Introduction
Because DNA replication strongly depends on the pool
of available deoxyribonucleoside triphosphates, the intracellular
metabolism of tumor cells adapts to facilitate rapid proliferation.
Accordingly, nucleic acid metabolism is one of the most upregulated
pathways in tumor tissues (1,2).
Thus, the pyrimidine synthesis pathway involving deoxythymidine
(dThd; Fig. 1) biosynthesis was
recognized in early studies as a target for solid tumor
chemotherapy with drugs, such as 5-fluorouracil (5-FU) and/or
antifolate agents (3,4).
Thymidylate synthase (TS), the target of 5-FU and
antifolate drugs, is highly expressed in various tumor tissues and
is the sole de novo enzyme of dThd synthesis. TS catalyzes
the methylation of deoxyuridine monophosphate (dUMP) to dTMP
(5–7). However, the dThd salvage pathway
involves multiple factors, such as nucleoside transporters and dThd
kinases (TK). TK1 is expressed in the cytoplasm during S phase
(8), while TK2 expression is
localized to mitochondria and is cell cycle independent (9). TK1 and TS are highly upregulated in
various tumor tissues (7) and may
serve as potential targets for cancer therapy. However, antitumor
agents targeting the dThd salvage pathway have yet to be developed
clinically.
Trifluridine (FTD; Fig.
1) is a thymidine-derived nucleoside first synthesized by
Heidelberger et al in 1964 as an antitumor agent (10), and clinical trials using FTD for
monotherapy have been conducted in US (11). However, these trials showed an
unexpected toxicity, and FTD was later repurposed as the ocular
antiviral drug Viroptic® (12). FTD is well absorbed, but it is
easily degraded by the hepatic enzyme thymidine phosphorylase (TP)
following oral administration. TAS-102 is an oral combination of
FTD and tipiracil hydrochloride (TPI) that prevents FTD degradation
by TP (13). Co-administration of
TPI and FTD increases the overall FTD concentration in the body,
leading to augmented antitumor activity (14).
Recently, TAS-102 treatment showed prolonged
survival in patients with metastatic colorectal cancer (mCRC) that
were refractory or intolerant to standard chemotherapies including
5-FU, oxaliplatin and CPT-11, in a KRAS mutation-independent
manner (15). Based on this phase
II result, TAS-102 was launched in Japan in May 2014 as an agent
for treating unresectable advanced and recurrent colorectal
cancers. The antitumor activity of FTD occurs via two distinct
mechanisms, namely, TS inhibition by the mononucleotide form of FTD
(F3dTMP) and DNA incorporation itself (16,17).
Previous studies have shown that the mechanism of TS inhibition of
FTD is different from that of 5-FU (18,19).
Moreover, in the phase II study mentioned above, TAS-102, showed
efficacy in patients who were progressive after treatment with
5-FU, confirming that FTD and 5-FU have different mechanisms of
cytotoxicity.
TS inhibition by the metabolites of FTD or FdUrd
(Fig. 1), a clinically active 5-FU
analog, has been described by Reyes and Heidelberger (20). Both nucleosides were reported to be
metabolized by dThd salvage pathway, involving the nucleoside
transporter family members hENT and TK1 (21–23).
However, the DNA incorporation profiles regarding substrate
specificities in DNA extension reactions by DNA polymerase were not
compared. Moreover, in terms of nucleoside triphosphate specificity
during DNA synthesis, deoxyUTPase (DUT) plays an important role in
DNA replication and 5-FU sensitivity. DUT functions as a gatekeeper
protein to prevent the misincorporation of
deoxyuridine-triphosphate (dUTP) into DNA by converting dUTP to
dUMP. DUT also converts FdUTP (FdUrd-triphosphate) to FdUMP
(FdUrd-monophosphate) and prevents FdUTP misincorporation, such
that high DUT expression causes 5-FU resistance (24).
These phenomena indicate that the incorporation of
5-FU metabolites and dUTP into DNA are important for 5-FU
cytotoxicity, but investigations regarding the DNA incorporation
profile of FTD have been limited (25). Therefore, we studied the levels of
FTD and FdUrd incorporation into DNA, as well as the substrate
specificities of hENT family members (hENT1 and hENT2), TK1, DUT
and DNA polymerase α.
Materials and methods
Chemical and reagents
FTD was obtained from Yuki Gosei Kogyo Co., Ltd.
(Tokyo, Japan). TPI was synthesized at Junsei Chemical Co., Ltd.
(Tokyo, Japan). dThd, FdUrd and dUrd were purchased from Wako Pure
Chemical Industries, Ltd. (Osaka, Japan). [5-methyl-3H]
dThd (25.0 Ci/mmol), [6-3H] dUrd (19 Ci/mmol),
[6-3H] FdUrd (13.5 C i/mmol), and [6-3H] FTD
(10.0 Ci/mmol) were purchased from Moravek Biochemicals (Brea, CA,
USA). dNTPs were obtained from Takara Bio (Otsu, Japan).
F3dTTP was synthesized in house. Single-stranded DNA
oligonucleotides were synthesized by Nihon Gene Research
Laboratories (Sendai, Japan).
Cell culture
The human HCT116 colorectal cancer cell line derived
from an adult male was obtained from the American Type Culture
Collection (ATCC; Manassas, VA, USA) and cultured at 37°C with 5%
CO2 in McCoy’s 5A modified medium supplemented with 10%
fetal bovine serum (FBS). Short tandem repeat profiling was
performed to confirm the origin and authenticity of the HCT116 cell
line.
Measurement of dThd, FTD and FdUrd
incorporation into DNA
HCT116 cells (1×107) were seeded
overnight in 175-cm2 culture flasks. Subsequently, cells
were incubated with drug mixtures containing tritium-labeled
nucleosides at a final concentration of 1 μmol/l. The cells were
harvested at 1, 2, 4, 10 and 24 h after treatment with each drug.
