby prevent sensitization to DNA damage
(19).
In this study, we observed that 5-FU induced S-phase
arrest only in p53-deficient cells and that 5-FU treatment induced
Chk1 activation. We also found that Chk1 inhibition by SB218078
significantly increased the population of sub-G1 cells only in the
presence of 5-FU. These results indicate that the development of
tumor-specific anticancer therapy with 5-FU and Chk1 inhibitors for
colorectal cancer, of which p53 is frequently mutated, is to be
expected.
In the 5FUR cell line, 5-FU treatment did not induce
Chk1 activation or S-phase arrest. Moreover, SB218078 in
combination with 5-FU did not induce a sub-G1 population. These
results revealed that in 5FUR, 5-FU-induced DNA damage induced
neither Chk1 activation nor S-phase arrest, and 5FUR proliferated
in the presence of 5-FU.
To understand the mechanisms underlying the
abrogation of Chk1 activation in 5FUR cells, we examined the
activation status of a major upstream regulator, Chk1, in these
cells. More specifically, Chk1 is a traditional target of ATR in
the DNA damage response. We observed that 5-FU treatment did not
induce the activation of ATR in 5FUR cells or parental cells,
although this treatment activated Chk1 in the parental cells. ATM
is also required for Chk1 activation under certain circumstances
(45–47). In this study, we found activation
of ATM by 5-FU in parental cells, which demonstrates another
example of ATM-Chk1 signaling mediating early S-phase arrest. In
contrast, in 5FUR, neither ATR nor ATM was activated by DNA damage
induced by 5-FU treatment, leading to no induction of Chk1
activation, although there was no difference in the cellular 5-FU
concentration or in the amount of free TS between the parental and
5FUR cells, indicating that 5-FU functioned in 5FUR cells as well
as in parental cells. The precise mechanism of ATR, ATM, and Chk1
inactivation observed in 5FUR cells remains to be elucidated.
DNA-damaging agents combined with a Chk1 inhibitor
cause tumor cells to undergo apoptosis in p53-deficient tumors
(48). Mice with Chk1 disruption
die during early development (49,50),
and the conditional deletion of Chk1 in proliferating mouse mammary
epithelial cells induces apoptosis and developmental defects. In
contrast to these studies, we did not observe increased apoptosis
in 5FUR cells, in which 5-FU treatment induced DNA damage but did
not induce Chk1 activation, cell cycle arrest, or an increased
sub-G1 fraction. These results indicated that the 5FUR cells
acquired apoptotic resistance. It has been reported that the
overexpression of Bcl-xL suppresses Chk1 inhibitory lethality
(51–53). The precise mechanisms of the
anti-apoptotic potentials remain to be elucidated; however, in this
study, we observed enhanced expression of Bcl-w and c-FLIP (data
not shown) in 5FUR cells.
This study re-evaluated the key role of Chk1 in
regulating the 5-FU-induced DNA damage checkpoint and the utility
of Chk1 inhibition in tumor-specific anticancer therapy by
enhancing the anticancer efficacy of 5-FU only in p53-deficient
cells. However, 5-FU resistance in p53-deficient colorectal cancer
cells abrogated 5-FU-induced Chk1 phosphorylation, S-phase arrest,
and sensitization of p53-deficient cancer cells to 5-FU by Chk1
inhibition. Therefore, Chk1 inhibition combined with 5-FU in
p53-deficient cells does not appear to be a promising approach in
the context of tolerance to 5-FU.
References
|
1
|
Marin JJ, Briz O, Monte MJ, Blazquez AG
and Macias RI: Genetic variants in genes involved in mechanisms of
chemo-resistance to anticancer drugs. Curr Cancer Drug Targets.
12:402–438. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnston PG and Kaye S: Capecitabine: a
novel agent for the treatment of solid tumors. Anticancer Drugs.
12:639–646. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martin RC II, Scoggins CR, Tomalty D, et
al: Irinotecan drug-eluting beads in the treatment of chemo-naive
unresectable colorectal liver metastasis with concomitant systemic
fluorouracil and oxaliplatin: results of pharmacokinetics and phase
I trial. J Gastrointest Surg. 16:1531–1538. 2012. View Article : Google Scholar
|
|
4
|
Feng QY, Wei Y, Chen JW, et al: Anti-EGFR
and anti-VEGF agents: important targeted therapies of colorectal
liver metastases. World J Gastroenterol. 20:4263–4275. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pinedo HM and Peters GF: Fluorouracil:
biochemistry and pharmacology. J Clin Oncol. 6:1653–1664. 1988.
|
|
6
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sampath D, Rao VA and Plunkett W:
Mechanisms of apoptosis induction by nucleoside analogs. Oncogene.