DNA was extracted from cell pellets using a DNeasy Blood &
Tissue kit (Qiagen, Hilden, Germany). The resulting DNA solutions
were dissolved in 10 ml of Ultima Gold AB liquid scintillation
fluid (Perkin-Elmer, Tokyo, Japan). Radioactivity in the samples
was measured with a Tri-Carb 2900TR liquid scintillation analyzer
(Perkin-Elmer), and the incorporation of tritium-labeled
nucleosides into DNA was quantitated. DNA concentrations were
determined using the Qubit® dsDNA BR assay kit (Life
Technologies, Carlsbad, CA, USA).
Transport experiments
Nucleoside transport was evaluated by the silicone
layer method (26). Briefly, cells
were harvested the day before experiments began and suspended in
transport medium containing 125 mmol/l NaCl, 4.8 mmol/l KCl, 5.6
mmol/l D-glucose, 1.2 mmol/l CaCl2, 1.2 mmol/l
KH2PO4, 12 mmol/l MgSO4, and 25
mmol/l HEPES (pH 7.4). Cell suspensions were pre-incubated for 20
min at 37°C in transport medium, centrifuged, and the resultant
cell pellets were resuspended in transport medium (pH 7.4)
containing a radiolabeled nucleoside or nucleoside analog to
initiate uptake. At appropriate times, 160-μl aliquots of cell
suspension were withdrawn, and cells were separated by
centrifugation through a layered mixture of silicone oil (SH550;
Dow Corning Toray, Tokyo, Japan) and liquid paraffin (Wako Pure
Chemicals Industries) with a density of 1.03 g/ml. Cell pellets
were solubilized in 3 mol/l KOH and then neutralized with HCl.
Next, cell-associated radioactivities were measured using a
Tri-Carb 2900TR liquid scintillation analyzer. The cellular protein
content was determined using the Qubit® Protein assay
kit (Life Technologies).
Active recombinant protein expression and
purification of TK1 and DUT
The pENTR221/TK1 and pENTR221/DUT constructs were
obtained from Life Technologies. A Gateway LR reaction was
performed to subclone the fusion partners into the Gateway
bacterial expression vector pEXP1/DEST. Transformed BL21
StarTM (DE3) pLysS cells were grown for 10 h at 37°C in
LB medium containing 100 μg/ml ampicillin. Gene expression was
induced by adding isopropyl β-D-1-thiogalactopyranoside (0.1
μmol/l) that was cooled to 10°C. After 18 h of induction at 25°C,
bacteria were collected by centrifugation, and pellets were stored
at −135°C until purification. Bacterial pellets were resuspended in
10 ml BugBuster® HT Protein Extraction reagent (Merck
KGaA, Darmstadt, Germany) and incubated for 30 min at room
temperature. Subsequently, the extract was clarified by centrifugal
filtration with a 0.4-μm filter and purified with an AKTAprime plus
with a HiTrap metal chelate column (GE Healthcare, Buckinghamshire,
UK).
Analysis of TK1 substrate specificity for
dThd, FTD, dUrd and FdUrd
Ten micromolar tritium-labeled nucleosides and
nucleoside analogs were incubated for 5 min at 37°C in 100-μl
reactions containing 150 mmol/l Tris-HCl (pH 7.6), 6 mmol/l ATP, 15
mmol/l MgCl2, 1% bovine serum albumin (BSA) and 20 ng
recombinant human TK1. Reactions were stopped by the addition of
200 μl of methanol and clarified by ultrafiltration. After
clarification, solutions were desiccated by exposure to
N2 gas, dissolved in 100 μl H2O, and analyzed
by radio-high-performance liquid chromatography (HPLC). HPLC was
performed using a 150TR Flow System analyzer (Perkin-Elmer) and a
Prominence Series HPLC system (Shimadzu Corp., Kyoto, Japan)
equipped with an ODS reverse-phase column (TSKgel ODS-100V, 250 mm
× 4.6 mm, 3 μm; Tosoh Corp., Tokyo, Japan), with the eluent [10
mmol/l phosphate buffer (pH 3.0):Acetonitrile = 9:1] set to a flow
rate 1.0 ml/min for 15 min. This procedure enabled quantitation of
residual nucleosides and the production of nucleoside
monophosphates formed in the reactions just described.
Analysis of DUT substrate specificity for
dThd, FTD, dUrd and FdUrd-triphosphate
dUTPase assays were performed as previously
described (27). Deoxynucleoside
triphosphates (dUTP, dTTP, F3dTTP and FdUTP; 30 mmol/l)
were incubated for 30 min at 37°C in 100-μl reactions composed of
50 mmol/l Tris-HCl (pH 7.4), 4 mmol/l MgCl2, 2 mmol/l
2-mercaptoethanol, 0.1% BSA and 100 ng human recombinant DUT.
Reactions were stopped by the addition of 11 μl 4.2 mol/l
perchloric acid per reaction. Samples were clarified by
centrifugation for 10 min at 13,000 rpm. Supernatants (70 μl) were
neutralized with 32 μl of 1 mol/l K2HPO4,
followed by a 10-min centrifugation at 13,000 rpm. Supernatants (10
μl) were resolved by HPLC as described above to quantify residual
dUTP and newly formed dUMP.
Substrate specificity of dThd, FTD, dUrd
and FdUrd-triphosphate for DNA polymerase α
Recombinant DNA polymerase α (CHIMERx, WI) activity
was assayed according to the manufacturer’s recommended protocol.
The substrates (dNTPs) were incubated for 30 min at 37°C with 60
mmol/l Tris-HCl (pH 7.0), 5 mmol/l MgCl2, 0.3 mg/ml BSA,
1 mmol/l DTT, 0.1 μmol/l, 0.5 U DNA polymerase α, fluorescein
isothiocyanate (FITC)-labeled primer (5′-FITCGTAAAACG ACGCC
AGT-3′), and 0.1 μmol/l synthetic DNA template (5′-TCG GACTGGCCG
TCG TTTTAC-3′, 5′-TCG CACTGGCCG TCG TTTTAC-3′, 5′-TCG AACTGGC CG
TCG TTTTAC-3′, or 5′-TCG TACTGGCCGTCGTTTT AC-3′). The underlined
sequences represent nucleotides that were modified partially to
confirm the sites where F3dTTP and FdUTP were
inserted.
After the enzyme reactions were complete, samples
were resolved by electrophoresis on an ERICA-S system (DRC, Tokyo,
Japan) at 300 V for 3.5 h, and FITC-fluorescence was detected using
an ImageQuant LAS 4010 imager (GE Healthcare).
Morphological analysis of HCT116 cell
nuclei treated with FTD and FdUrd
For electron microscopy studies, samples were fixed
with 2% paraformaldehyde and 2% glutaraldehyde, after which they
were exposed to 2% osmium tetroxide. Following dehydration with a
graded ethanol series, samples were transferred to resin quetol 812
(Nisshin EM, Tokyo, Japan) and polymerized at 60°C for 48 h. The
blocks were sectioned at 70 nm with an Ultracut UCT ultramicrotome
(Leica Microsystems, Wetzlar, Germany), and sections were placed on
copper grids and stained with 2% uranyl acetate and lead stain
solution (Sigma-Aldrich, Carlsbad, CA, USA). The grids were
evaluated using a JEM-1200EX transmission electron microscope
(JEOL, Tokyo, Japan).
Results
Incorporation and elimination of dThd,
FTD and FdUrd from DNA in HCT116 cells
Initially, we compared the rates of FTD and FdUrd
accumulation into DNA in HCT116 cells with that of the native
substrate dThd, using final nucleoside concentrations of 1 μmol/l.
Fig. 2A shows the time course for
nucleoside accumulation at 1, 2, 4, 10 and 24 h post-exposure.
Tritium-labeled nucleosides were used as substrates and were
quantified by liquid scintillation counting (LSC). dThd and FTD
were incorporated into DNA at comparable levels at the 1- and 2-h
time-points. dThd incorporation increased linearly until 10 h,
while saturation of FTD incorporation occurred by 4 h, reaching
approximately half that observed with dThd (dThd, 41.5 pmol/μg DNA;
FTD, 17.6 pmol/μg DNA). However, the level of FdUrd incorporation
into DNA was relatively low. For example, after a 24-h incubation,
FdUrd incorporation was one sixteenth of that observed with FTD
(FdUrd, 1.1 pmol/μg DNA), which is close to the limit of
detection.
 | Figure 2Comparison of dThd, FTD and FdUrd
incorporation (A) into DNA and elimination of dThd and FTD (B) from
DNA after a washout step. (A) HCT116 cells were treated for 1, 2,
4, 10 or 24 h with each compound at 1 μmol/l, and DNA incorporation
was measured by LSC. (B) Following 24-h treatment with dThd or FTD,
these compounds were washed out. At 24, 48 and 72 h following the
washout step, the amount of dThd or FTD remaining incorporated into
DNA was measured by liquid scintillation counting. Open circles,
triangles and squares represent the incorporation of dThd, FTD and
FdUrd, respectively. |
Next, we compared the susceptibility of FTD to
elimination from DNA with that of dThd. In this assay, cells were
treated with 1 μmol/l dThd and FTD for 24 h, after which drug-free
medium was added and the cells were incubated for an additional 24,