22:9063–9074. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartek J, Lukas C and Lukas J: Checking on
DNA damage in S phase. Nat Rev Mol Cell Biol. 5:792–804. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aziz K, Nowsheen S, Pantelias G, Iliakis
G, Gorgoulis VG and Georgakilas AG: Targeting DNA damage and
repair: embracing the pharmacological era for successful cancer
therapy. Pharmacol Ther. 133:334–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou BB and Elledge SJ: The DNA damage
response: putting checkpoints in perspective. Nature. 408:433–439.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Levesque AA and Eastman A: p53-based
cancer therapies: is defective p53 the Achilles heel of the tumor?
Carcinogenesis. 28:13–20. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tao ZF and Lin NH: Chk1 inhibitors for
novel cancer treatment. Anticancer Agents Med Chem. 6:377–388.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma CX, Janetka JW and Piwnica-Worms H:
Death by releasing the breaks: CHK1 inhibitors as cancer
therapeutics. Trends Mol Med. 17:88–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maugeri-Saccà M, Bartucci M and De Maria
R: Checkpoint kinase 1 inhibitors for potentiating systemic
anticancer therapy. Cancer Treat Rev. 39:525–533. 2013.PubMed/NCBI
|
|
17
|
Sangster-Guity N, Conrad BH, Papadopoulos
N and Bunz F: ATR mediates cisplatin resistance in a p53
genotype-specific manner. Oncogene. 30:2526–2533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Z, Xiao Z, Gu WZ, et al: Selective
Chk1 inhibitors differentially sensitize p53-deficient cancer cells
to cancer therapeutics. Int J Cancer. 119:2784–2794. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiao Z, Xue J, Sowin TJ, Rosenberg SH and
Zhang H: A novel mechanism of checkpoint abrogation conferred by
Chk1 down-regulation. Oncogene. 24:1403–1411. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Y and Hunter T: Roles of Chk1 in
cell biology and cancer therapy. Int J Cancer. 134:1013–1023. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar
|
|
22
|
Lu Y, Morimoto K and Nakayama K: Health
practices and leukocyte DNA damage in Japanese hard-metal workers.
Prev Med. 43:140–144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yoshikawa R, Kusunoki M, Yanagi H, et al:
Dual antitumor effects of 5-fluorouracil on the cell cycle in
colorectal carcinoma cells: a novel target mechanism concept for
pharmacokinetic modulating chemotherapy. Cancer Res. 61:1029–1037.
2001.
|
|
24
|
Okita N, Minato S, Ohmi E, Tanuma S and
Higami Y: DNA damage-induced CHK1 autophosphorylation at Ser296 is
regulated by an intramolecular mechanism. FEBS Lett. 586:3974–3979.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sriuranpong V, Mutirangura A, Gillespie
JW, et al: Global gene expression profile of nasopharyngeal
carcinoma by laser capture microdissection and complementary DNA
microarrays. Clin Cancer Res. 10:4944–4958. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cho SH, Toouli CD, Fujii GH, Crain C and
Parry D: Chk1 is essential for tumor cell viability following
activation of the replication checkpoint. Cell Cycle. 4:131–139.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Madoz-Gúrpide J, Cañamero M, Sanchez L,
Solano J, Alfonso P and Casal JI: A proteomics analysis of cell
signaling alterations in colorectal cancer. Mol Cell Proteomics.
6:2150–2164. 2007.PubMed/NCBI
|
|
28
|
Verlinden L, Vanden Bempt I, Eelen G, et
al: The E2F-regulated gene Chk1 is highly expressed in
triple-negative estrogen receptor/progesterone receptor/HER-2
breast carcinomas. Cancer Res. 67:6574–6581. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yao H, Yang Z and Li Y: Expression of
checkpoint kinase 1 and polo-like kinase 1 and its
clinicopathological significance in benign and malignant lesions of
the stomach. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 35:1080–1084.