48 or 72 h. Fig. 2B shows the
residual ratios of dThd to FTD at 24, 48 and 72 h after the washout
steps. Both dThd and FTD incorporation into DNA decreased gradually
after the washout step, and only minor differences were observed
between dThd and FTD (dThd 34.1%, FTD: 29.0%). We also attempted to
assess FdUrd elimination from DNA after the washout step; however,
it was nearly undetectable in this assay and could not be
evaluated.
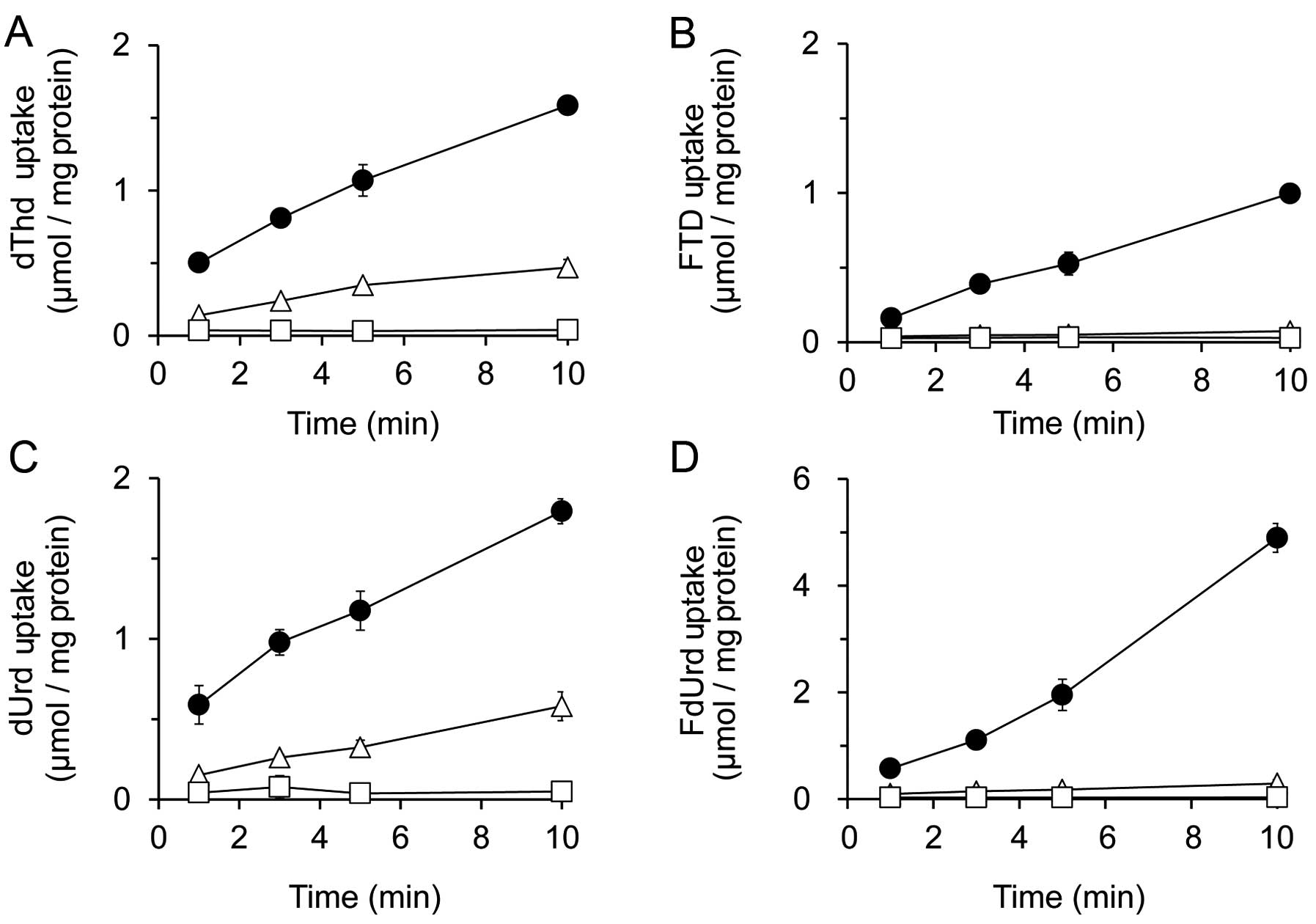
Intracellular uptake of dThd, FTD, dUrd
and FdUrd in HCT116 cells
To monitor the transport of FTD and FdUrd into the
cytoplasm and their intracellular uptake, the inhibitory effects of
the nucleoside transporter inhibitors 6-[(4-nitrobenzyl)
thio]-9-(β-D-ribofuranosyl) purine (NBMPR) and dipyridamole (DPM)
were analyzed and compared to the native substrates dThd and dUrd,
respectively (Fig. 3). The
concentration of inhibitors used was based on the concentration of
NBMPR required to inhibit ENT1, or that required for DPM inhibition
of both ENT1 and ENT2 (28).
After incubating HCT116 cells with dThd or FTD for
10 min, we found that the inhibitory effect of 1 μmol/l NBMPR was
less than that of 10 μmol/l DPM. For instance, the levels of
cellular dThd uptake at 10-min post-treatment with a vehicle
control, 1 μmol/l NBMPR, or 10 μmol/l DPM were 1.59±0.06, 0.47±0.06
or 0.04±0.01 nmol/mg protein, respectively, while those for FTD
uptake were 1.00±0.04, 0.07±0.01 and 0.03±0.02 nmol/mg protein,
respectively (Fig. 3A and B).
HCT116 cells were also incubated with dUrd and FdUrd
for 10 min, and inhibition by 1 μmol/l NBMPR was again lower than
that by 10 μmol/l DPM. The levels of dUrd uptake at 10-min
post-incubation with a vehicle control, 1 μmol/l NBMPR, or 10
μmol/l DPM were 1.80±0.08, 0.58±0.09 and 0.05±0.01 nmol/mg protein,
respectively, and those of FdUrd were 4.90±0.27, 0.29±0.07 and
0.03±0.00 nmol/mg protein, respectively (Fig. 3C and D). These results indicate
that all nucleosides tested in these experiments could be
recognized and transported into cells by both ENT1 and ENT2.
Analysis of TK1 affinities for dThd, FTD
dUrd and FdUrd
To evaluate the substrate specificity of FTD, dUrd
and FdUrd for TK1, recombinant human TK1 was incubated with
tritium-labeled substrates. The Km values determined for
each tested nucleoside revealed that dThd analogs had a higher
affinity for TK1 than for dUrd analogs (Table I). The Vmax values of
dThd-based compounds exceeded those of the dUrd-based compounds.
FTD and FdUrd showed higher affinities compared to the native form
of these nucleosides, namely, dThd and dUrd.
 | Table IThe affinity of TK1 for dThd, FTD,
dUrd and FdUrd. |
Table I
The affinity of TK1 for dThd, FTD,
dUrd and FdUrd.
| Substrate | Km
(μmol/l) | Vmax
(pmol/min/ng protein) | kcat
(min) |
kcat/Km
(x106/min·mol) |
|---|
| dThd | 1.16 | 0.70 | 17.78 | 15.28 |
| FTD | 2.34 | 0.93 | 10.26 | 19.26 |
| dUrd | 4.23 | 0.30 | 7.65 | 1.81 |
| FdUrd | 3.33 | 0.51 | 12.97 | 3.86 |
To evaluate the catalytic efficiency of TK1 in
phosphorylating dThd, FTD, dUrd and FdUrd,
kcat/Km ratios were calculated. The
kcat/Km values of dThd and its analog FTDs
tended to be higher than those of dUrd and its analog FdUrd
(Table I). TK1 showed higher
catalytic efficiency in phosphorylating FTD than it did with dThd,
and the catalytic efficiency of FTD phosphorylation by TK1 was
~4-fold higher than that of FdUrd.
Substrate specificity of DUT for the
triphosphate forms of dThd, FTD, dUrd and FdUrd
Pyrimidine triphosphate incorporation into DNA is
strictly regulated by DUT (29,30)
and high DUT expression is reported in various types of cancer,
suggesting that DUT may confer resistance to 5-fluoropyrimidines
(24,30). To determine whether FTD is
substrate of DUT, we performed enzymatic assays with recombinant
human DUT and either FdUTP or F3dTTP (Table II). Although DUT efficiently
converted dUTP and FdUTP to their monophosphate forms (dUMP and
FdUMP, respectively), dTTP and F3dTTP were not found to
be substrates for DUT.
| Table IISubstrate specificities of
triphosphate form of dThd, FTD, dUrd and FdUrd for DUT. |
Table II
Substrate specificities of
triphosphate form of dThd, FTD, dUrd and FdUrd for DUT.
| Substrate | Production of
5′-monophosphate (pmol/min/100 ng protein) |
|---|
| dTTP | N.D. |
|
F3dTTP | N.D. |
| dUTP | 41.4 |
| FdUTP | 34.8 |
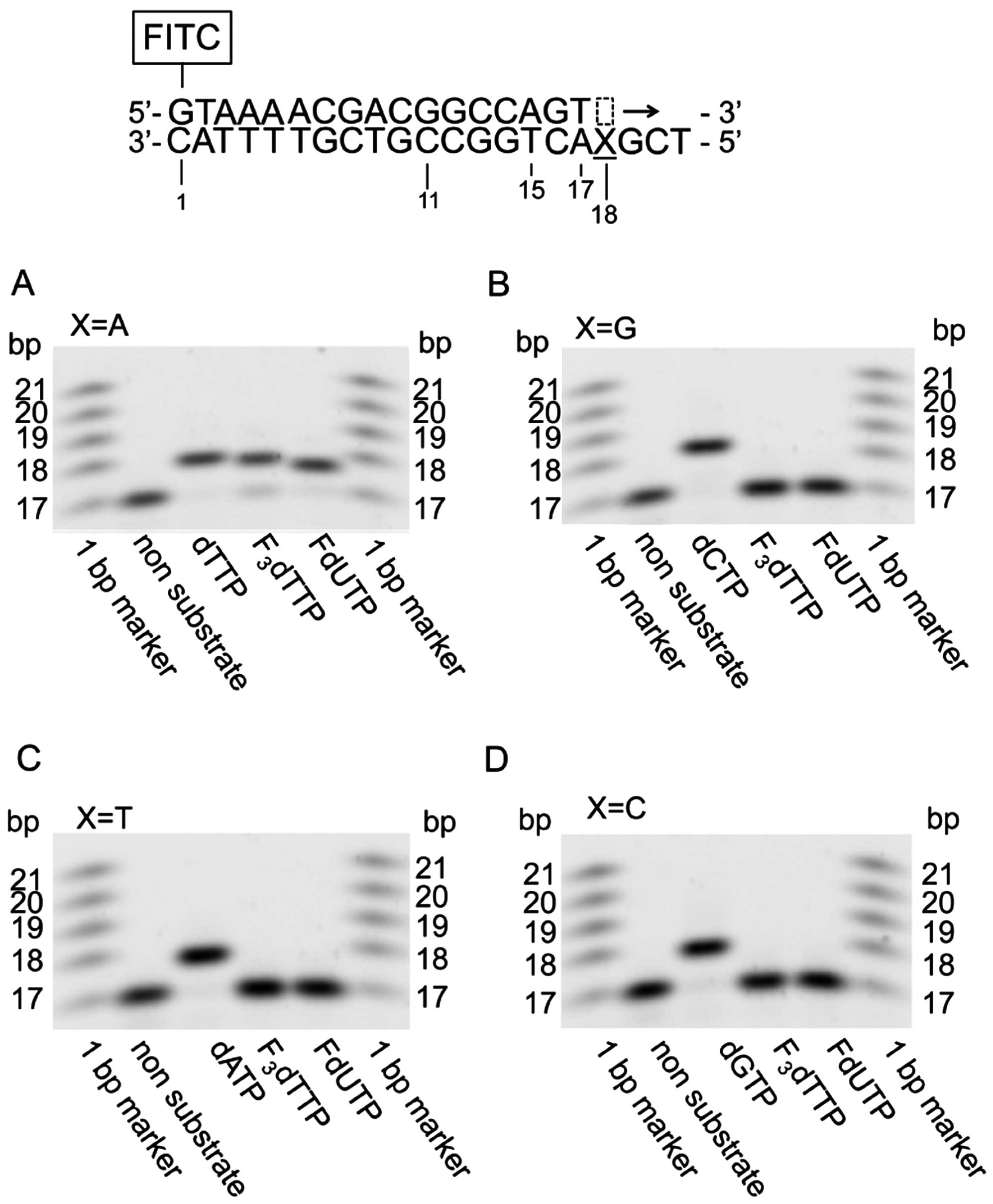
Substrate specificity of DNA polymerase α
for dThd, FTD, dUrd and FdUrd-triphosphate
Next, we investigated whether F3dTTP and
FdUTP serve as substrates for DNA polymerase α, the responsible
enzyme for catalyzing DNA replication. We analyzed extension
reactions by DNA polymerase α using dNTPs (as control substrates)
and the triphosphate forms of dUrd, FTD and FdUrd, and a synthetic
single-stranded DNA template (Fig.
4). F3dTTP and FdUTP, dThd and dUrd were
incorporated into the nascent strand at sites matching adenine on
the template strand, but not at guanine, cytosine or thymine
sites.