2010.(In Chinese).
|
|
30
|
Hong J, Hu K, Yuan Y, et al: CHK1 targets
spleen tyrosine kinase (L) for proteolysis in hepatocellular
carcinoma. J Clin Invest. 122:2165–2175. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu J, Li Y, Wang F, et al: Suppressed
miR-424 expression via upregulation of target gene Chk1 contributes
to the progression of cervical cancer. Oncogene. 32:976–987. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lundgren K, Holm K, Nordenskjöld B, Borg A
and Landberg G: Gene products of chromosome 11q and their
association with CCND1 gene amplification and tamoxifen resistance
in premenopausal breast cancer. Breast Cancer Res. 10:R812008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Perego P, Gatti L, Righetti SC, et al:
Development of resistance to a trinuclear platinum complex in
ovarian carcinoma cells. Int J Cancer. 105:617–624. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bao S, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cavelier C, Didier C, Prade N, et al:
Constitutive activation of the DNA damage signaling pathway in
acute myeloid leukemia with complex karyotype: potential importance
for checkpoint targeting therapy. Cancer Res. 69:8652–8661. 2009.
View Article : Google Scholar
|
|
36
|
Zhang YW, Brognard J, Coughlin C, et al:
The F box protein Fbx6 regulates Chk1 stability and cellular
sensitivity to replication stress. Mol Cell. 35:442–453. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bartucci M, Svensson S, Romania P, et al:
Therapeutic targeting of Chk1 in NSCLC stem cells during
chemotherapy. Cell Death Differ. 19:768–778. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang X, Ma Z, Xiao Z, et al: Chk1
knockdown confers radiosensitization in prostate cancer stem cells.
Oncol Rep. 28:2247–2254. 2012.PubMed/NCBI
|
|
39
|
Brody LC: Treating cancer by targeting a
weakness. N Engl J Med. 353:949–950. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Audeh MW, Carmichael J, Penson RT, et al:
Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients
with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a
proof-of-concept trial. Lancet. 376:245–251. 2010. View Article : Google Scholar
|
|
41
|
Bouwman P and Jonkers J: The effects of
deregulated DNA damage signalling on cancer chemotherapy response
and resistance. Nat Rev Cancer. 12:587–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dar AC, Das TK, Shokat KM and Cagan RL:
Chemical genetic discovery of targets and anti-targets for cancer
polypharmacology. Nature. 486:80–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Petitjean A, Mathe E, Kato S, et al:
Impact of mutant p53 functional properties on TP53 mutation
patterns and tumor phenotype: lessons from recent developments in
the IARC TP53 database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tu YS, Kang XL, Zhou JG, Lv XF, Tang YB
and Guan YY: Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in
simvastatin induced S-phase cell cycle arrest and apoptosis in
multiple myeloma cells. Eur J Pharmacol. 670:356–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sahu RP, Batra S and Srivastava SK:
Activation of ATM/Chk1 by curcumin causes cell cycle arrest and
apoptosis in human pancreatic cancer cells. Br J Cancer.
100:1425–1433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jazayeri A, Falck J, Lukas C, et al: ATM-
and cell cycle-dependent regulation of ATR in response to DNA
double-strand breaks. Nat Cell Biol. 8:37–45. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cuadrado M, Martinez-Pastor B, Murga M, et
al: ATM regulates ATR chromatin loading in response to DNA
double-strand breaks. J Exp Med. 203:297–303. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eastman A: Cell cycle checkpoints and
their impact on anti-cancer therapeutic strategies. J Cell Biochem.
91:223–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu Q, Guntuku S, Cui XS, et al: Chk1 is
an essential kinase that is regulated by Atr and required for the
G(2)/M DNA damage checkpoint. Genes Dev. 14:1448–1459.
2000.PubMed/NCBI
|
|
50
|
Takai H, Naka K, Okada Y, et al:
Chk2-deficient mice exhibit radioresistance and defective
p53-mediated transcription. EMBO J. 21:5195–5205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bhonde MR, Hanski ML, Magrini R, et al:
The broad-range cyclin-dependent kinase inhibitor UCN-01 induces
apoptosis in colon carcinoma cells through transcriptional
suppression of the Bcl-x(L) protein. Oncogene. 24:148–156. 2005.
View Article : Google Scholar
|
|
52
|
Mitchell C, Park M, Eulitt P, Yang C,
Yacoub A and Dent P: Poly(ADP-ribose) polymerase 1 modulates the
lethality of CHK1 inhibitors in carcinoma cells. Mol Pharmacol.
78:909–917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tang Y, Dai Y, Grant S and Dent P:
Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol
Ther. 13:379–388. 2012. View Article : Google Scholar : PubMed/NCBI
|