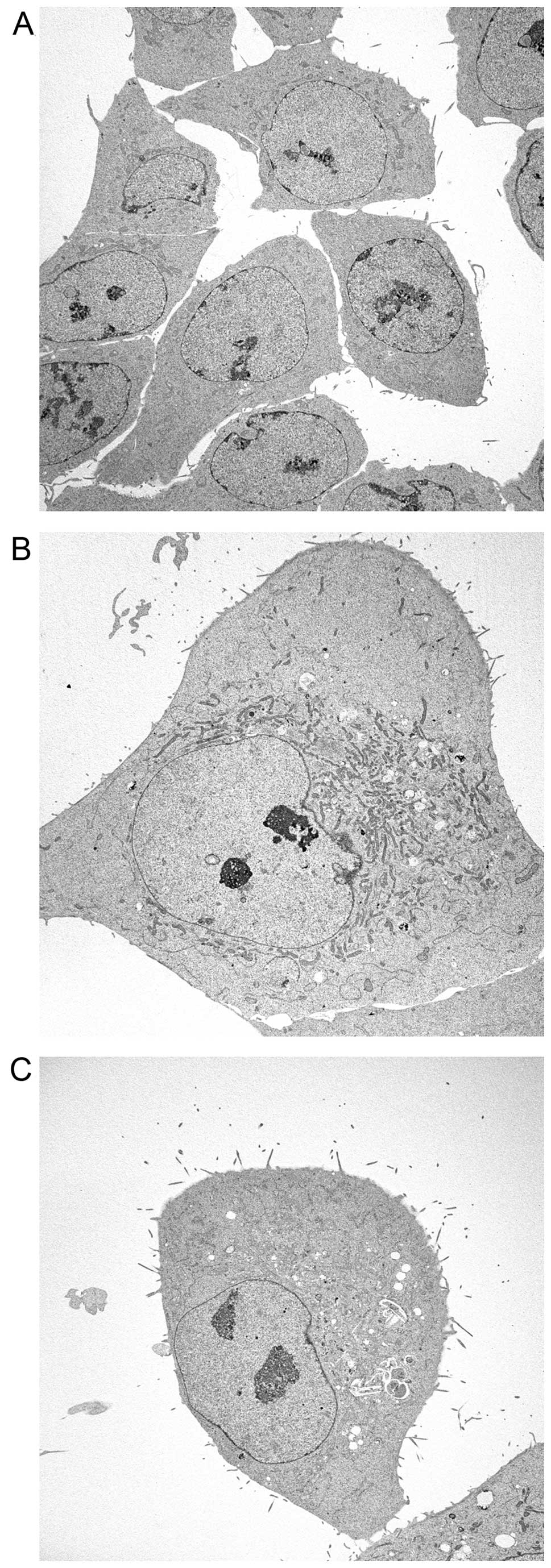
Morphological analyses of HCT116 cells
treated with FTD and FdUrd
Finally, to compare DNA incorporation data with
observable phenotypes, we performed morphological analyses of
HCT116 cells treated with FTD and FdUrd by electron microscopy
(Fig. 5). Swollen nuclei and
structural abnormalities of nucleoli were observed in both FTD- and
FdUrd-treated cells (relative to untreated control cells), with the
degree of alteration caused by FTD exposure much stronger than that
caused by FdUrd. Furthermore, the perinuclear heterochromatin
content was decreased by both FTD and FdUrd treatment.
Discussion
In the present study, we demonstrated that FTD is
incorporated into DNA with greater efficacy than is FdUrd (Fig. 6), which resulted from differences
in the affinity of TK1 and DUT for these nucleosides. These
differences may overcome resistance to FdUrd. The catalytic
efficiency of FTD phosphorylation by TK1 is 4-fold higher than that
of FdUrd and dUrd. These results imply that TK1 may recognize FTD
as dThd, while FdUrd may mimic dUrd. A similar phenomenon was
observed for the substrate specificity of DUT. The triphosphate
forms of both dUrd and FdUrd were degraded to their monophosphate
forms by DUT, while the triphosphate forms of dThd and FTD did not
serve as DUT substrates. As a result, FTD-triphosphate was not only
produced at a high level, but was also retained for a relatively
long duration in DNA (16),
compared with FdUrd. In contrast, the monophosphate form of FdUrd
may accumulate at a high level, and TS inhibition may be
potentiated by DUT.
Regarding the relationship between DNA incorporation
and cytotoxicity, we previously reported that one factor promoting
higher FTD incorporation than FdUrd incorporation is TS inhibition
(17). Indeed, FdUMP derived from
FdUrd irreversibly inhibits TS through the formation of a covalent
bond, whereas F3dTMP inhibits TS in a reversible manner.
Short-term TS inhibition by F3dTMP is not sufficient to
cause cytotoxicity, but may still deplete dTMP and dTTP, enabling
F3TTP to be incorporated into DNA at a higher level.
FTD is resistant to degradation by DUT, suggesting
that TAS-102 could be effective against tumors resistant to 5-FU
because of elevated DUT expression (31). In addition, several investigators
have shown that patients with high TK1 expression have a relatively
poor prognosis with some tumor types (32). These findings indicate that TK1
overexpression represents a cancer-specific pathway and that
TAS-102 treatment can improve the poor prognosis of metastatic
colorectal cancer patients who are refractory to standard
chemotherapies, including 5-FU.
From the viewpoint of FTD resistance, the
downregulation of nucleoside transporters and TK1 involved in the
dThd salvage pathway results in ineffective therapy against cancer
cells (23). These factors
indicate that FTD-resistant cells may rely on de novo dThd
synthesis as an alternative means of dThd production. A TS
inhibitor cocktail involving 5-fluoropyrimidines and/or
anti-folates may be a good choice in treating FTD-resistant cancer.
Modulating dThd synthesis by 5-FU and FTD could be an effective
cancer therapy.
The DNA repair system removes DNA damage sites from
the genome, making it one of the determinants of 5-fluoropyrimidine
efficacy (33). We could not
analyze FdUrd efflux from DNA because of its low level of DNA
incorporation. However, the elimination ratio of FTD was similar to
that observed with the natural substrate dThd. This phenomenon
implies that FTD is stably incorporated into DNA at the same level
as dThd. It is reported that uracil DNA glycosylases, thymine DNA
glycosylase, and the methyl-CpG binding domain 4 protein recognize
5-FU (but not FTD) and that they may contribute to FTD
incorporation into DNA. However, further studies should be
performed to confirm the associated FTD antitumor mechanisms at
play following DNA incorporation, including potential involvements
of DNA repair systems.
In conclusion, evidence presented here suggests that
TAS-102 consisting of FTD and TPI is the first dThd-based antitumor
agent whose main mechanism is the inhibition of DNA incorporation
itself. Further studies regarding mechanisms occurring after DNA
incorporation, as well as combination therapies with other
antitumor agents may be needed to improve targeted cancer
chemotherapy.
Acknowledgements
The authors are indebted to Professor Godefridus J.
Peters of the Department of Oncology of the VU University Medical
Center for providing scientific advice. We also thank Tokai
Electron Microscopy (Nagoya, Japan) for providing technical
assistance for the electron microscopy experiments. We would like
to thank Editage (www.editage.jp) for English language
editing.
Abbreviations:
|
5-FU
|
5-fluorouracil
|
|
DPM
|
dipyridamole
|
|
dThd
|
deoxythymidine
|
|
dUrd
|
deoxyuridine
|
|
dUMP
|
deoxyuridine-monophosphate
|
|
dUTP
|
deoxyuridine-triphosphate
|
|
DUT
|
deoxy UTPase
|
|
FTD
|
trifluridine
|
|
F3dTMP
|
FTD-monophosphate
|
|
F3dTTP
|
FTD-triphosphate
|
|
FITC
|
fluorescein isothiocyanate
|
|
FdUrd
|
2′-deoxy-5-fluorouridine
|
|
FdUMP
|
FdUrd-monophosphate
|
|
FdUTP
|
FdUrd-triphosphate
|
|
hENT
|
human equilibrative nucleoside
transporter
|
|
HPLC
|
high performance liquid
chromatography
|
|
LSC
|
liquid scintillation counting
|
|
NBMPR
|
6-[(4-nitrobenzyl)
thio]-9-(β-D-ribofuranosyl) purine
|
|
TK
|
dThd kinase
|
|
TS
|
thymidylate synthase
|
References
|
1
|
Hu J, Locasale JW, Bielas JH, O’Sullivan
J, Sheahan K, Cantley LC, Vander Heiden MG and Vitkup D:
Heterogeneity of tumor-induced gene expression changes in the human
metabolic network. Nat Biotechnol. 31:522–529. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weber G: Biochemical strategy of cancer
cells and the design of chemotherapy: G. H. A. Clowes Memorial
Lecture. Cancer Res. 43:3466–3492. 1983.PubMed/NCBI
|
|
3
|
Heidelberger C, Chaudhuri NK, Danneberg P,
Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E and
Scheiner J: Fluorinated pyrimidines, a new class of
tumour-inhibitory compounds. Nature. 179:663–666. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muggia FM, Peters GJ and Landolph JR Jr:
XIII International Charles Heidelberger Symposium and 50 Years of
Fluoropyrimidines in Cancer Therapy; September 6 to 8, 2007; New
York University Cancer Institute, Smilow Conference Center. Mol
Cancer Ther. 8. pp. 992–999. 2009
|
|
5
|
Johnston PG, Benson AB III, Catalano P,
Rao MS, O’Dwyer PJ and Allegra CJ: Thymidylate synthase protein
expression in primary colorectal cancer: Lack of correlation with
outcome and response to fluorouracil in metastatic disease sites. J
Clin Oncol. 21:815–819. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leichman CG: Predictive and prognostic
markers in gastrointestinal cancers. Curr Opin Oncol. 13:291–299.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shintani M, Urano M, Takakuwa Y, Kuroda M
and Kamoshida S: Immunohistochemical characterization of pyrimidine
synthetic enzymes, thymidine kinase-1 and thymidylate synthase, in
various types of cancer. Oncol Rep. 23:1345–1350. 2010.PubMed/NCBI
|
|
8
|
Sherley JL and Kelly TJ: Regulation of
human thymidine kinase during the cell cycle. J Biol Chem.
263:8350–8358. 1988.PubMed/NCBI
|
|
9
|
Arnér ES and Eriksson S: Mammalian
deoxyribonucleoside kinases. Pharmacol Ther. 67:155–186. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Heidelberger C, Parsons DG and Remy DC:
Syntheses of 5-trifluoromethyluracil and
5-trifluoromethyl-2′-Deoxyuridine. J Med Chem. 7:1–5. 1964.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ansfield FJ and Ramirez G: Phase I and II
studies of 2′-deoxy-5-(trifluoromethyl)-uridine (NSC-75520). Cancer
Chemother Rep. 55:205–208. 1971.PubMed/NCBI
|
|
12
|
Skevaki CL, Galani IE, Pararas MV,
Giannopoulou KP and Tsakris A: Treatment of viral conjunctivitis
with antiviral drugs. Drugs. 71:331–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fukushima M, Suzuki N, Emura T, Yano S,
Kazuno H, Tada Y, Yamada Y and Asao T: Structure and activity of
specific inhibitors of thymidine phosphorylase to potentiate the
function of antitumor 2′-deoxyribonucleosides. Biochem Pharmacol.
59:1227–1236. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Temmink OH, Emura T, de Bruin M, Fukushima
M and Peters GJ: Therapeutic potential of the dual-targeted TAS-102
formulation in the treatment of gastrointestinal malignancies.
Cancer Sci. 98:779–789. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoshino T, Mizunuma N, Yamazaki K, Nishina
T, Komatsu Y, Baba H, Tsuji A, Yamaguchi K, Muro K, Sugimoto N, et
al: TAS-102 monotherapy for pretreated metastatic colorectal
cancer: A double-blind, randomised, placebo-controlled phase 2
trial. Lancet Oncol. 13:993–1001. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Emura T, Nakagawa F, Fujioka A, Ohshimo H,
Yokogawa T, Okabe H and Kitazato K: An optimal dosing schedule for
a novel combination antimetabolite, TAS-102, based on its
intracellular metabolism and its incorporation into DNA. Int J Mol
Med. 13:249–255. 2004.PubMed/NCBI
|
|
17
|
Tanaka N, Sakamoto K, Okabe H, Fujioka A,
Yamamura K, Nakagawa F, Nagase H, Yokogawa T, Oguchi K, Ishida K,
et al: Repeated oral dosing of TAS-102 confers high trifluridine
incorporation into DNA and sustained antitumor activity in mouse
models. Oncol Rep. 32:2319–2326. 2014.PubMed/NCBI
|
|
18
|
Eckstein JW, Foster PG, Finer-Moore J,
Wataya Y and Santi DV: Mechanism-based inhibition of thymidylate
synthase by 5-(trifluoromethyl)-2′-deoxyuridine 5′-monophosphate.
Biochemistry. 33:15086–15094. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Temmink OH, Comijn EM, Fukushima M and
Peters GJ: Intracellular thymidylate synthase inhibition by
trifluorothymidine in FM3A cells. Nucleosides Nucleotides Nucleic
Acids. 23:1491–1494. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reyes P and Heidelberger C: Fluorinated
pyrimidines. XXVI. Mammalian thymidylate synthetase: Its mechanism
of action and inhibition by fluorinated nucleotides. Mol Pharmacol.
1:14–30. 1965.PubMed/NCBI
|
|
21
|
Belt JA and Noel LD: Isolation and
characterization of a mutant of L1210 murine leukemia deficient in
nitrobenzylthioinosine-insensitive nucleoside transport. J Biol
Chem. 263:13819–13822. 1988.PubMed/NCBI
|
|
22
|
Heidelberger C: Fluorinated pyrimidines.
Prog Nucleic Acid Res Mol Biol. 4:1–50. 1965.PubMed/NCBI
|
|
23
|
Temmink OH, Bijnsdorp IV, Prins HJ,
Losekoot N, Adema AD, Smid K, Honeywell RJ, Ylstra B, Eijk PP,
Fukushima M, et al: Trifluorothymidine resistance is associated
with decreased thymidine kinase and equilibrative nucleoside
transporter expression or increased secretory phospholipase A2. Mol
Cancer Ther. 9:1047–1057. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ladner RD, Lynch FJ, Groshen S, Xiong YP,
Sherrod A, Caradonna SJ, Stoehlmacher J and Lenz HJ: dUTP
nucleotido-hydrolase isoform expression in normal and neoplastic
tissues: Association with survival and response to 5-fluorouracil
in colorectal cancer. Cancer Res. 60:3493–3503. 2000.PubMed/NCBI
|
|
25
|
Temmink OH, de Bruin M, Comijn EM,
Fukushima M and Peters GJ: Determinants of trifluorothymidine
sensitivity and metabolism in colon and lung cancer cells.
Anticancer Drugs. 16:285–292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kobayashi D, Nozawa T, Imai K, Nezu J,
Tsuji A and Tamai I: Involvement of human organic anion
transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport
across intestinal apical membrane. J Pharmacol Exp Ther.
306:703–708. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Williams MV and Cheng Y: Human
deoxyuridine triphosphate nucleotidohydrolase. Purification and
characterization of the deoxyuridine triphosphate
nucleotidohydrolase from acute lymphocytic leukemia. J Biol Chem.
254:2897–2901. 1979.PubMed/NCBI
|
|
28
|
Ward JL, Sherali A, Mo ZP and Tse CM:
Kinetic and pharmacological properties of cloned human
equilibrative nucleoside transporters, ENT1 and ENT2, stably
expressed in nucleoside transporter-deficient PK15 cells. Ent2
exhibits a low affinity for guanosine and cytidine but a high
affinity for inosine. J Biol Chem. 275:8375–8381. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parsels LA, Parsels JD, Wagner LM, Loney
TL, Radany EH and Maybaum J: Mechanism and pharmacological
specificity of dUTPase-mediated protection from DNA damage and
cytotoxicity in human tumor cells. Cancer Chemother Pharmacol.
42:357–362. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wilson PM, LaBonte MJ, Lenz HJ, Mack PC
and Ladner RD: Inhibition of dUTPase induces synthetic lethality
with thymidylate synthase-targeted therapies in non-small cell lung
cancer. Mol Cancer Ther. 11:616–628. 2012. View Article : Google Scholar
|
|
31
|
Nobili S, Napoli C, Landini I, Morganti M,
Cianchi F, Valanzano R, Tonelli F, Cortesini C, Mazzei T and Mini
E: Identification of potential pharmacogenomic markers of clinical
efficacy of 5-fluorouracil in colorectal cancer. Int J Cancer.
128:1935–1945. 2011. View Article : Google Scholar
|
|
32
|
Aufderklamm S, Todenhöfer T, Gakis G,
Kruck S, Hennenlotter J, Stenzl A and Schwentner C: Thymidine
kinase and cancer monitoring. Cancer Lett. 316:6–10. 2012.
View Article : Google Scholar
|
|
33
|
Meyers M, Wagner MW, Hwang HS, Kinsella TJ
and Boothman DA: Role of the hMLH1 DNA mismatch repair protein in
fluoropyrimidine-mediated cell death and cell cycle responses.
Cancer Res. 61:5193–5201. 2001.PubMed/NCBI
|